

Abstract
Ras/MAPK signaling is often aberrantly activated in human cancers. The downstream effectors are transcription factors, including those encoded by the ETS gene family. Using cell-based assays and biophysical measurements, we have determined the mechanism by which Ras/MAPK signaling affects the function of Ets1 via phosphorylation of Thr38 and Ser41. These ERK2 phosphoacceptors lie within the unstructured N-terminal region of Ets1, immediately adjacent to the PNT domain. NMR spectroscopic analyses demonstrated that the PNT domain is a four-helix bundle (H2–H5), resembling the SAM domain, appended with two additional helices (H0–H1). Phosphorylation shifted a conformational equilibrium, displacing the dynamic helix H0 from the core bundle. The affinity of Ets1 for the TAZ1 (or CH1) domain of the coactivator CBP was enhanced 34-fold by phosphorylation, and this binding was sensitive to ionic strength. NMR-monitored titration experiments mapped the interaction surfaces of the TAZ1 domain and Ets1, the latter encompassing both the phosphoacceptors and PNT domain. Charge complementarity of these surfaces indicate that electrostatic forces act in concert with a conformational equilibrium to mediate phosphorylation effects. We conclude that the dynamic helical elements of Ets1, appended to a conserved structural core, constitute a phospho-switch that directs Ras/MAPK signaling to downstream changes in gene expression. This detailed structural and mechanistic information will guide strategies for targeting ETS proteins in human disease.
Keywords: MAP kinase, protein structure/dynamics, transcriptional regulation, protein-protein interaction, Ets2
The Ras/MAP kinase signaling pathway regulates cell proliferation. A large number of human tumors have genetic alterations in the Ras signaling pathway that drive constitutive signaling. In spite of this importance to human disease, no therapeutics currently disrupt the nuclear effectors of this pathway, which offer the potential for both tissue and disease specificity. Several known effectors belong to the ETS transcription factor family (1), but even in this case only a few specific target genes have been identified. Examples include the serum-response-element (SRE) of the c-Fos promoter, at which the ETS protein Elk1 plays a central role (2). Other Ras-responsive elements that have been characterized include tandem ETS or ETS/AP1 composite sites associated with Ets1 and Ets2 (3–5). In Drosophila, a genetic link between the ETS family and Ras/MAPK signaling implicates Pnt-P2, the ortholog of vertebrate Ets1 and Ets2, and Yan, the apparent ortholog of vertebrate Tel (6). Phosphorylation affects nuclear export (Yan) (7) and CBP (CREB-binding protein)/p300 recruitment (Elk1, Ets1, and Ets2) (2, 8, 9). Ets1 bears one characterized MAP kinase ERK2 phosphoacceptor, Thr38, found in an unstructured region N-terminal to its structured PNT domain (10–12). Although both these regions are critical for Ras/MAPK-dependent enhanced transcriptional regulation (8), the molecular mechanism of this signaling pathway has not been elucidated.
Here we describe the coupled roles of the PNT domain and an adjacent disordered region bearing both Thr38 and a previously unrecognized phosphoacceptor, Ser41, in the ERK2-enhanced binding of the CBP TAZ1 domain by Ets1. Phosphorylation plays a dual role by altering a conformational equilibrium of a dynamic helix H0 that tethers the flexible phosphorylated region of Ets1 to the core PNT domain and by augmenting electrostatic forces that drive TAZ1 recognition. In this integrated model, structured elements with intrinsic flexibility provide a scaffold for responsiveness to a signaling pathway. The overall mechanism represents an evolutionary development within a gene family, whereby dynamic elements are appended to conserved core folds to increase the capacity for biological regulation.
Results
ERK2 Phosphoacceptors Thr38 and Ser41 Contribute to Ets1 Transactivation.
To investigate the mechanism by which phosphorylation affects the function of Ets1, we developed an in vitro system using ERK2 to covalently modify purified Ets1. Phosphorylation of both the known ERK2 acceptor site, Thr38, and a previously unrecognized nonconsensus site, Ser41, was detected by mass spectrometry and 31P-NMR experiments (13). The in vivo relevance of phosphorylation was tested in a cell-based assay for Ras/MAPK enhancement of Ets1 activity by the coexpression of a constitutively active form of MEK1 (11). Mutation of both phosphoacceptor sites was necessary to reduce relative luciferase activity (RLA) to vector-only control levels (Fig. 1). To provide better quantification through replica experiments, we measured superactivation as defined by (RLAWT or mutant)/(RLAempty vector) in the presence of MEK1. Wild-type Ets1 caused two- to threefold (2.4 ± 0.1) superactivation. Mutation of Thr38 or Ser41 to alanine partially reduced the effect (1.2 ± 0.08 and 1.4 ± 0.09, respectively), whereas Ets1 with a double phosphoacceptor site mutation displayed no superactivation (1.1 ± 0.06). Substitution of glutamic acid residues at positions 38 and 41 could only partially reconstitute the effects of phosphorylation (1.4 ± 0.2). Phosphorylation of Thr38 in vivo was confirmed for wild-type and the S41A mutant with phospho-specific antibodies (Fig. 1). Thus, the full effect of the signaling required the two phosphoacceptors.
Fig. 1.

Ras/MAPK signaling enhances Ets1 transactivation. NIH 3T3 cells were transfected with an ETS site-driven firefly luciferase reporter, an expression vector for FLAG-tagged Ets1 (wild type or the indicated mutant), and either an expression vector for constitutively active MEK1 (+) or an empty vector control (-). In addition, Renilla luciferase control vector was included to provide RLA. The bar graph (Upper) shows a representative experiment with RLA as mean and standard deviation for triplicate transfections. MEK1-dependent activation, observed in the absence of transfected Ets1, is possibly due to ERK2 effects on other elements of the transcription machinery (35). Gel panels are immunoblots showing Ets1 species from transfected cells, which were immunoprecipitated with FLAG-specific antibodies, then probed by immunoblotting with pThr38-Ets1 (Upper) or Ets1 (Lower) specific antibodies. Dash, 50 kDa marker. Additional expression controls are shown in Fig. S8G.
Phosphorylation Increases the Affinity of the Ets1 PNT Domain for the CBP TAZ1 Domain.
The CBP interaction domain(s) for binding to Ets1 and Ets2 was mapped by pull-down assays with GST-tagged CBP fragments. An ∼50 kDa N-terminal fragment of CBP, which contains three transcription factor interaction domains, displayed binding and was subjected to further analysis (Fig. S1). The TAZ1 domain-bearing fragments bound full length Ets1, as previously shown in cell extracts (14, 15). Furthermore, the deletion mutants Ets11-138 and the analogous Ets21-172, which contain PNT domains, also bound the TAZ1 domain. In contrast, the isolated N-terminal domain of CBP, which binds nuclear receptors, and the KIX domain, best known for CREB binding, did not measurably associate with Ets1. The phosphorylation effect on the interaction of Ets11-138 with the isolated TAZ1 domain was quantified by isothermal titration calorimetry (ITC). The affinity of unmodified Ets11-138 for TAZ1 (Kd = 58 ± 12 μM) was enhanced 34 ± 1-fold upon phosphorylation.
Ets1 PNT Domain Structure Couples Dynamic Phosphoacceptors and Helix H0 to a Core Helical Bundle.
To develop a mechanistic model for Ets1/CBP binding, we undertook additional biochemical and biophysical approaches. A high-resolution structural ensemble of Ets129-138 was determined from extensive NMR spectroscopically derived dihedral angle, distance, and orientational restraints via a semiautomated ARIA/CNS protocol (16) (Fig. 2A and Table S1). In addition to a four-helix bundle (H2–H5) with a SAM domain fold (Fig. S2A), a predominantly polar helix H1 (residues 54–62) is well positioned along helices H2 and H5 via packing of Phe53 and Phe56 with Trp80, His128, and Ile131. More importantly, the refined model demonstrated an Ets1 PNT domain-specific helix, designated H0 (residues 42–52), which is also present in Ets2 (Fig. S2). This amphipathic helix packs against helix H5 and the end of helix H2 via hydrophobic interactions involving Met44, Met45, and Leu49 with Phe88, Phe120, and Ile124, as well as potential salt bridges between Lys42-Asp123 and Lys50-Glu127. Helix H0 is preceded by an unstructured region (residues 29–41) bearing the phosphoacceptors Thr38 and Ser41.
Fig. 2.

Superimposed structural ensembles of (A Right) Ets129-138 and (B) 2P-Ets129-138 showing the helical bundle PNT domain preceded by a flexible region containing the ERK2 phosphoacceptors Thr38 and Ser41. The α-helices (H0, residues 42–52; H1, 54–62; H2, 74–87; H3, 102–107; H4, 109–116; H5, 119–134) and a 310-helix (H2′, 95–98) are identified by rainbow coloring, and Thr38 (cyan) and Ser41 (blue) phosphates as spheres. Disordered residues 29–37 are omitted for clarity. (A Left) Expanded view of the interface between the dynamic helix H0 and helices H2 and H5 of the Ets129-138 core PNT domain, colored by helix identity. Dashes show potential salt bridges (K42-D123, K50-E127). Residues examined by mutation are underlined (see Fig. 4 and Fig. S8). (C) The phospho-switch model for the interaction of Ets1 and CBP. The PNT domain exists in a conformational equilibrium with the dynamic helix H0 in closed and open states (as well as being frayed or fully unfolded; not drawn). Phosphorylation shifts the population distribution to the open state, which is favored for TAZ1 binding via complementary electrostatic interactions. (D) This model is supported by colinear, population-weighted amide chemical shifts of residues at the helix H0/H1 junction (Gly55) and the H0 interface along H5 (Leu125 and His128) in superimposed spectra of a series of deletion fragments. The shifts of 2P-Ets11-138 (magenta) are intermediate between those of the Ets11-138 (black) and Ets129-138 (green), which are preferentially closed, and those of Ets151-138 (red), which lacks H0 and thus models fully open. The shifts of Gly55 indicate that Ets142-138 (blue) may adopt an even more closed state (see Figs. S5 and S8 for details).
Although clearly defined in the structural ensemble of Ets129-138, helix H0 is marginally stable and conformationally dynamic by several criteria. Using the algorithm SSP (secondary structure propensity) to predict secondary structure from main chain chemical shifts (17), H0 exhibits helical scores less than those of the core PNT domain (Fig S3A). 15N relaxation measurements confirmed that the backbone of helix H0 is mobile on the sub-nsec time scale (Fig. S3 B–E). In addition, residues in this helix undergo facile amide hydrogen exchange (HX), exhibiting protection factors of only ∼2 relative to a corresponding random coil polypeptide (Fig. S3F). This behavior, indicative of extensive local unfolding, was supported by partial proteolysis measurements. Cleavage at Lys42, Lys50, and Arg62 within helices H0 and H1 revealed at least transient accessibility of these residues to trypsin, whereas lysines/arginines within the core PNT domain were resistant to proteolysis under the same conditions (Fig. S4 A and B).
Phosphorylation Shifts a Conformational Equilibrium Involving Helix H0.
The structural ensemble of dually phosphorylated 2P-Ets129-138 was also determined by NMR methods (Fig. 2B and Table S1). The core PNT domain and helix H1 superimpose closely on that of the unmodified protein. In contrast, although residues 42–52 continue to form helix H0 as evidenced by chemical shifts (Fig. S3A) and sequential main-chain NOE interactions, the position of this helix is no longer restrained by long-range NOE interactions (Fig. S4D). Rather, helix H0 adopts a broad distribution, extending roughly outward from helix H1 and away from the core PNT domain. This distribution is not experimentally unrestricted, but limited by short-range NOE- and chemical shift-derived restraints around the H0/H1 junction and by 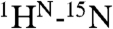 and
and 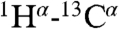 residual dipolar couplings. This creates an open conformation that contrasts to the more compact, closed conformation observed in the unmodified Ets129-138 ensemble. Helix H0 has similar SSP scores in Ets129-138 and 2P-Ets129-138, yet residues at the H0/H1 junction have slightly higher scores in the phosphorylated species (Fig. S3A). As indicated by CD measurements (Fig. S4C), this may reflect an increased average helical character of these residues, possibly forming a more continuous H0-H1 helix. The protection factors against HX increased slightly (∼2- to 5-fold) for amides near pThr38 and pSer41, including those at the N terminus of helix H0 (Fig. S3 F and G). This is suggestive of modest local stabilization of this dynamic helix, as also detected by its reduced sensitivity to proteolysis (Fig. S4B). In contrast, the sequences preceding helix H0, including Thr38 and Ser41, remain disordered after phosphorylation as evidenced by chemical shift, 15N relaxation, and HX measurements (Fig. S3).
residual dipolar couplings. This creates an open conformation that contrasts to the more compact, closed conformation observed in the unmodified Ets129-138 ensemble. Helix H0 has similar SSP scores in Ets129-138 and 2P-Ets129-138, yet residues at the H0/H1 junction have slightly higher scores in the phosphorylated species (Fig. S3A). As indicated by CD measurements (Fig. S4C), this may reflect an increased average helical character of these residues, possibly forming a more continuous H0-H1 helix. The protection factors against HX increased slightly (∼2- to 5-fold) for amides near pThr38 and pSer41, including those at the N terminus of helix H0 (Fig. S3 F and G). This is suggestive of modest local stabilization of this dynamic helix, as also detected by its reduced sensitivity to proteolysis (Fig. S4B). In contrast, the sequences preceding helix H0, including Thr38 and Ser41, remain disordered after phosphorylation as evidenced by chemical shift, 15N relaxation, and HX measurements (Fig. S3).
The NMR-derived ensembles of Ets129-138 and 2P-Ets129-138, as well as changes observed in chemical shifts, HX, protease sensitivity, and CD molar ellipticity, indicated that a substantial conformational transition accompanies phosphorylation. However, in recognizing that helix H0 is dynamic even in the absence of modification, we hypothesized that Ets129-138 also exhibits a conformational equilibrium between these closed and open states (Fig. 2C). To explore this model, PNT-domain-bearing fragments of Ets1 missing the phosphoacceptors (Ets142-138) or the entire H0 (Ets151-138) were also analyzed by NMR. Comparison of their 15N-HSQC spectra to those of Ets11-138 and 2P-Ets11-138 revealed a striking pattern of colinear chemical shift perturbations. This is exemplified by the amide signals of Gly55, at the H0/H1 junction, and His128 and Leu125, which lie at the interface of helix H0 and the core PNT domain (Fig. 2D and Fig. S5). Colinear chemical shift changes are a signature of a fast conformational equilibrium between two states, with the intermediate chemical shift of a given species representing its population-weighted average of these states. Thus, we propose that phosphorylation shifts the population distribution toward the state with helix H0 displaced from the core PNT domain, creating an open conformation necessary for the interface with CBP (Fig. 2C).
NMR Titrations Map the TAZ1/Ets129-138 Interface.
To dissect the molecular bases for the recruitment of CBP by Ets1, 15N- and 13C-HSQC monitored titrations were used to detect spectral changes, reflective of structural perturbations, accompanying the interaction of TAZ1 with Ets11-138 and 2P-Ets11-138. Upon addition of unlabeled TAZ1, a subset of amide, methyl, and aromatic groups showed a progressive loss of 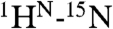 and 1H-13C peak intensity from the free protein, without the concomitant appearance of any new signals from the resulting complex (Fig. S6). Even under near saturating conditions, achieved in the case of 2P-Ets11-138, peaks were not recovered. This behavior, which is reported in other cases such as the transactivation region of p53 interacting with CBP domains (18), is indicative of intermediate time scale exchange broadening between multiple conformations within the higher molecular mass complex and confirms that the two protein domains bind specifically. The residues showing the most pronounced loss in intensity were mapped onto the structural ensembles of Ets129-138 and 2P-Ets129-138 (Fig. 3A and Fig. S6). Similar amides, including (p)Thr38 and (p)Ser41, were perturbed in the phosphorylated vs. unmodified Ets1 fragment, yet to a greater degree in the former at the same molar ratio of TAZ1. This could be due to the increased affinity of TAZ1 for the phosphorylated Ets1 fragment, as well as possible additional contacts. The altered residues cluster within a region of the PNT domain where the helices H0, H2, and H5 converge in the closed conformation, yet are exposed in the open state. We conclude that this region functions in binding TAZ1 and propose that the most perturbed residues directly contribute to the intermolecular interface.
and 1H-13C peak intensity from the free protein, without the concomitant appearance of any new signals from the resulting complex (Fig. S6). Even under near saturating conditions, achieved in the case of 2P-Ets11-138, peaks were not recovered. This behavior, which is reported in other cases such as the transactivation region of p53 interacting with CBP domains (18), is indicative of intermediate time scale exchange broadening between multiple conformations within the higher molecular mass complex and confirms that the two protein domains bind specifically. The residues showing the most pronounced loss in intensity were mapped onto the structural ensembles of Ets129-138 and 2P-Ets129-138 (Fig. 3A and Fig. S6). Similar amides, including (p)Thr38 and (p)Ser41, were perturbed in the phosphorylated vs. unmodified Ets1 fragment, yet to a greater degree in the former at the same molar ratio of TAZ1. This could be due to the increased affinity of TAZ1 for the phosphorylated Ets1 fragment, as well as possible additional contacts. The altered residues cluster within a region of the PNT domain where the helices H0, H2, and H5 converge in the closed conformation, yet are exposed in the open state. We conclude that this region functions in binding TAZ1 and propose that the most perturbed residues directly contribute to the intermolecular interface.
Fig. 3.

Identification of the Ets1/CBP binding interface. (Left) Residues in 2P-Ets11-138 and TAZ1 showing large 15N-HSQC intensity reductions in the presence of equimolar TAZ1 or 2P-Ets11-138, respectively (∼90% saturation; Figs. S6 and S7), are mapped on ribbon diagrams of low-energy members of the (A) 2P-Ets129-138 (representing 2P-Ets11-138) and (B) TAZ1 (PDB ID code 1u2n) structural ensembles. Increasing cyan sphere size corresponds to greater signal loss upon complex formation (intensity ratios: 0 < large sphere < 0.05, 0.05 < medium < 0.10, 0.10 < small < 0.15), and qualitatively defines the binding interface between the Ets1 and CBP fragments. Zn+2 ions, magenta balls. (Center) Surface representations of 2P-Ets129-138 and TAZ1 with residues shown as spheres now identified by physicochemical properties (red, Asp, Glu; blue, Arg, Lys, His; yellow, neutral polar; green, hydrophobic). Residues examined by mutation (Fig. 4 and Fig. S8) are underlined. (Right) Electrostatic surfaces of 2P-Ets129-138 and TAZ1, calculated with MolMol, are complementary (default parameters with “simple charge”; red, negative; blue, positive).
Complementary 15N-HSQC monitored titrations of 15N-labeled TAZ1 were carried out with unlabeled Ets11-138 and 2P-Ets11-138 (Fig. 3B and Fig. S7). Again, a progressive loss of signal intensity from selected amides in TAZ1 resulted upon addition of either Ets1 species, confirming specific binding. Consistent with the higher affinity of TAZ1 for phosphorylated Ets1, similar, yet more pronounced, spectral changes occurred in the presence of an equimolar amount of 2P-Ets11-138 relative to Ets11-138. Mapping the residues showing the largest amide intensity changes on the structure of TAZ1 revealed that the binding interface encompasses an extended region, including primarily helices H1, H3, and H4.
The interaction surfaces identified by NMR titrations are net negatively charged for Ets1 and net positively charged for TAZ1 (Fig. 3), suggesting that binding is driven electrostatically. To test this hypothesis, the interaction detected by NMR was challenged using buffers containing 20 to 500 mM NaCl. Consistent with a role for electrostatic forces, binding was disrupted with increasing ionic strength (Fig. S6B).
Role of Newly Identified Structural Components in MAPK Signaling.
A subset of residues that showed NMR spectral changes due to phosphorylation or TAZ1 association was tested for a role in responsiveness to MAPK signaling in cell-based transcription assays (Fig. 4 and Fig. S8). The hypothesized role of electrostatic forces in binding implicates several Ets1 residues in CBP recognition. In support of this hypothesis, negatively charged Asp119, Asp123, or Glu127, lying along helix H5, gave severe reductions in superactivation when mutated to arginines, but not alanines. Mutation of an interior residue, Trp126, showed no effect. The proposed conformational change of the PNT domain predicts that residues packed in the folded state might also be mutation sensitive. Indeed, mutation of Leu49, which is buried in the H0–H5 interface of this closed state, but exposed in the open conformation, to a glutamate or arginine abrogated MAPK induction. Likewise, mutations of Thr52, Phe53, and Ser54, which are located near the H0-H1 junction, reduced superactivation. In sum, these disrupted phenotypes, in combination with controls demonstrating proper folding, in vivo expression, and phosphorylation, as well as reduced TAZ1 binding in vitro (Fig. S8), indicate that these residues play a role, either directly at the interface or indirectly via helix H0 packing, in complex formation. In conclusion, these functional assays corroborate the structural and mechanistic model of the Ets1/CBP regulated interaction.
Fig. 4.

Residues of Ets1 functional in transcriptional superactivation. Superactivation (RLAWT or mutant)/(RLAempty vector) is reported as mean values for three to six independent experiments ± the standard error of the mean, except mutants W126A and D123A, which had two replicates. Fig. S8 presents controls for expression levels, phosphorylation state of Thr38, and folding competence (L49R, D123R, and E127R only). Structural environments of the residues are shown in Fig. 2.
Discussion
We have described how phosphorylation enhances the constitutive binding of Ets1 to CBP. Our mechanistic findings build upon the refined structure of Ets129-138, in which two helices tether a flexible region containing the phosphoacceptors to the core PNT domain. We found that ERK2 phosphorylation increases the binding affinity of Ets11-138 for the CBP TAZ1 domain by shifting a conformational equilibrium toward a proposed binding-competent open state and by augmenting the electrostatic complementarity of the Ets1/CBP interface. The general principles of this phospho-switch illustrate evolution of signaling-dependent regulation.
Role of Protein Dynamics in Phosphorylation-Regulated Ets1/CBP Interaction.
Phosphorylation-enhanced binding relies on two flexible components that contrast with the structured, conserved core PNT domain. First, Thr38 and Ser41 are conformationally disordered, even when posttranslationally modified. Second, helix H0 is only marginally stable undergoing substantial conformational fluctuations to locally frayed or unfolded state(s). Upon phosphorylation, helix H0 is slightly stabilized locally, yet adopts a broad distribution of positions displaced from the core PNT. We propose that the unmodified protein also samples these conformations. This leads to the regulatory mechanism in which phosphorylation shifts the population distribution of a conformational equilibrium between closed and open states of the PNT domain. We speculate that the displacement of helix H0 to the open state is favored by phosphorylation due to the proximity of (p)Thr38 and (p)Ser41 to numerous charged aspartate and glutamate residues in helices H2 and H5.
Role of Electrostatics in Phosphorylation-Regulated Ets1/CBP Interaction.
Based on the patterns of intensity changes in NMR-monitored titrations, a net negative-charged surface of the PNT domain of Ets1 interacts with a positive-charged region of TAZ1. Phosphorylation increases the negative charge of Ets1, thus reinforcing a complimentary electrostatic binding interface for TAZ1. This binding mechanism is supported by mutational analyses, in which addition of arginines to Ets1 at the TAZ1 interface disrupted ERK2-dependent transcriptional activation and shifted the conformational equilibrium, as well as by the ionic strength sensitivity of the interaction. In sum, MAPK phosphorylation promotes CBP binding to Ets1 via electrostatic effects combined with a conformational transition involving dynamic elements of the PNT domain.
Distinct Features of Ets1/CBP Binding.
CBP and the closely related p300 are widely used coactivators, reputed to associate with over 300 DNA binding transcription factors (19, 20). The involvement of a prestructured module (the core PNT domain) in combination with a flexible, yet structured region (helix H0) distinguishes the CBP/Ets1 interaction from other known CBP complexes. In this report mutational studies and NMR titrations demonstrate a mobile, folded helix in conjunction with a highly stable structured domain provides the CBP macromolecular interface. Previous structural studies demonstrated that disordered peptides corresponding to the negatively charged transactivation domains of several transcription factors, including HIF1α, CITED2, STAT1/2, and p53 fold as induced helices, wrapping around the extended binding clefts of the TAZ1 and TAZ2 domains (21–26). Despite these differences, the TAZ1 residues mapped as contacts for Ets1 overlap with those involved in binding these other transcriptions factors.
The affinities of known phosphorylation-regulated CBP interactions including phosphorylated Ets11-138 for TAZ1 (Kd = 1.7 μM, this paper), KIX binding to phosphorylated KID (Kd = 10 μM) (27) and the phosphorylated p53 binding to TAZ2 (Kd = 17.6 μM) (26) are relatively weak in comparison with HIF1α binding to TAZ1 with Kd = 10 nM (24). This suggests that regulated macromolecular interfaces function best through weaker interactions that enable responsiveness to signaling pathways.
ETS Family PNT Domains and Cellular Signaling.
The PNT domain, which is present in ∼1/3 of ETS proteins, displays close structural similarity to the SAM domain, a widespread protein-protein and -RNA interaction module (28) (Fig. S2). Ets1, Ets2, and their Drosophila ortholog Pnt-P2, as well as GABPα and SPDEF share an appended N-terminal helix H1. The highly similar proteins Ets1 and Ets2 also bear the dynamic helix H0 (29). These helices are preceded with conserved spacing by an ERK2 consensus site only in Ets1, Ets2, and Pnt-P2. An ERK2 docking site is located on the Ets1 and Ets2 PNT domains (11, 12). The conformational flexibility of helix H0 may contribute to the accessibility of Thr38 and Ser41 to the catalytic site of ERK2 while the PNT domain is docked at an ancillary site on the enzyme (30). We conclude that the conservation in spacing between the core PNT domain and phosphoacceptors sites is likely explained by the role of helix H0 in both CBP binding and potentially ERK2 docking. Furthermore, unique regulation of Ets1, Ets2, and Pnt-P2 is enabled by the two helices appended to the core PNT domain.
The addition of helices N-terminal to the PNT domain provides functional diversity to ETS family members. The PNT domain of GABPα, which binds TAZ2, but not TAZ1, contains only the analogous helix H1 and shows no regulation by phosphorylation (31). However, this ETS protein has a structured OST domain that also contributes to CBP binding via TAZ1 and TAZ2 (31). In the cases of vertebrate Tel and Drosophila Yan, the core PNT domain displays homopolymerization, which is implicated in transcriptional repression (32, 33). Thus, the broader ETS family illustrates additional ways in which structural extensions of the core PNT domain provide routes to specific biological regulation.
Materials and Methods
The experimental procedures are provided in detail as SI Materials and Methods.
In Vivo Transcription Assays.
Transient expression assays and expression controls (Fig. S8G) were performed in NIH3T3 mouse fibroblasts as previously described (8). Superactivation levels (RLAWT or mutant)/(RLAempty vector) in the presence of active MEK1 presented in the text were derived from three to six replica experiments, and except otherwise noted, presented as mean ± standard error of the mean.
Protein Purification.
Unlabeled and uniformly 15N- or 15N/13C-labeled murine Ets1 constructs were prepared as described previously (10, 11, 13, 29). Ets11-138 and Ets129-138 were phosphorylated in vitro using 50 mM ATP and a 1∶20 molar ratio of ERK2∶Ets1 (8). Phosphorylation states were verified by electrospray ionization mass spectrometry (ESI-MS). The gene encoding TAZ1 (CBP340–439) was cloned from the murine CBP gene (GenBank release no. 70995311) into the pET28b (Novagen) vector for expression as a His-tagged protein in Escherichia coli BL21(λDE3) cells (31).
Isothermal Titration Calorimetry.
ITC experiments were conducted on a VP-ITC Microcalorimeter, MicroCal LLC. To block reactive cysteines, Ets11-138, S26A and 2P-Ets11-138, S26A were treated with 50 mM iodoacetamide at pH 8.0 and 25 °C for 1 hr, followed by quenching with 150 mM DTT at 25 °C for 30 min. Mass spectrometry (ESI-MS Quatro-II) revealed three acetamide modifications, on four possible cysteines, in both species. Kd values (mean and standard deviations of four to five measurements) were 58 ± 12 μM (Ets11-138, S26A) and 1.7 ± 1.5 μM (2P-Ets11-138, S26A).
NMR Spectroscopy.
NMR spectra of the Ets1 and CBP constructs were recorded using Varian 500 MHz Unity and 600 MHz Inova spectrometers. Resonance assignments were obtained via standard heteronuclear correlation experiments. NMR-monitored titrations were carried out by recording HSQC spectra of ∼0.1 mM-labeled protein in 20–50 mM Tris pH 7.0, 20 mM NaCl, 2 mM DTT, and ∼10% D2O at 25 °C, to which the unlabeled protein partner (∼1.2–2.8 mM stock solution in the same buffer) was added in small aliquots. Rapid amide exchange rates at 30 °C and pH 6.0 were determined for 15N-labeled Ets129-138 and 2P-Ets129-138 by the CLEANEX-PM method (34). The chemical shifts of Ets129-138 [BioMagResBank (BMRB) accession code 4205] and 2P-Ets129-138 (BMRB accession code 16426) have been deposited in the Biological Magnetic Resonance Data Bank.
Structure Calculations.
Structure calculations were performed using extensive NMR-derived distance, dihedral angle, and orientation restraints (Table S1). The atomic coordinates of the Ets129-138 (PDB ID code 2jv3) and 2P-Ets129-138 (PDB ID code 2kmd) ensembles have been deposited in the Protein Data Bank.
Supplementary Material
Acknowledgments.
We thank Martin Blackledge, H. Jerome Coyne III, Charles Foulds, David Goldenberg, Ira Kraft, Mark Okon, and Manuela Schärpf for advice and help. This research was supported by grants from the National Institutes of Health, R01GM38663 (to B.J.G.), T32GM08537 (training grant support to M.L.N.), and P30CA42014 (to the Huntsman Cancer Institute for support of core facilities), and the Canadian Cancer Society (to L.P.M.). BJG acknowledges funding from the Huntsman Cancer Institute/Huntsman Cancer Foundation. Instrument support was provided by the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, the British Columbia Knowledge Development Fund, the UBC Blusson Fund, and the Michael Smith Foundation for Health Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2jv3 and 2kmd). The NMR chemical shifts and restraints have been deposited in the BioMagResBank, www.bmrb.wisc.edu (accession nos. 4205 and 16426).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0915137107/-/DCSupplemental.
References
- 1.Tootle TL, Rebay I. Post-translational modifications influence transcription factor activity: A view from the ETS superfamily. Bioessays. 2005;27:285–298. doi: 10.1002/bies.20198. [DOI] [PubMed] [Google Scholar]
- 2.Li QJ, et al. MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300. EMBO J. 2003;22:281–291. doi: 10.1093/emboj/cdg028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassuk AG, Leiden JM. A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity. 1995;3:223–237. doi: 10.1016/1074-7613(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 4.Stacey KJ, Fowles LF, Colman MS, Ostrowski MC, Hume DA. Regulation of urokinase-type plasminogen activator gene transcription by macrophage colony-stimulating factor. Mol Cell Biol. 1995;15:3430–3441. doi: 10.1128/mcb.15.6.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang BS, et al. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol. 1996;16:538–547. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill EM, Rebay I, Tjian R, Rubin GM. The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell. 1994;78:137–147. doi: 10.1016/0092-8674(94)90580-0. [DOI] [PubMed] [Google Scholar]
- 7.Tootle TL, Lee PS, Rebay I. CRM1-mediated nuclear export and regulated activity of the Receptor Tyrosine Kinase antagonist YAN require specific interactions with MAE. Development. 2003;130:845–857. doi: 10.1242/dev.00312. [DOI] [PubMed] [Google Scholar]
- 8.Foulds CE, Nelson ML, Blaszczak AG, Graves BJ. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol. 2004;24:10954–10964. doi: 10.1128/MCB.24.24.10954-10964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao F, et al. Mae inhibits Pointed-P2 transcriptional activity by blocking its MAPK docking site. EMBO J. 2006;25:70–79. doi: 10.1038/sj.emboj.7600924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slupsky CM, et al. Structure of the Ets-1 pointed domain and mitogen-activated protein kinase phosphorylation site. Proc Natl Acad Sci USA. 1998;95:12129–12134. doi: 10.1073/pnas.95.21.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seidel JJ, Graves BJ. An ERK2 docking site in the Pointed domain distinguishes a subset of ETS transcription factors. Genes Dev. 2002;16:127–137. doi: 10.1101/gad.950902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waas WF, Dalby KN. Transient protein-protein interactions and a random-ordered kinetic mechanism for the phosphorylation of a transcription factor by extracellular-regulated protein kinase 2. J Biol Chem. 2002;277:12532–12540. doi: 10.1074/jbc.M110523200. [DOI] [PubMed] [Google Scholar]
- 13.McIntosh LP, et al. Detection and assignment of phosphoserine and phosphothreonine residues by 13C-31P spin-echo difference NMR spectroscopy. J Biomol NMR. 2009;43:31–37. doi: 10.1007/s10858-008-9287-6. [DOI] [PubMed] [Google Scholar]
- 14.Yang C, Shapiro LH, Rivera M, Kumar A, Brindle PK. A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol. 1998;18:2218–2229. doi: 10.1128/mcb.18.4.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayaraman G, et al. p300/cAMP-responsive element-binding protein interactions with Ets-1 and Ets-2 in the transcriptional activation of the human stromelysin promoter. J Biol Chem. 1999;274:17342–17352. doi: 10.1074/jbc.274.24.17342. [DOI] [PubMed] [Google Scholar]
- 16.Rieping W, et al. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 17.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: Implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci USA. 2007;104:7009–7014. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasper LH, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freedman SJ, et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freedman SJ, et al. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol. 2003;10:504–512. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- 24.De Guzman RN, Martinez-Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: Regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J Biol Chem. 2004;279:3042–3049. doi: 10.1074/jbc.M310348200. [DOI] [PubMed] [Google Scholar]
- 25.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009;28:948–958. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng H, et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure. 2009;17:202–210. doi: 10.1016/j.str.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuno H, Furusawa H, Okahata Y. Kinetic study of phosphorylation-dependent complex formation between the kinase-inducible domain (KID) of CREB and the KIX domain of CBP on a quartz crystal microbalance. Chemistry. 2004;10:6172–6178. doi: 10.1002/chem.200400030. [DOI] [PubMed] [Google Scholar]
- 28.Kim CA, Bowie JU. SAM domains: Uniform structure, diversity of function. Trends Biochem Sci. 2003;28:625–628. doi: 10.1016/j.tibs.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Mackereth CD, et al. Diversity in structure and function of the Ets family PNT domains. J Mol Biol. 2004;342:1249–1264. doi: 10.1016/j.jmb.2004.07.094. [DOI] [PubMed] [Google Scholar]
- 30.Abramczyk O, Rainey MA, Barnes R, Martin L, Dalby KN. Expanding the repertoire of an ERK2 recruitment site: Cysteine footprinting identifies the D-recruitment site as a mediator of Ets-1 binding. Biochemistry. 2007;46:9174–9186. doi: 10.1021/bi7002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang HS, et al. Identification and structural characterization of a CBP/p300-binding domain from the ETS family transcription factor GABP alpha. J Mol Biol. 2008;377:636–646. doi: 10.1016/j.jmb.2008.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim CA, et al. Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 2001;20:4173–4182. doi: 10.1093/emboj/20.15.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiao F, et al. Derepression by depolymerization; structural insights into the regulation of Yan by Mae. Cell. 2004;118:163–173. doi: 10.1016/j.cell.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Hwang TL, van Zijl PC, Mori S. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J Biomol NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 35.Gusterson R, et al. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J Biol Chem. 2002;277:2517–2524. doi: 10.1074/jbc.M104626200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.