

Abstract
It is currently considered that idiopathic minimal change nephrotic syndrome (I-MCNS) is an immune-mediated glomerular disease. Its association with classical Hodgkin lymphoma (cHL-MCNS) suggests a molecular link between these two diseases, which remains to be elucidated. We analyzed the expression of c-mip (cmaf inducing protein) in lymphomatous tissues and kidney biopsies of patients with cHL-MCNS (n=8) and in lymphomatous tissues of isolated cHL (n=9). Because c-mip affects the regulatory loop involving Fyn, we investigated possible structural defects in this signaling pathway, using laser capture microdissection, RT-PCR and Western-blotting. We found that c-mip was selectively expressed in Hodgkin and Reed-Sternberg (HRS) cells and podocytes of patients with cHL-MCNS but is undetectable in patients with isolated cHL. We demonstrated that c-mip was specifically involved in the negative regulation of early proximal signaling through its interaction with PAG and Fyn. We showed that the upregulation of c-mip in cHL-MCNS was associated with a possible Fyn defect in HRS cells and podocytes, while Fyn was normally expressed in isolated cHL and normal podocytes. Moreover, we showed that c-mip was upregulated in Fyn-deficient podocytes. C-mip may be a useful marker of cHL-MCNS and its induction reflects the dysregulation of proximal signaling.
Introduction
Classical Hodgkin lymphoma (cHL) is one of the most common malignant lymphoma in Western countries. It is characterized by the presence of malignant cells called Hodgkin and Reed-Sternberg (HRS) cells, which are embedded in a reactive cellular background consisting of T-cells, histiocytes, eosinophils, and plasma cell infiltrates.1 HRS cells are not all derived from the same cell type. The vast majority (> 98%) are derived from germinal center or post-germinal center B-cells, with a very small minority (< 2%) from T cells.2,3
Minimal change nephrotic syndrome (MCNS) is the most frequent glomerular disease, which can occur during the course of cHL.4 MCNS is an acquired glomerular disease of unknown origin, characterized by heavy proteinuria without inflammatory lesions or cell infiltrations. The pathogenesis of this disorder remains poorly understood; however, experimental studies and clinical observations suggest an immune origin.5 It is currently thought that MCNS is caused by a putative circulating factor, which increases glomerular capillary permeability and leads to podocyte cytoskeleton disorganization and proteinuria.6 However, in the case of cHL-associated MCNS (cHL-MCNS), the nature of such a putative glomerular permeability factor of HRS cell or reactive T-lymphocyte origin remains elusive.4
In a previous study of the molecular mechanisms underlying idiopathic MCNS (I-MCNS), we have isolated a new gene named c-mip (for c-maf inducing protein).7 The naturally occurring isoform encodes an 86-kDa protein. The predicted protein structure of c-mip includes an N-terminal region containing a pleckstrin homology domain (PH), a middle region containing several interacting docking sites including a 14-3-3 module, a PKC domain, and an SH3 domain similar to the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K), and a C-terminal region containing a leucin-rich repeat (LRR) domain. The functional role of c-mip appears complex and is not clearly understood. We have recently shown that c-mip interacts with RelA and inhibits its nuclear translocation, resulting in downregulation of NF-kB activity.8 We have also reported that c-mip interacts with filamin A, suggesting its involvement in cytoskeleton organization.9
In a recent retrospective study, we described the medical history of 21 patients with cHL-MCNS. Extensive immunohistochemical analysis of the lymph nodes for eight of these patients did not provide any evidence for a B- or T- cell origin for HRS cells. The co-occurrence of MCNS and cHL, although rare, is not fortuitous but the mechanisms by which cHL induces podocyte disease remains unknown.10 In light of recent results suggesting a crucial role for c-mip in the pathogenesis of MCNS, we studied its presence in patients with Hodgkin lymphoma with or without associated MCNS.
Materials and Methods
Patients
Of the eight cHL-MCNS described previously,10 seven had available renal biopsy and lymphomatous tissue samples and were thus included in this study. MCNS and cHL occurred simultaneously in two cases (Patient N° 1 and 2); MCNS occurred before cHL in three patients (Patient N°3 to N° 5) and after cHL in the two remaining patients (Patient N°6 and 7). The clinical, biological and histological characteristics of these patients are summarized in table 1. An additional patient with the simultaneous occurrence of both diseases was included in this study (patient N°8). This patient was treated with a VBVP chemotherapy regimen for cHL (IIA,a) revealed by MCNS. A binephrectomy was performed three years after initial presentation because of steroid resistant MCNS, rapid deterioration of renal function and major denutrition, requiring the beginning of periodic haemodialysis. This patient did not have a previous history of opportunistic infection before the occurrence of cHL-MCNS but experienced multiple severe bacterial, fungal and viral infections occurring after starting of chemotherapy. All experiments were conducted with approval from the INSERM research ethics committee in accordance with international ethics codes and guidelines.
Table 1.
Clinical, biological and pathological data for patients with cHL-MCNS
| Patients# | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Sex (M/F) | M | M | M | F | M | F | F | M |
| MCNS occurrence (month/year | 07/1999 | 12/1990 | 06/1995 | 05/1995 | 03/1999 | 02/2000 | 06/2002 | 12/2000 |
| cHL occurrence (month/year) | 07/1999 | 12/1990 | 09/1995 | 11/1997 | 10/1999 | 08/1997 | 04/2002 | 12/2000 |
| Urinary protein (g/day) | 16 | 13.3 | 17 | 13 | 5.36 | 4.5 | 3 | 9 |
| Serum albumin (g/l) | 8.9 | 14.8 | 19 | 10 | 12 | 10 | 13.8 | 12.8 |
| Ann Arbor Staging | I | III | II | III | II | II | III | II |
| Systemic symptoms (A/B) | A | B | A | A | B | A | A | A |
| Inflammatory Syndrome (a/b) | b | b | b | b | b | a | b | a |
| WHO classification | ns | ns | ns | ns | ns | ns | ns | ns |
| MCNS remission | yes | yes | SRNS | yes | SRNS | SDNS | yes | SRNS |
| cHL remission | yes | yes | yes | yes | yes | yes | yes | yes |
MCNS: Minimal change nephrotic syndrome; cHL: Classical Hodgkin lymphoma; ns: nodular sclerosis; SRNS: Steroid Resistant Nephrotic Syndrome; SD: Steroid Dependent Nephrotic Syndrome.
The control group consisting of nine patients with cHL without known renal disease was matched for age, Ann Arbor staging, and histological subtype with the study group. All patients in the control and study groups were negative for the human immunodeficiency virus. Control cases were obtained from patient files of the department of Pathology, Henri Mondor Hospital. All patients underwent a histological study of the lymph nodes to confirm the diagnosis of cHL, which was based on the presence of HRS cells in an appropriate cellular background of reactive leucocytes, histiocytes and in some case, fibrosis. The histological subtypes were defined according to the WHO classification of cHL (nodular sclerosis, mixed cellularity, lymphocyte-rich and lymphocyte depleted cHL).11 Laboratory tests for features of inflammatory syndrome including C-reactive protein, sedimentation rate and fibrinogen levels were carried out (a: absence / b: presence). Systemic symptoms - fever, weight loss and night sweats - were recorded for each patient (A: absence / B: presence). Diagnostic criteria for MCNS required the presence of nephrotic syndrome associated with minimal change glomerular lesions identified by light microscopy and negative immunofluorescence or the presence of IgM deposits in the mesangium upon histological examination of kidneys biopsy specimens. Microdissected glomeruli were obtained from frozen kidney tissue for patient N° 8, and from normal kidney tissue provided by our pathological department for control subjects.
In situ hybridization (ISH) and Immunohistochemistry
Reactive lymphoid tissue, normal adult thymus and spleen were used as control tissues. Fyn-deficient mice (B6;129S7-Fyntm1Sor/j; stock number 2385) were obtained from Jackson Laboratory (Bar Harbor, Me). In situ hybridization (ISH) experiments were performed as previously described.12 The c-mip probe corresponds to positions 313–1072 of the cDNA coding sequence. For immunohistochemistry analysis, kidney samples were fixed for 16 hours in Dubosq Brazil, then dehydrated and embedded in paraffin. Antigen retrieval was performed by immersing the slides in boiling 0.01 M citrate buffer in a 500 W microwave oven for 15 min. Endogenous peroxidase activity was blocked with 0,3% H2O2 in methanol for 30 min. Slides were incubated with the blocking reagents containing avidin–biotin solution for 30 min and normal blocking serum for 20 min. For detection of c-mip, slides were incubated overnight with polyclonal antibody at a final concentration of 15 μg/ml, then with biotinylated secondary antibody. Incubation with anti-Fyn antibody (Abcam PLC, Cambridge, United Kington) was performed according to the instructions provided by the manufacturer. We used an avidin-biotinylated horseradish peroxidase complex (Vectastain ABC Reagent, Vector Laboratories; Burlingame, CA) with 3,3′-diaminobenzidine (Sigma Biochemicals; St Louis, MO) as a chromogen, for visualization of the immunoreaction. Slides were counterstained with hematoxylin. Primary antibody was omitted for negative control.
Laser capture microdissection
A series of 10 μm-thick sections were cut from frozen tissue specimens using a Leica CM3050 cryostat at −20°C, and mounted onto slides coated with a thermoplastic membrane (Glass PEN-membrane slides; Leica Microsystems, Rueil-Malmaison, France). Nuclei were stained with Mayer’s hematoxylin solution. Glomerular structures were selectively dissected using the AS LMD laser microdissection microscope (Leica Microsystems, Rueil-Malmaison, France). Five 10 μm cryosections from each patient were used to microdissect 80 glomeruli each. Microdissected fragments were dropped into screw-cap vials containing 50 μl of extraction buffer. RNA was extracted from microdissected glomeruli with a picopure RNA isolation kit (Arcturus, Alphelys, Plaisir, France).
Cell line culture and transfection
We used two well characterized HRS cell lines originating from patients with nodular sclerosis (L428) or mixed cellularity subtype (KMH2), which we obtained from the Deutsche Culture Collection (DSMZ, Braunschweig, Germany). Previous phenotypic and genotypic studies of HRS cell lines have shown that the L428 and KMH2 cell lines have a B-cell origin.13 All cell lines were grown in RPMI-1640 medium with Glutamax 1 supplemented with penicillin, streptomycin, 1% pyruvate and 10% fetal calf serum (FCS), at 37°C in an atmosphere containing 5% CO2.
HRS cells (3×106 per condition) were transiently transfected with c-mip expression plasmid (1 μg/106 cells) or empty vector by electroporation using a Gene pulser X cell (Bio Rad, Paris, France) set at 950 μF and 230 V. Cells were allowed to recover overnight and were harvested 20h after transfection for protein extraction and immunochemical analysis.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated using an RNeasy kit (Qiagen, Chatsworth, CA). RT-PCR was performed as described previously.14 cDNA was prepared from whole lymphomatous tissue of patients with cHL with or without MCNS and from HRS cell lines. The transcripts Fyn, c-mip, FAT, Neuropilin (NPI) and GAPDH were amplified using the primers listed in table 2. PCR products were resolved on a 1.5% agarose gel and stained with SYBR green.
Table 2.
Sequence of primers and PCR conditions
| mRNA | Primer | Accession n° | Expected size (bp) | Ann ealing T (°C) | PCR cycles |
|---|---|---|---|---|---|
| GAPDH | Forward: ACCACAGTCCATGCCATCAC Reverse: TCCACCACCCTGTTGCTGTA |
AF261085 | 446 | 60 | 32 |
| Fyn | Forward: GAATTTAGATAATGGGCTGTGTGCAATGTAAGGAT Reverse: ACCCGGTTACAGGTTTTCACCAGGT |
NM_153047 | 1605 | 60 | 34 |
| C-mip | Forward: GGCCATGGATGTGACCAGCAGCTC Reverse: TGGGAGCTTCACCAGGCTTCGGTGTAGC |
AK096598 | 2330 | 60 | 34 |
| FAT | Forward: CAGCGACTCCATCCAGAAGCCTAGCTG Reverse: CGAGGCATGTCTCTAGGAGGGTGGATG |
NM_005245 | 450 | 60 | 35 |
| Neuropilin-1 | Forward: CTGATTCAGGCTCCGGACCCATACCAGAG Reverse: CTCCTGATTCCATGCCCAGAGCTTCCA |
BC007737 | 684 | 60 | 35 |
Preparation of glomerular extracts, immunoprecipitations and Western blot analysis
Primary antibodies used in this study included, anti-CBP/PAG (sc-25748, Santa Cruz Biotechnology), anti-Csk (Cell Signaling), anti-Fyn (BD Biosciences), and anti-phosphotyrosine clone 4G10 (Upstate, Biotechnology, Inc, Lake placid, NY). The anti-c-mip polyclonal antibody was produced in our lab by immunizing rabbits with acrylamide gel sections containing the c-mip protein.
Kidney fractions enriched in glomeruli were isolated using a graded sieving method. Cell protein extracts were prepared in lysis buffer (NaCl 150 mM, 10 mM tris Hcl pH 7.5, 2 mM DTT, 10% glycerol, 1 mM EDTA, 1% NP40, 1 mM of protease inhibitors, 1 mM NaF et 1 mM sodium orthovanadate). Glomerular protein extracts were prepared in lysis buffer (50 mM Tris Hcl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% X NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM of protease inhibitors, 1 mM NaF et 1 mM sodium orthovanadate). Immunoprecipitation was performed overnight at 4° C. Immunoprecipitates were resolved by SDS-PAGE and analyzed by Western blot with the indicated antibodies.
Results
Expression of c-mip in podocytes from patients with cHL and MCNS
We have previously reported the clinical study of a series of patients with cHL-MCNS.10 In this present study, we examined whether a possible molecular link may exist between I-MCNS and cHL-MCNS. Our initial studies of c-mip expression in glomerular diseases showed that c-mip is induced in podocytes of patients with MCNS disease. The availability of biopsy specimens from patients with cHL-MCNS led us to investigate whether c-mip could be expressed in this entity. We performed in situ hybridization (ISH) analyses on kidney biopsies from eight patients with cHL-MCNS and twelve patients with I-MCNS. Incubation with a c-mip antisense probe revealed an intense signal in the glomeruli, which was mainly restricted to cells surrounding the capillary loops, suggestive of labeling in podocytes (Figure 1A), while no signal was seen with a c-mip sense probe. We also observed intense signal in parietal epithelial cells in eight cHL-MCNS cases. Staining intensity appeared to be higher than in I-MCNS samples, with labeling detected throughout each glomerulus. Immunohistochemistry analysis showed that the distribution of c-mip protein correlated with the location of podocytes (Figure 1B). Within podocytes, c-mip was detected in the nuclear and cytoplasmic compartments. We conclude that, as observed for I-MCNS, podocytes in cHL-MCNS exhibited an upregulation of c-mip at the mRNA and protein levels.
Figure 1. C-mip is upregulated in the glomeruli of eight patients with cHL-MCNS.
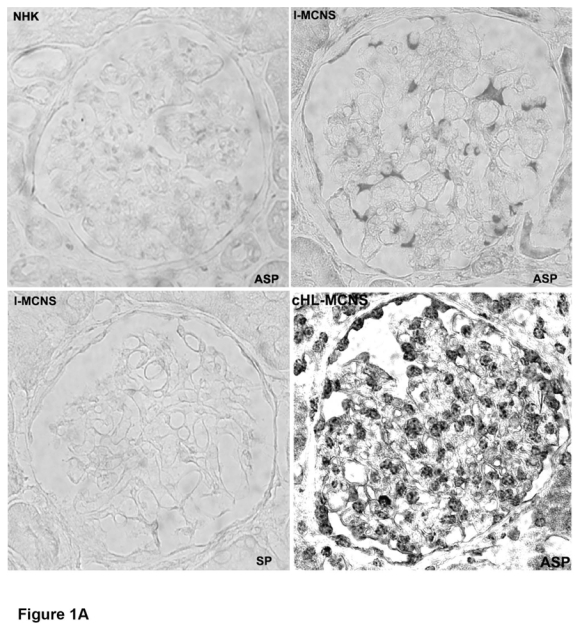
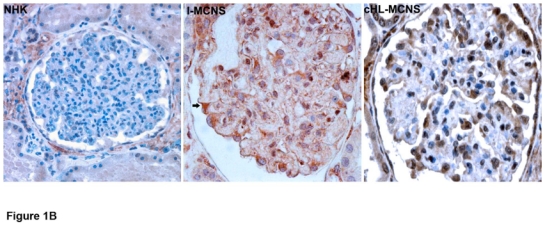
A, Upper panel, Representative in situ hybridization of c-mip transcript in serial sections from normal human kidney (NHK) and kidney biopsies of patients with I-MCNS and cHL-MCNS (ASP: antisense probe; SP: sense probe); B, Representative immunohistochemistry on serial sections of normal human kidney and kidney biopsies from patients with I-MCNS and cHL-MCNS (original magnification, X40). C-mip upregulation was detected in the podocytes of all patients with cHL-MCNS.
Expression of c-mip in lymphomatous tissue from patients with cHL, with or without associated MCNS
In light of our previous findings consistent with changes in T cell function in I-MCNS,15 we investigated whether c-mip was produced in lymphoid organs. In normal lymphoid tissues, the expression of c-mip was mostly negative in the thymus and in the red pulp of the spleen, with only a few scattered positive lymphoid cells observed (Figure 2A and data not shown). By contrast, c-mip was detected in lymph nodes and in lymphoid follicles within the white pulp of spleen (Figure 2A). Signal was mainly restricted to the transitional zone of the lymphatic follicle, which corresponds to the area of B- and T-cell cooperation.16 In lymphoid follicles, c-mip was selectively produced in the core of the follicle, a B-cell-dependent area (Figure 2A). We then analyzed c-mip in lymphomatous tissue from eight patients with cHL-MCNS and nine patients with isolated cHL. We did not detect c-mip in lymph nodes from isolated cHL patients (n=9), either in HRS cells, or in the reactive lymphocytes surrounding them (Figures 2B and 2D). On the other hand, c-mip labeling was particularly intense in HRS cells of eight patients with cHL-MCNS (100%), with no significant signal detected in the surrounding reactive cellular infiltrates (Figures 2C and 2D). The percentage of positive HRS cells in cHL-MCNS patients varied from 10% to 90%. The highest level of c-mip immunoreactivity in HRS cells was observed in lymph nodes of patients with simultaneous occurrence of cHL and MCNS (table 1). Within HRS cells, c-mip was either detected in the cytoplasm in the Golgi area of neoplastic cells, or at the plasma membrane (Figures 2C and 2D). Altogether, these findings show that cHL-MCNS is associated with an overexpression of c-mip in both HRS cells and podocytes and may represent a molecular signature of cHL-MCNS.
Figure 2. Upregulation of c-mip in lymph nodes of eight patients with cHL-MCNS is restricted to Hodgkin and Reed-Sternberg (HRS) cells.


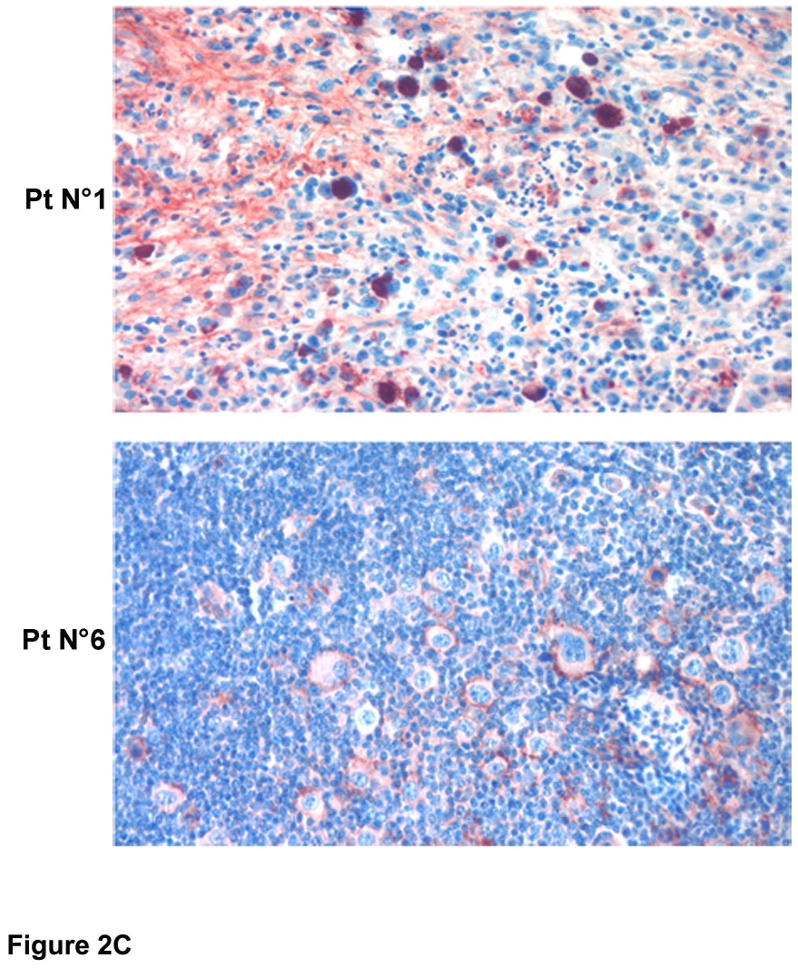

A, Immunohistochemistry analysis of c-mip in normal lymphoid tissues (thymus, lymphoid follicle of the spleen and lymph nodes); note that c-mip is not seen in normal tissues, except in the transitional zone of node tissue and in the lymphoid follicle (indicated by arrows) (original magnification, X20); B, Representative image showing c-mip in lymphomatous tissues from two patients with isolated cHL (original magnification, X20) (HRS cells are indicated by arrows); note that c-mip is undetectable in lymphomatous tissues from isolated cHL; C, Representative expression of c-mip in lymphomatous tissues from two patients with cHL-MCNS (original magnification, X20). c-mip shows intense staining in cHL-MCNS, restricted to HRS cells. D, Localization of c-mip in HRS cells from four patients with cHL-MCNS; no staining was detected in the HRS cells of two patients with isolated cHL (original magnification, X100). C-mip upregulation in HRS cells was found in all patients with cHL-MCNS, whereas no immunostaining was observed for c-mip in 9 control cases with isolated cHL
Given the malignant origin of the HRS cells, we sought to determine whether the c-mip gene exhibited any sequence changes. The gene encoding c-mip spans 268 kb on chromosom 16 and includes 21 exons. encoding for a messenger RNA (mRNA) of 4.6 Kb. The coding sequence consists of 2319 nucleotides. We purified c-mip mRNA from the podocytes and lymph node tissues of one patient with cHL-MCNS with high c-mip expression levels (patient N°8). Analysis of the coding sequence of the transcript did not reveal any mutation (data not shown).
C-mip affects proximal signaling pathways in HRS cells
Proteins containing a PH domain, such as c-mip, are recruited into lipid rafts (LR) following the activation of membrane receptors involved in several signaling pathways including those of protein tyrosine kinase (PTK) and Ras.17 We have previously demonstrated that c-mip interacts with Fyn in vitro and in vivo in various cell systems, including HEK cells and podocytes (Zhang et al., manuscript submitted). Our understanding of the proximal regulation of Fyn comes primarily from T-cell studies; little is known about this regulation in podocytes.18,19
In physiological situations, T cells are maintained in a resting state by an active mechanism involving the phosphorylation of PAG (protein associated with glycosphingolipid-enriched microdomains) by Fyn within the lipid rafts.20,21 Activated PAG recruits Csk, a cytoplasmic PTK, which phosphorylates the major pool of Fyn on tyrosine 528 (Fyn-Y528), enabling the intra-molecular folding of the protein into its inactive conformation.22 The PAG/Fyn interaction appears to be essential for the function of PAG. Indeed, levels of PAG phosphorylation and subsequent interaction with Csk are dramatically reduced in Fyn-deficient T cells.21 Upon T-cell activation, CD45 moves into lipid rafts and dissociates the PAG-Fyn complex, resulting in PAG dephosphorylation, dissociation of the PAG-Csk complex and inactivation of Csk.23 Fyn released from its Csk inhibitor, binds to membrane receptors, such as the TCR δ chain, and activates downstream targets, including PI3 kinase and Akt. Thus, depending on its partners, Fyn may have a dual effect on signaling, exerting inhibitory effects on unstimulated cells through its interaction with and phosphorylation of PAG, and stimulatory effects upon its dissociation from PAG and its interaction with activated receptors. These considerations along with previous results regarding the functional effects of c-mip on podocyte signaling led us to investigate whether c-mip interacts with PAG and/or Csk.
HRS cell lines, which do not constitutively produce c-mip (Figure 3a), were transfected with c-mip or empty vector, and then processed for immunoprecipitation and Western blot analyses. We found that c-mip interacted with endogenous Fyn in the HRS cell line (Figure 3b). Western blotting with the phosphotyrosine antibody 4G10 showed that c-mip also interacted with endogenous PAG and increased its phosphorylation (Figure 3c). On the other hand, we did not detect any interaction between c-mip and Csk. These results suggest that c-mip facilitates the negative regulation of early proximal signaling through activation of PAG. Given that active Fyn is required for PAG activation and that c-mip enhances the inhibitory role of Fyn, upregulation of c-mip in HRS cells and podocytes may be caused by a break in the negative feedback loop, due to the inability of Fyn to exert its function. We therefore hypothesized that the regulation of the Fyn-PAG axis may be altered in HRS cells and in podocytes from patients with cHL-MCNS. To explore this possibility, we firstly analyzed the expression of PAG and Csk in podocytes. We found that Fyn, PAG and Csk were present in human and rat glomerular extracts, suggesting that like in lymphocytes, the regulatory loop is operational in human podocytes (Figure 4).
Figure 3. C-mip interacts with PAG and Fyn in HRS cell lines.

a, Western blot detection of c-mip protein in lysates from HRS and Jurkat cell lines. HEK cells were transfected with a c-mip expression plasmid and were used as positive controls. Immunoprecipitation of endogenous Fyn (b) and PAG/Cbp (c) from HRS cell lines co-transfected with the c-mip expression plasmid or empty vector (Ev).
Figure 4. Expression of Fyn signaling regulatory loop in human and rat glomeruli.
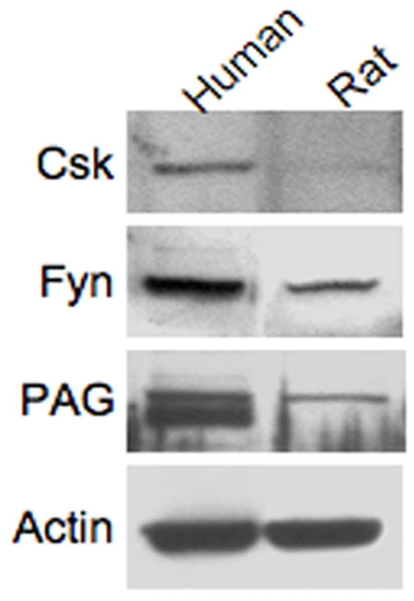
The presence of Csk, Fyn and PAG was analyzed by Western blotting on glomerular extracts.
Upregulation of c-mip in cHL-MCNS patients is associated with Fyn deficiency in both HRS cells and podocytes
We hypothesized that c-mip induction may be associated with a defect of Fyn, inasmuch as c-mip is a partner of Fyn, which is expressed in both podocytes and HRS cells.24,25
To test this hypothesis, we studied the expression of Fyn transcript using semi-quantitative RT-PCR. Fyn transcript levels did not differ between lymph node tissue from patients with cHL and peripheral blood mononuclear cells (PBMC) from normal subjects (Figure 5A). However, we were unable to detect Fyn transcript in lymph node tissue from a patient with cHL-MCNS (patient N° 8), whereas it was found in HRS cell lines and in PBMC from two patients with an I-MCNS relapse (Figure 5A). We next analyzed the expression of Fyn in microdissected glomeruli from patient N°8. Again, we did not detect any Fyn transcripts, whereas expression was easily observed in microdissected glomeruli from normal human kidneys (Figure 5B). By contrast, neuropilin-1 and FAT transcripts were produced normally in podocytes from this patient (Figure 5B). These results suggest that c-mip was recruited in response to changes in the proximal signaling negative regulatory loop in cHL-MCNS patients, which may involve a defect in Fyn expression. To confirm our observations, we performed immunohistochemistry analysis on kidney biopsy from this patient using anti-Fyn antibody (Figure 5C). By contrast to normal glomeruli, which exhibited strong immunostaining in podocytes with Fyn antibody, we did not detect any Fyn protein expression in glomeruli from this patient with cHL-MCNS. Altogether, these findings suggest that changes in proximal signaling induced by a potential defect in Fyn are likely to be involved in the upregulation of c-mip in podocyte.
Figure 5. Absence of Fyn detection in lymphomatous tissue and in microdissected glomeruli from one patient with cHL-MCNS.


a, Analysis of Fyn expression by RT-PCR in normal PBMC, in lymphomatous tissue from patient N° 8 and a control patient with isolated cHL, in the PBMC of two patients with an I-MCNS relapse and in HRS cell lines. b, Analysis of Fyn expression in microdissected glomeruli from the patient N° 8 and a control patient without glomerular disease. Notably, transcripts for FAT and neuropilin1 were easily detected in the microdissected glomeruli. c, Immunohistochemistry analysis of Fyn protein in normal human kidney (left panel) and in a kidney biopsy specimen from patient N° 8 (right panel). Note the absence of Fyn protein from podocytes of patient N° 8.
Fyn deficient mice exhibited an upregulation of c-mip in podocytes
The results presented here suggest that upregulation of c-mip in the podocytes of patients with cHL-MCNS, may be due to a Fyn defect. We thus analyzed c-mip protein in Fyn-deficient mice.26 These mice were proteinuric at one month of age and previous ultrastructural studies have shown that podocyte foot processes are altered.24 Immunohistochemistry analysis showed significant induction of c-mip in one-month-old Fyn null mice, but not in wild type littermates (Figure 6). This result supports our in vivo data from patient 8.
Figure 6. C-mip is upregulated in Fyn-deficient mice.
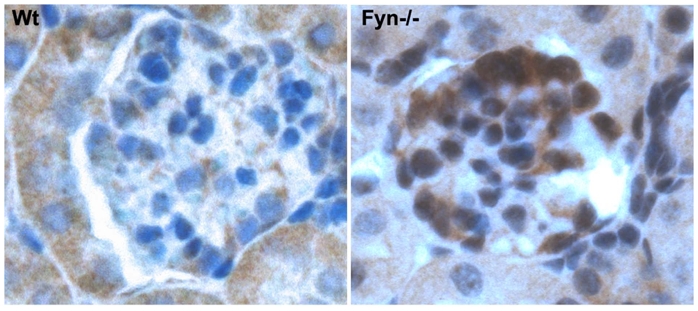
Immunohistochemistry analysis of c-mip in serial sections from normal and Fyn-deficient mice kidneys (original magnification, X40).
Discussion
It is currently considered that MCNS is an immune-mediated glomerular disease. Its association with cHL suggests there that is a molecular link between these two disorders. In this study, we demonstrated, for the first time, that c-mip is selectively induced both in podocytes and in HRS cells in patients with cHL-MCNS but is not detected in patients with isolated cHL, suggesting its potential involvement in the pathophysiology of this association. We showed that: i) c-mip interacted with PAG and Fyn; ii) c-mip transcript from lymphomatous tissue and podocytes did not carry any mutation in the coding sequence; and iii) the upregulation of c-mip in cHL-MCNS may be associated with a defect in Fyn, which is normally expressed in isolated cHL.25
Although the expression of c-mip in I-MCNS is clearly established, it is important to note that its expression level in podocytes and HRS cells of patients with cHL-MCNS is much higher than in the cells of patients with I-MCNS. This suggests that the changes of proximal signaling are more severe in cHL-MCNS and may result from a protein defect.
Fyn plays a major role in podocyte proximal signaling. However, as for the nephrin signaling pathway, the mechanisms that regulate its activity in these cells are largely unknown. In lymphocytes, many studies have shown that Fyn is inactivated upon its dissociation from PAG and subsequent binding with Csk.26–28 It has been shown that Fyn-deficient mice display proteinuria even in the rag−/− genetic background, suggesting that podocyte damage is independent of immune mechanisms.29
The role of c-mip in the negative regulation of proximal signaling is underscored by its ability to interact with Fyn and PAG. We show that c-mip, like active Fyn, increases the phosphorylation of PAG. In cHL-MCNS, it is possible that c-mip compensates for the deficiency in Fyn by reducing cell activity through enhanced negative proximal signaling. C-mip is mainly restricted to the transitional zone of B-cell follicles and lymphoid follicles in normal hematopoietic tissues, suggestive of a role in B-cell/helper T-cell cooperation and in B-cell function. Despite previous clinical and experimental studies suggesting a disorder of a T function, recent data suggest that B cells may be involved in the pathogenesis of MCNS. Rituximab, a chimeric monoclonal antibody, which inhibits B cell proliferation and differentiation, has recently been used successfully in patients with steroid-dependent MCNS.30 Although speculative, we cannot exclude that B cells may be involved in the pathogenesis of MCNS. Early studies have demonstrated an impairment of immunoglobulin production during MCNS relapse, in which IgG levels are severely reduced with unbalance of IgG subclasses, suggesting that intrinsic B cells properties and class switch recombination (CSR) may be primarily altered.31
This patient cohort shows clear upregulation of c-mip in cHL-MCNS. However, only one patient, with a demonstrable defect in Fyn, likely to account for the steroid resistance seen in MCNS, offers us a clue as to the underlying mechanisms. We cannot exclude the possibility that another protein involved in the negative regulation of proximal signaling could be defective in other patients. In a previous study, Fyn was found to be expressed in HRS cells in 42% of the cHL cases analyzed.25 We therefore screened for Fyn expression in the lymphocytes of patients with I-MCNS. We found that the transcript expression is normal in these patients, contrasting with observations for our patient with cHL-MCNS. Nevertheless, c-mip upregulation probably reflects a defect in the negative regulation of proximal signaling. The identification of Fyn and c-mip binding partners in podocytes will be essential for a thorough understanding of the pathophysiology of this disease.
Acknowledgments
We are grateful to Dr François Combarnous, Dr Raymonde Bouvier and Dr Aurelia Liutkus (Nephrology department, Lyon, France) for their help with the care and follow-up of patient 8. We thank Dr Yves Allory and Dr Karen Leroy (Pathology department, Henri Mondor Hospital) for providing us with control renal tissues and mRNA samples from isolated cHL patients.
This work was supported in part by an Avenir Program from INSERM, a grant from the French Kidney Foundation and the Association pour l’Utilisation du Rein Artificiel (AURA).
Footnotes
Author contribution statement
VA, SYZ, CCB, CRM, VO, MC, and MB performed the experiments
DS, AP designed the research
PL has contributed to management of the project
VA, DS, AP, SYZ and CCB analyzed the data
DS and VA wrote the paper
Conflict of Interest Disclosures
We have no conflict of interest to disclose
VA is a recipient of an INSERM Poste d’Accueil (Institut National de la Santé et de la Recherche Médicale).
References
- 1.Yung L, Linch D. Hodgkin’s lymphoma. Lancet. 2003;361:943–951. doi: 10.1016/S0140-6736(03)12777-8. [DOI] [PubMed] [Google Scholar]
- 2.Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;184:1495–1505. doi: 10.1084/jem.184.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9:15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 4.Ronco PM. Paraneoplastic glomerulopathies: new insights into an old entity. Kidney Int. 1999;56:355–377. doi: 10.1046/j.1523-1755.1999.00548.x. [DOI] [PubMed] [Google Scholar]
- 5.Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet. 1974;2:556–560. doi: 10.1016/s0140-6736(74)91880-7. [DOI] [PubMed] [Google Scholar]
- 6.Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629–639. doi: 10.1016/S0140-6736(03)14184-0. [DOI] [PubMed] [Google Scholar]
- 7.Grimbert P, Valanciute A, Audard V, et al. Truncation of C-mip (Tc-mip), a new proximal signaling protein, induces c-maf Th2 transcription factor and cytoskeleton reorganization. J Exp Med. 2003;198:797–807. doi: 10.1084/jem.20030566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamal M, Valanciute A, Dahan K, et al. C-mip interacts physically with RelA and inhibits nuclear factor kappa B activity. Mol Immunol. 2009;46:991–998. doi: 10.1016/j.molimm.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 9.Grimbert P, Valanciute A, Audard V, Lang P, Guellaen G, Sahali D. The Filamin-A is a partner of Tc-mip, a new adapter protein involved in c-maf-dependent Th2 signaling pathway. Mol Immunol. 2004;40:1257–1261. doi: 10.1016/j.molimm.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 10.Audard V, Larousserie F, Grimbert P, et al. Minimal change nephrotic syndrome and classical Hodgkin’s lymphoma: report of 21 cases and review of the literature. Kidney Int. 2006;69:2251–2260. doi: 10.1038/sj.ki.5000341. [DOI] [PubMed] [Google Scholar]
- 11.Pileri SA, Ascani S, Leoncini L, et al. Hodgkin’s lymphoma: the pathologist’s viewpoint. J Clin Pathol. 2002;55:162–176. doi: 10.1136/jcp.55.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang SY, Marlier A, Gribouval O, et al. In vivo expression of podocyte slit diaphragm-associated proteins in nephrotic patients with NPHS2 mutation. Kidney Int. 2004;66:945–954. doi: 10.1111/j.1523-1755.2004.00840.x. [DOI] [PubMed] [Google Scholar]
- 13.Jungnickel B, Staratschek-Jox A, Brauninger A, et al. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin’s lymphoma. J Exp Med. 2000;191:395–402. doi: 10.1084/jem.191.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahali D, Pawlak A, Valanciute A, et al. A novel approach to investigation of the pathogenesis of active minimal-change nephrotic syndrome using subtracted cDNA library screening. J Am Soc Nephrol. 2002;13:1238–1247. doi: 10.1681/ASN.V1351238. [DOI] [PubMed] [Google Scholar]
- 15.Valanciute A, le Gouvello S, Solhonne B, et al. NF-kappa B p65 antagonizes IL-4 induction by c-maf in minimal change nephrotic syndrome. J Immunol. 2004;172:688–698. doi: 10.4049/jimmunol.172.1.688. [DOI] [PubMed] [Google Scholar]
- 16.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 17.DiNitto JP, Lambright DG. Membrane and juxtamembrane targeting by PH and PTB domains. Biochim Biophys Acta. 2006;1761:850–867. doi: 10.1016/j.bbalip.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Huber TB, Kwoh C, Wu H, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest. 2006;116:1337–1345. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shima T, Nada S, Okada M. Transmembrane phosphoprotein Cbp senses cell adhesion signaling mediated by Src family kinase in lipid rafts. Proc Natl Acad Sci U S A. 2003;100:14897–14902. doi: 10.1073/pnas.2432139100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasuda K, Nagafuku M, Shima T, et al. Cutting edge: Fyn is essential for tyrosine phosphorylation of Csk-binding protein/phosphoprotein associated with glycolipid-enriched microdomains in lipid rafts in resting T cells. J Immunol. 2002;169:2813–2817. doi: 10.4049/jimmunol.169.6.2813. [DOI] [PubMed] [Google Scholar]
- 22.Filipp D, Zhang J, Leung BL, et al. Regulation of Fyn through translocation of activated Lck into lipid rafts. J Exp Med. 2003;197:1221–1227. doi: 10.1084/jem.20022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidson D, Schraven B, Veillette A. PAG-associated FynT regulates calcium signaling and promotes anergy in T lymphocytes. Mol Cell Biol. 2007;27:1960–1973. doi: 10.1128/MCB.01983-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verma R, Wharram B, Kovari I, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. 2003;278:20716–20723. doi: 10.1074/jbc.M301689200. [DOI] [PubMed] [Google Scholar]
- 25.Marafioti T, Pozzobon M, Hansmann ML, Delsol G, Pileri SA, Mason DY. Expression of intracellular signaling molecules in classical and lymphocyte predominance Hodgkin disease. Blood. 2004;103:188–193. doi: 10.1182/blood-2003-05-1487. [DOI] [PubMed] [Google Scholar]
- 26.Kawabuchi M, Satomi Y, Takao T, et al. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000;404:999–1003. doi: 10.1038/35010121. [DOI] [PubMed] [Google Scholar]
- 27.Brdicka T, Pavlistova D, Leo A, et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J Exp Med. 2000;191:1591–1604. doi: 10.1084/jem.191.9.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horejsi V, Zhang W, Schraven B. Transmembrane adaptor proteins: organizers of immunoreceptor signalling. Nat Rev Immunol. 2004;4:603–616. doi: 10.1038/nri1414. [DOI] [PubMed] [Google Scholar]
- 29.Yu CC, Yen TS, Lowell CA, DeFranco AL. Lupus-like kidney disease in mice deficient in the Src family tyrosine kinases Lyn and Fyn. Curr Biol. 2001;11:34–38. doi: 10.1016/s0960-9822(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 30.Guigonis V, Dallocchio A, Baudouin V, et al. Rituximab treatment for severe steroid-or cyclosporine-dependent nephrotic syndrome: a multicentric series of 22 cases. Pediatr Nephrol. 2008;23:1269–1279. doi: 10.1007/s00467-008-0814-1. [DOI] [PubMed] [Google Scholar]
- 31.Mathieson PW. Minimal change nephropathy and focal segmental glomerulosclerosis. Semin Immunopathol. 2007;29:415–426. doi: 10.1007/s00281-007-0094-z. [DOI] [PubMed] [Google Scholar]