

Abstract
Transforming growth factor-β-activated kinase 1 (TAK1) is a member of the NF-κB pathway and regulates inflammatory responses. We previously showed that TAK1 also regulates keratinocyte growth, differentiation, and apoptosis. However, it is unknown whether TAK1 has any role in epithelial–mesenchymal interactions. To examine this possibility, we studied the role of TAK1 in mouse hair follicle development and cycling as an instructive model system. By comparing keratinocyte-specific TAK1-deficient mice (Map3k7 fl/flK5-Cre) with control mice, we found that the number of hair germs (hair follicles precursors) in Map3k7 fl/flK5-Cre mice was significantly reduced at E15.5, and that subsequent hair follicle morphogenesis was retarded. Next, we analyzed the role of TAK1 in the cyclic remodeling in follicles by analyzing hair cycle progression in mice with a tamoxifen-inducible keratinocyte-specific TAK1 deficiency (Map3k7 fl/flK14-Cre-ERT2). After active hair growth (anagen) was induced by depilation, TAK1 was deleted by topical tamoxifen application. This resulted in significantly retarded anagen development in TAK1-deficient mice. Deletion of TAK1 in hair follicles that were already in anagen induced premature, apoptosis-driven hair follicle regression, along with hair follicle damage. These studies provide the first evidence that the inflammatory mediator TAK1 regulates hair follicle induction and morphogenesis, and is required for anagen induction and anagen maintenance.
Introduction
The NF-κB pathway mediates innate immune or pro-inflammatory responses, such as signaling by Toll-like receptors (TLRs), the IL-1 receptor (IL-1R), and tumor necrosis factor receptor (TNFR) [1], [2]. Transforming growth factor-β-activated kinase 1 (TAK1) is a member of the MAP3 kinase family [3] and an important member of the NK-κB pathway, involved in IL-1 and TNF-α-induced activation of NF-κB and MAP kinases [1]. Upon ligand binding, TNF-receptor-associated factor (TRAF) 6 or TRAF2 [4], [5], [6] activates TAK1, which then phosphorylates IκB kinases (IKKs), resulting in NF-κB activation.
Because members of the NF-κB pathway are increasingly recognized as important in the regulation of epithelial–mesenchymal interaction systems, ranging from tooth development to hair follicle induction and morphogenesis [7], [8], [9], [10], [11], we were interested in learning whether TAK1 also played a role in the biology of the hair follicle, a prototypic epithelial–mesenchymal interaction system. This interest was further fueled by our previous discovery in keratinocyte-specific TAK1-deficient mice (Map3k7 fl/flK5-Cre) that TAK1 regulates keratinocyte growth, differentiation, and apoptosis [12]. However, the role of TAK1 in hair follicles has not been previously studied.
Induction and morphogenesis of the hair follicle [13] is controlled by complex signaling networks within the skin epithelium and between the epithelium and specialized inductive fibroblasts in its adjacent mesenchyme [7], [11]. Among these signaling networks, the NF-κB pathway and Wnt/β-catenin signaling provide central controls [7], [8]; however, the exact relationship between these signaling networks is not fully understood. Binding of EdaA1 to its receptor EdaR in the embryo is essential for the development of ectodermal appendages [14], and mutations in these genes cause reduced or absent ectodermal appendages [15], [16], [17], [18], [19]. Subsequently, the EdaA1/EdaR pathway activates the downstream NF-κB pathway [20]. A recent report showed that Wnt/β-catenin signaling lies both upstream and downstream of the EdaR/NF-κB pathway [8]. Wnt/β-catenin signaling within the epithelial cells is required for activation of the Eda/EdaR/NF-κB pathway at an early stage of hair follicle development [8], and the expression of Eda and EdaR requires Wnt/β-catenin signaling [8]. At a later stage, maintenance of Wnt signaling and elevated Wnt10a, Wnt10b, and Dkk4 expression requires the Eda/EdaR/NF-κB pathway [8].
The postnatal hair cycle in mice begins with catagen induction around P17, followed by the first telogen. Recently, the EdaR pathway has been shown to be involved in the hair cycle [9], [21], [22]. The expression of EdaA1 and EdaR increases in the anagen-catagen phase [21]. Furthermore, EdaA1 prolongs the anagen phase [22]. Thus, in addition to its well-established role in hair follicle morphogenesis, the EdaR pathway is also involved in hair cycle control.
Since TAK1 is a member of NF-κB pathway, we hypothesized that TAK1 is involved in hair follicle morphogenesis and hair cycle control. To explore this, we studied hair follicle development in keratinocyte-specific TAK1-deficient (Map3k7 fl/flK5-Cre) mice and subsequent hair follicle cycling in tamoxifen-inducible keratinocyte-specific TAK1 deficient mice (Map3k7 fl/flK14-Cre-ERT2) to avoid gene-targeting in embryonic development because this might damage the hair follicle, impairing its later capacity to cycle. These studies provide the first evidence that TAK1 regulates hair follicle induction, morphogenesis, and cycling.
Materials and Methods
Ethics Statement
The protocol for generating Map3k7 fl/flK5-Cre mice and Map3k7 fl/flK14-Cre-ERT2 mice was approved by the Institutional Review Board of Ehime University Graduate School of Medicine (#I-20-13 and #NE-27-16).
Generation of keratinocyte-specific TAK1-deficient mice (Map3k7 fl/fl K5-Cre)
TAK1 is encoded by the Map3k7 gene. The targeting construct has been described previously [1]. We generated keratinocyte-specific TAK1-deficient mice (Map3k7 fl/flK5-Cre) by breeding Map3k7 fl/fl mice (C57Bl/6 background) with K5-Cre mice (C57Bl/6 background) [23], as previously described [12].
Generation of tamoxifen-inducible keratinocyte-specific TAK1-deficient mice (Map3k7 fl/fl K14-Cre-ERT2)
Map3k7K fl/fl mice were bred with K14-Cre-ERT2 mice (C57Bl/6 background) [24], [25] to generate Map3k7 fl/flK14-Cre-ERT2 mice. We applied 100 µL of 4-hydroxytamoxifen (Sigma-Aldrich Co., St. Louis, MO) in ethanol at a concentration of 1 mg/mL topically to the dorsal skin of 8-week-old female Map3k7 fl/flK14-Cre-ERT2 mice for 5 consecutive days [24].
Wax depilation
The hair cycle was synchronized in the dorsal skin of 8-week-old female mice by wax (SURGI-WAX™, Ardell International, Los Angeles, CA) depilation, as described previously [26].
Histological analysis
The stages of hair follicle morphogenesis, cycling, and dystrophic catagen were morphologically defined using dorsal skin of the mice and the score was defined as follows.
The hair morphogenesis stage of each hair follicle in each mouse group was evaluated as described previously [13]. At least 40 hair follicles or all hair follicles in the mice were evaluated in each mouse group (two mice/group). The score of each hair morphogenesis stage was defined as follows: stage 1 = 1, stage 2 = 2, stage 3 = 3, stage 4 = 4, stage 5 = 5, stage 6 = 6, stage 7 = 7, and stage 8 = 8. Then, the rate (%) of a certain hair morphogenesis stage in the total hair follicles and the median score in each mouse group were determined. The score of Map3k7 fl/flK5-Cre mice was compared with Map3k7 fl/fl mice. Statistical significance was determined using a Mann-Whitney's U-test. A difference of *P<0.01 was considered statistically significant.
The hair cycle stage of each hair follicle in each mouse group was evaluated as described previously [27]. At least 40 hair follicles were evaluated in each mouse group (two mice/group). The score of each hair cycle stage was defined as follows: catagen I = 1, catagen II = 2, catagen III = 3, catagen IV = 4, catagen V = 5, catagen VI = 6, catagen VII = 7, catagen VIII = 8, telogen = 9, anagen I = 10, anagen II = 11, anagen IIIa = 12, anagen IIIb = 13, anagen IIIc = 14, anagen IV = 15, anagen V = 16, and anagen VI = 17. Then, the rate (%) of a certain hair cycle stage in the total hair follicles and the median score in each mouse group were determined. The score of tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice was compared with the control mice. Statistical significance was determined using a Mann-Whitney's U-test. A difference of *P<0.01 was considered statistically significant.
Dystrophic catagen was defined according to a previous report [28] as early dystrophic catagen, mid dystrophic catagen, late dystrophic catagen, and dystrophic telogen. Then, the rate (%) of a certain hair follicle stage per total hair follicles was calculated in each mouse group. At least 40 hair follicles were evaluated in each mouse group (two mice/group).
Results
Impaired hair follicle morphogenesis in keratinocyte-specific TAK1-deficient mice
Keratinocyte-specific TAK1-deficient (Map3k7 fl/flK5-Cre) mice were generated, as previously described [12]. Map3k7 fl/fl mice were used as controls. Histological analysis of hair follicle development is shown in Figure 1A. Although hair germs (follicle precursors) and dermal condensations appeared in both types of mice at E15.5, the number of hair germs in Map3k7 fl/flK5-Cre mice was significantly lower than that in Map3k7 fl/fl mice (Fig. 1A). At E16.5, the hair germ further progressed into the hair peg stage of hair follicle morphogenesis in Map3k7 fl/fl mice at the rate expected for wild-type mice [29], while hair pegs were essentially absent in Map3k7 fl/flK5-Cre mice. Similarly, at P1-6, postnatal hair follicle development was severely impaired in Map3k7 fl/flK5-Cre mice (Fig. 1A).
Figure 1. Impaired hair follicle morphogenesis in keratinocyte-specific TAK1-deficient mice.

(A) Histological analysis of hair follicle development from E14.5 to P6. Map3k7 fl/flK5-Cre were keratinocyte-specific TAK1-deficient mice [12]. Map3k7 fl/fl mice were used as controls. Arrows indicate hair germs or hair pegs. Scale bar, 100 µm. (B) The hair morphogenesis stage of each hair follicle in each mouse group was evaluated as described previously [13]. Then, the rate (%) of a certain hair morphogenesis stage in the total hair follicles (left panel) and the median score in each mouse group (right panel) were determined. The score of Map3k7 fl/flK5-Cre mice was compared with Map3k7 fl/fl mice. Statistical significance was determined using a Mann-Whitney's U-test. *P<0.01.
Hair follicle morphogenesis was quantitatively analyzed. The morphogenesis stage and the median score of each mouse group are shown in Fig. 1B. Hair follicle morphogenesis indicators were significantly delayed in Map3k7 fl/flK5-Cre mice, as evident from their lower hair morphogenesis score, compared with Map3k7 fl/fl mice at P3 and P6 (Fig. 1B).
Generation of tamoxifen-inducible keratinocyte-specific TAK1-deficient mice
Because germline targeting of TAK1 greatly disrupted hair morphogenesis and, thus, precluded a meaningful analysis of subsequent hair follicle cycling, we next used tamoxifen-inducible keratinocyte-specific TAK1-deficient mice (Map3k7 fl/flK14-Cre-ERT2), in which Cre-ERT2 was expressed in the epidermis under the control of the K14 promoter [24]. Southern blot analysis demonstrated efficient deletion of the floxed allele in the epidermis of tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice (Fig. 2).
Figure 2. Southern blot analysis.
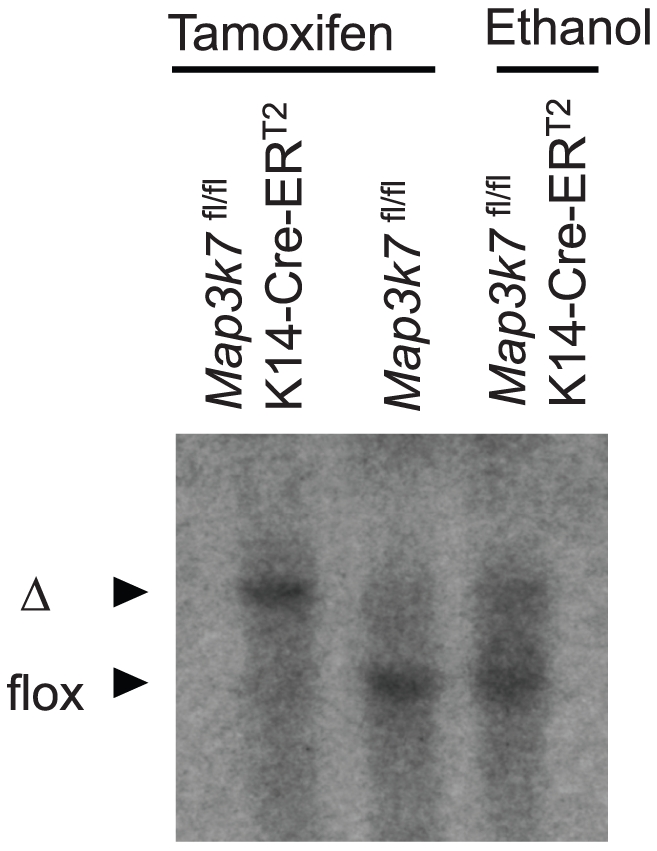
Genomic DNA prepared from the ear skin of mice treated with tamoxifen solution (15 µL/ear) or ethanol for 5 consecutive days was digested with XbaI and EcoRI. Southern blot analysis for the deletion of the floxed Tak1 allele was performed as described previously [1]. Cre expression resulted in excision of the floxed allele (flox) and generated the deleted allele (Δ) of Map3k7.
Although, the skin sample for Southern blot analysis contained non-keratinocyte cells, such as Langerhans cells, melanocytes, or fibroblasts, the band of tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice was a single-recombined band. Since, the majority of this skin sample consists of keratinocytes, flox band of non-keratinocyte cells may not be apparent in this blot. Ethanol-treated Map3k7 fl/flK14-Cre-ERT2 mice also show a minor recombination band, presumably due to slightly leaky Cre- ERT2 activity.
Keratinocyte-specific TAK1 deletion results in hair loss in adolescent mice
In the first experiment, tamoxifen was simply applied to the dorsal skin of 8 weeks old mice for 5 consecutive days. Two weeks after the application, the tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice started to lose their hair shafts, and this process continued for more than 4 weeks (Fig. 3). This phenotype suggested the involvement of TAK1 in hair follicle cycling.
Figure 3. Keratinocyte-specific TAK1 deletion results in hair loss in adolescent mice.

Tamoxifen was topically applied to the dorsal skin of the Map3k7 fl/flK14-Cre-ERT2 mice for 5 consecutive days to delete TAK1. The clinical appearance of the mice 4 weeks after the application is shown. As controls, Map3k7 fl/fl mice or Map3k7 fl/flK14-Cre-ERT2 mice were treated with tamoxifen or ethanol, respectively.
Keratinocyte-specific TAK1 deletion delays hair cycle progression in adolescent mice
To further dissect the role of TAK1 in hair follicle cycling, synchronized, active hair growth (anagen) was induced in resting (telogen) hair follicles by wax depilation [26]. This was followed by topical tamoxifen application to the dorsal skin, to delete TAK1 (Fig. 4A). At 2 weeks after synchronized anagen induction, hair shaft formation was noted in the control mice, as a macroscopic indicator of well-advanced anagen development, while hair shaft growth was not seen in the tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice (Fig. 4B). Histological analysis revealed that anagen progression was severely delayed in TAK1-deleted mice at 1–3 weeks (Fig. 4C).
Figure 4. Keratinocyte-specific TAK1 deletion delays hair cycle progression in adolescent mice.

(A) Schedule of tamoxifen application. The hair cycle was synchronized to anagen phase by wax depilation, and tamoxifen was topically applied to the dorsal skin of Map3k7 fl/flK14-Cre-ERT2mice to delete TAK1. As controls, Map3k7 fl/fl mice or Map3k7 fl/flK14-Cre-ERT2 mice were treated with tamoxifen or ethanol, respectively. (B) Clinical appearance at the indicated time point after the depilation. At 2 weeks, hair shaft formation was noted in the control mice, while hair shaft growth was not seen in the tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice. (C) Histological analysis at the indicated time point after the depilation. Anagen progression was severely delayed in TAK1-deleted mice at 1–3 weeks. Scale bar, 100 µm.
Quantitative hair cycle histomorphometry and hair cycle score calculation (Fig. 5) confirmed that depilation-induced anagen progression was severely delayed in TAK1-deleted mice at 1–3 weeks, while anagen development progressed as expected in TAK1-competent mice. Taken together, these data suggest that TAK1 is an important regulator of early anagen development in telogen hair follicles, although TAK1 does not appear to be indispensable for anagen induction.
Figure 5. Quantitative hair cycle analysis.

Hair cycle stage of each hair follicle in each mouse group in Fig. 4 was evaluated according to our previous report [27]. Then, the rate (%) of a certain hair cycle stage in the total hair follicles (left panel) and the median score in each mouse group (right panel) were determined. The score of tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice was compared with tamoxifen-treated Map3k7 fl/fl mice or ethanol-treated Map3k7 fl/flK14-Cre-ERT2 mice. Statistical significance was determined using a Mann-Whitney's U-test. *P<0.01.
Although leaky Cre- ERT2 activity was noted in ethanol-treated Map3k7 fl/flK14-Cre-ERT2 mice (Fig. 2), there was no significant difference between ethanol-treated Map3k7 fl/flK14-Cre-ERT2 mice and tamoxifen-treated Map3k7 fl/fl mice. Therefore, leaky activity of Cre- ERT2 seems to have a minimum effect in this experiment.
Keratinocyte-specific TAK1 deletion causes a transition from anagen to dystrophic catagen in adolescent mice
In the third experimental setup, TAK1 was deleted only 1 week after anagen induction (Fig. 6A). At 2 weeks after depilation, hair regrowth was reduced in the tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice (Fig. 6B), compared with controls. Histological analysis revealed that almost all of the hair follicles in tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice had prematurely entered the apoptosis-driven regression stage of hair follicle cycle (i.e., catagen; Fig. 6C).
Figure 6. Keratinocyte-specific TAK1 deletion causes a transition from anagen to dystrophic catagen in adolescent mice.

(A) Schedule of tamoxifen application. The hair cycle was synchronized to anagen phase by wax depilation. At 7 days after depilation, tamoxifen was applied for 5 days. (B) Clinical appearance of the mice 2 weeks after depilation. (C) Histological analysis of the mice 2 weeks after depilation. (D) Higher magnification of the tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice in (C). Scale bar, 100 µm. (E) Dystrophic catagen was defined according to a previous report [28]. Then, the rate (%) of a certain hair follicle stage per total hair follicles was calculated. Quantitative analyses confirmed that most of the hair follicles in tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice were in late dystrophic catagen, while those of controls were in anagen.
Interestingly, however, this accelerated catagen development was associated with striking pigmentary signs of hair follicle damage (dystrophy): many large, ectopically located melanin clumps, often larger than keratinocyte nuclei, were found not only in their normal location (i.e., the precortical hair matrix), but also eccentrically in the hair bulb periphery and in the epithelial strand of the involuting catagen hair follicles (Fig. 6D). Thus, TAK1 deletion induced “dystrophic catagen,” an indicator of major hair follicle damage [28]. Quantitative analyses (Fig. 6E) confirmed that most of the hair follicles in tamoxifen-treated Map3k7 fl/flK14-Cre-ERT2 mice were in late dystrophic catagen, while those of controls were in anagen. These data suggest that TAK1 is essential for maintaining a functional anagen phase.
Discussion
Here, we show by mouse genomics and targeted deletion experiments that TAK1, a member of the NF-κB pathway that has chiefly been recognized as a mediator of innate and adaptive immunity [1], [2], [4], [5], [6], [30], is also a key component of the molecular machinery that controls murine hair growth. Consistent with our previous discovery that TAK1 regulates keratinocyte growth, differentiation, and apoptosis [12], we now show that the selective deletion of TAK1 in keratinocytes retards hair follicle induction, morphogenesis, and anagen development, and is required for the maintenance of normal anagen. This newly identified role of TAK1 in hair follicle development and cycling implicates TAK1 as a novel player in complex organ remodeling events and epithelial-mesenchymal interactions, which can be modeled by murine hair follicles [11].
The TAK1-NF-κB pathway regulates not only immune responses [1], [30], but also epithelial function [12]. Because the NF-κB pathway controls pro-inflammatory responses, deletion of this pathway was expected to suppress epithelial inflammation. Unexpectedly, however, deletion of TAK1, IKK-β, or IKK-γ was found to result in severe skin inflammation (including abscess formation) [12], [31], [32], [33]. Similarly, a lack of NF-κB signaling produced by the conditional ablation of IKKγ or IKKα and IKKβ in the intestinal epithelium caused severe chronic intestinal inflammation in mice [34]. This suggests that a continuous, basal level of NF-κB activation may be required to maintain epithelial integrity and homeostasis and to suppress excessive skin inflammation. In the current study, we add to the established role of TAK1 in murine skin the novel function of hair growth control.
Since TAK1 regulates keratinocyte function [12], there is a possibility that hair follicle defects in TAK1-deficient mice might be attributed to such abnormal capacities of keratinocytes rather than reflecting the specific function of TAK1 in hair follicle regulation. However, clinical skin pheno types and histological abnormalities were not apparent at birth and started to appear at P2 in Map3k7 fl/flK5-Cre mice [12]. On the other hand, E15.5 is the time point when the defect of hair follicle development became apparent (Fig. 1). Therefore, the defect of hair follicle development is primarily due to the defect of TAK1 signaling during embryogenesis, rather than the functional defect of keratinocytes.
Recently, TAK1 binding protein (TAB) 2 has been identified as a binding partner of EdaR-associated death domain protein (EDARADD) using a yeast two-hybrid screening [35]. In 293 cells, endogenous and overexpressed TAB2, TRAF6 and TAK1 were co-immunoprecipitated with EDARADD [35]. Furthermore, dominant negative forms of TAB2, TRAF6 and TAK1 blocked the NF-κB activation induced by EDARADD in 293 cells[35]. Therefore, it is suggested that TAK1 is involved in hair follicle development. However, the actual role of TAK1 in hair follicle has not been studied before.
The coats of mice contain four major hair follicle subpopulations: guard hairs, awl and auchene hairs, and zigzag hairs. Formation of each kind of hair follicle starts at E14, E16, and E18-P3, respectively and the regulatory mechanisms of hair follicle development are slightly different among them [7], [36]. Epidermal NF-κB activity is first observed in the placodes of primary guard hairs at E14.5 [36]. In the absence of NF-κB activity, downstream events, such as maintenance of Wnt signaling and an increase of Wnt10a, Wnt10b, and Dkk4 expression, are impaired [8] and further placode down-growth does not occur in primary guard hair follicles [36]. In Map3k7 fl/flK5-Cre mice, the number of hair germs was significantly reduced at E15.5, indicating that the development of primary guard hair follicles was greatly impaired. Impaired development of primary guard hair follicles at E15.5 can be explained by the absence of NF-κB activity, due to TAK1 deficiency, consistent with a model in which TAK1 is involved in the EdaA1/EdaR/NF-κB pathway [35] (see Fig. 7). The appearance of a few primary hair placodes might be explained by incomplete TAK1 deletion at E15.5.
Figure 7. Model for the signaling pathways involved in hair follicle development.

In the development of primary hair placodes, TAK1 is involved in the Edar/NF-κB pathway. In secondary hair placodes, NF-κB (TAK1)-independent pathways regulate hair placode development.
In contrast, EdaR/NF-κB activity is dispensable for the induction of awl/auchene hair follicles, as seen in tabby, downless, and cIκBαΔN mice, even though EdaR/NF-κB-defective, awl/auchene hair follicles subsequently produce abnormal awl-like hair shafts [36], [37]. EdaR/NF-κB independent Wnt/β-catenin signaling is required for this process [8]. Thus, the placodes that became visible in Map3k7 fl/flK5-Cre mice at E16.5 are likely to represent placodes of awl/auchene hair follicles. In a recent study, analyses of Eda and EdaR homologue Troy double-mutant mice revealed that, in addition to primary guard hair follicles, awl/auchene hair follicles were defective in these mice [38]. This study suggested that EdaR and Troy redundantly activate an NF-κB independent pathway, via TRAF6, to develop awl/auchene hair follicle placodes. Therefore, it is conceivable that placode development in Map3k7 fl/flK5-Cre mice is controlled by this NF-κB- (and TAK1)-independent pathway (Fig. 7).
Besides NF-κB signaling, recent studies indicate the implication of TAK1 in multiple signaling pathways such as MAP kinases and AP1 signaling [1], [39], supporting the alternative possibility that, in addition to the NF-κB signaling pathway, multiple signaling pathways may be also involved in hair follicle regulation downstream to TAK1. Although some TNF receptor family activate JNK pathway in addition to NF-κB, Edar shows only weak activation of JNK/AP-1 pathway [14], [20]. In contrast to Edar, Troy [40] leads to a strong activation of JNK pathway, but weak activation of NF-κB [20], [40]. Therefore, it is possible Troy/TAK1/JNK/AP-1 pathway is involved in hair morphogenesis. However, this point should be further clarified.
An interesting difference was observed between Map3k7 fl/flK5-Cre mice and tabby, downless, and cIκBαΔN mice. While tabby, downless, and cIκBαΔN mice produced abnormal awl-like hair shafts, Map3k7 fl/flK5-Cre mice exhibited a prolonged morphogenesis period and did not develop hair shafts, even after skin transplantation onto normal control mice (unpublished, preliminary findings). This suggests that TAK1 deletion also affects the morphogenesis of the secondary hair follicles.
Although numerous molecular players have been identified as powerful regulators of hair follicle cycling, the exact molecular machinery that drives the elusive “hair cycle clock” remains unclear [29], [41], [42], [43]. For example, IGF-1, HGF, glial-derived neurotrophic factor, and VEGF are known to prolong the duration of anagen, while fibroblast growth factor 5, TGFβ1 and TGFβ2, IL-1β, NT-3, estrogen receptor-mediated signaling, and IFN-γ are all known to induce the anagen-catagen transition [29], [41], [43], [44], [45]. The expression of regulatory molecules is controlled by upstream signals, such as those provided by the NF-κB, Wnt/β-catenin, bone morphogenetic protein (BMP), and Shh-Gli pathways [29], [41], [42]. On the basis of our findings, TAK1 should now be considered a part of the molecular machinery of the anagen induction.
In the present study, we have shown that TAK1 deletion severely delays the telogen-anagen transition, although it is not completely suppressed, while deletion of TAK1 in anagen follicles prematurely induces catagen and damages normal hair follicle function. Thus, TAK1 activity is important, but not essential, for anagen initiation and progression, yet is essential for the maintenance of mature anagen follicles, with loss of TAK1 activity resulting in catagen induction. The next challenge is to dissect the upstream and downstream signals of TAK1. Because NF-κB is thought to be a downstream signal of TAK1 in hair morphogenesis, and because strong NF-κB activity is detected in the anagen matrix of pelage follicles of adult mice [37], NF-κB is a major candidate downstream signal of TAK1 in anagen induction. Conversely, EdaR is a plausible candidate upstream signal of TAK1 because EdaA1 and EdaR have already been shown to be involved in hair follicle cycling [9], [21], [22].
After chemical, biological, or physical damage, hair follicles develop abnormalities that are collectively called hair follicle dystrophy [28]. Low follicular damage induces the “dystrophic anagen” response. Severe follicular damage induces the “dystrophic catagen” response, characterized by an immediate anagen termination. In Fig. 6, the hair follicle stage was defined as “dystrophic catagen”. The development of “dystrophic catagen” indicates TAK1-deletion-induced follicular damage is comparatively higher and similar to those of high dose cyclophosphamide-induced alopecia. These suggest that TAK1-deletion-induced dystrophic catagen could be a model of chemotherapy-induced alopecia.
The evidence reported here, that TAK1, a critical mediator of inflammation [1], [30], is also involved in hair morphogenesis and anagen induction.
Acknowledgments
We thank Manabu Ohyama (Keio University, Tokyo, Japan), Teruko Tsuda, and Eriko Tan (Ehime University) for providing significant technical assistance.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was supported in part by grants from the Ministries of Education, Culture, Sports, Science, and Technology of Japan to K.S. and K.H., by Health and Labor Sciences Research Grants (Research on Intractable Diseases) from the Ministry of Health, Labor, and Welfare of Japan to K.H., by the Deutsche Forschungsgemeinschaft (DFG 345 Pa/12-2) to R.P., and by the College de France to P.C. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 2.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 4.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 5.Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, et al. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003;326:105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 6.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, et al. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. Embo J. 2003;22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt-Ullrich R, Paus R. Molecular principles of hair follicle induction and morphogenesis. Bioessays. 2005;27:247–261. doi: 10.1002/bies.20184. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell. 2009;17:49–61. doi: 10.1016/j.devcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong X, Coulombe PA. Keratin 17 modulates hair follicle cycling in a TNFalpha-dependent fashion. Genes Dev. 2006;20:1353–1364. doi: 10.1101/gad.1387406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courtney JM, Blackburn J, Sharpe PT. The Ectodysplasin and NFkappaB signalling pathways in odontogenesis. Arch Oral Biol. 2005;50:159–163. doi: 10.1016/j.archoralbio.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Schneider MR, Schmidt-Ullrich R, Paus R. The hair follicle as a dynamic miniorgan. Curr Biol. 2009;19:R132–142. doi: 10.1016/j.cub.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Sayama K, Hanakawa Y, Nagai H, Shirakata Y, Dai X, et al. Transforming growth factor-beta-activated kinase 1 is essential for differentiation and the prevention of apoptosis in epidermis. J Biol Chem. 2006;281:22013–22020. doi: 10.1074/jbc.M601065200. [DOI] [PubMed] [Google Scholar]
- 13.Paus R, Muller-Rover S, Van Der Veen C, Maurer M, Eichmuller S, et al. A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J Invest Dermatol. 1999;113:523–532. doi: 10.1046/j.1523-1747.1999.00740.x. [DOI] [PubMed] [Google Scholar]
- 14.Mikkola ML, Thesleff I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003;14:211–224. doi: 10.1016/s1359-6101(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 15.Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13:409–416. doi: 10.1038/ng0895-409. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson BM, Brockdorff N, Formstone E, Ngyuen T, Kronmiller JE, et al. Cloning of Tabby, the murine homolog of the human EDA gene: evidence for a membrane-associated protein with a short collagenous domain. Hum Mol Genet. 1997;6:1589–1594. doi: 10.1093/hmg/6.9.1589. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava AK, Pispa J, Hartung AJ, Du Y, Ezer S, et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc Natl Acad Sci U S A. 1997;94:13069–13074. doi: 10.1073/pnas.94.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan M, Wang LC, Hymowitz SG, Schilbach S, Lee J, et al. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science. 2000;290:523–527. doi: 10.1126/science.290.5491.523. [DOI] [PubMed] [Google Scholar]
- 19.Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature. 2001;414:913–916. doi: 10.1038/414913a. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, Eby MT, Sinha S, Jasmin A, Chaudhary PM. The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J Biol Chem. 2001;276:2668–2677. doi: 10.1074/jbc.M008356200. [DOI] [PubMed] [Google Scholar]
- 21.Fessing MY, Sharova TY, Sharov AA, Atoyan R, Botchkarev VA. Involvement of the Edar signaling in the control of hair follicle involution (catagen). Am J Pathol. 2006;169:2075–2084. doi: 10.2353/ajpath.2006.060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mustonen T, Pispa J, Mikkola ML, Pummila M, Kangas AT, et al. Stimulation of ectodermal organ development by Ectodysplasin-A1. Dev Biol. 2003;259:123–136. doi: 10.1016/s0012-1606(03)00157-x. [DOI] [PubMed] [Google Scholar]
- 23.Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, et al. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci U S A. 1997;94:7400–7405. doi: 10.1073/pnas.94.14.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Indra AK, Warot X, Brocard J, Messaddeq N, et al. Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature. 2000;407:633–636. doi: 10.1038/35036595. [DOI] [PubMed] [Google Scholar]
- 25.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paus R, Stenn KS, Link RE. Telogen skin contains an inhibitor of hair growth. Br J Dermatol. 1990;122:777–784. doi: 10.1111/j.1365-2133.1990.tb06266.x. [DOI] [PubMed] [Google Scholar]
- 27.Muller-Rover S, Handjiski B, van der Veen C, Eichmuller S, Foitzik K, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 28.Hendrix S, Handjiski B, Peters EM, Paus R. A guide to assessing damage response pathways of the hair follicle: lessons from cyclophosphamide-induced alopecia in mice. J Invest Dermatol. 2005;125:42–51. doi: 10.1111/j.0022-202X.2005.23787.x. [DOI] [PubMed] [Google Scholar]
- 29.Paus R, Foitzik K. In search of the “hair cycle clock”: a guided tour. Differentiation. 2004;72:489–511. doi: 10.1111/j.1432-0436.2004.07209004.x. [DOI] [PubMed] [Google Scholar]
- 30.Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, et al. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol. 2006;18:1405–1411. doi: 10.1093/intimm/dxl082. [DOI] [PubMed] [Google Scholar]
- 31.Stratis A, Pasparakis M, Markur D, Knaup R, Pofahl R, et al. Localized inflammatory skin disease following inducible ablation of I kappa B kinase 2 in murine epidermis. J Invest Dermatol. 2006;126:614–620. doi: 10.1038/sj.jid.5700092. [DOI] [PubMed] [Google Scholar]
- 32.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, et al. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 34.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 35.Morlon A, Munnich A, Smahi A. TAB2, TRAF6 and TAK1 are involved in NF-kappaB activation induced by the TNF-receptor, Edar and its adaptator Edaradd. Hum Mol Genet. 2005;14:3751–3757. doi: 10.1093/hmg/ddi405. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt-Ullrich R, Tobin DJ, Lenhard D, Schneider P, Paus R, et al. NF-kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development. 2006;133:1045–1057. doi: 10.1242/dev.02278. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt-Ullrich R, Aebischer T, Hulsken J, Birchmeier W, Klemm U, et al. Requirement of NF-kappaB/Rel for the development of hair follicles and other epidermal appendices. Development. 2001;128:3843–3853. doi: 10.1242/dev.128.19.3843. [DOI] [PubMed] [Google Scholar]
- 38.Pispa J, Pummila M, Barker PA, Thesleff I, Mikkola ML. Edar and Troy signalling pathways act redundantly to regulate initiation of hair follicle development. Hum Mol Genet. 2008;17:3380–3391. doi: 10.1093/hmg/ddn232. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai H, Nishi A, Sato N, Mizukami J, Miyoshi H, et al. TAK1-TAB1 fusion protein: a novel constitutively active mitogen-activated protein kinase kinase kinase that stimulates AP-1 and NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2002;297:1277–1281. doi: 10.1016/s0006-291x(02)02379-3. [DOI] [PubMed] [Google Scholar]
- 40.Kojima T, Morikawa Y, Copeland NG, Gilbert DJ, Jenkins NA, et al. TROY, a newly identified member of the tumor necrosis factor receptor superfamily, exhibits a homology with Edar and is expressed in embryonic skin and hair follicles. J Biol Chem. 2000;275:20742–20747. doi: 10.1074/jbc.M002691200. [DOI] [PubMed] [Google Scholar]
- 41.Stenn KS, Paus R. Controls of hair follicle cycling. Physiol Rev. 2001;81:449–494. doi: 10.1152/physrev.2001.81.1.449. [DOI] [PubMed] [Google Scholar]
- 42.Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119:391–393. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- 43.Ohnemus U, Uenalan M, Inzunza J, Gustafsson JA, Paus R. The hair follicle as an estrogen target and source. Endocr Rev. 2006;27:677–706. doi: 10.1210/er.2006-0020. [DOI] [PubMed] [Google Scholar]
- 44.Mecklenburg L, Tobin DJ, Muller-Rover S, Handjiski B, Wendt G, et al. Active hair growth (anagen) is associated with angiogenesis. J Invest Dermatol. 2000;114:909–916. doi: 10.1046/j.1523-1747.2000.00954.x. [DOI] [PubMed] [Google Scholar]
- 45.Lindner G, Menrad A, Gherardi E, Merlino G, Welker P, et al. Involvement of hepatocyte growth factor/scatter factor and met receptor signaling in hair follicle morphogenesis and cycling. Faseb J. 2000;14:319–332. doi: 10.1096/fasebj.14.2.319. [DOI] [PubMed] [Google Scholar]