

Abstract
The trimeric human copper transporter 1 (hCTR1) is essential for copper uptake and is implicated in sensitivity to chemotherapy drugs. Using the cryoelectron microscopy (cryoEM) map of hCTR1 and evolutionary data, we constructed a Cα-trace model of the membrane region. The model structure, supported by mutagenesis data, was used to investigate global dynamics through elastic network models. Identified as dominant hinge regions, hCTR1’s MxxxM and GxxxG motifs were shown to have significant roles in functional movements characterized by the two slowest modes of motion. Both modes predicted significant changes at the wide cytoplasmic pore region; the slowest mode introduced a rotational motion around the pore central axis, whereas in the following mode the cytoplasmic parts of the helices approached and moved away from the pore center. In the most cooperative mode, the MxxxM motif in the extracellular narrow region remained static. The second mode of motion, however, predicted a cooperative rotational motion of this copper-binding motif, possibly reflecting activation at the pore’s extracellular entrance. We suggest a molecular mechanism of copper transport in which this motif serves both as a gate and as a selectivity filter. We also suggest residues that are responsible for pH activation.
Keywords: dynamics, hCTR1, mechanism, model structure, structural bioinformatics
Copper ions are vitally important in key biological processes such as clearance of free radicals and respiration in eukaryotes (1). Increased cellular concentration of copper, however, is toxic and causatively linked to Menkes and Wilson diseases (2, 3). Furthermore, copper was found to induce the formation of amyloidic protein structures, and excess copper has also been proposed to play roles in prion disorders (3, 4). Uptake of Cu+ is mediated by membrane proteins of the copper transporter (CTR) family, ubiquitously present in eukaryotic organisms (5). Human CTR1 (hCTR1) is considered the main copper uptake machinery (6) and is of particular interest owing to its role in accumulation of platinum-based chemotherapy agents (7). In the cytoplasm, Atox1 and other specialized chaperone proteins deliver the closely regulated copper ions to target proteins, including ATP7A/B (6). Interestingly, when hCTR1 was transfected in human cells, its activity was shown to be elevated in acidic pH relative to alkaline pH; more details regarding this detected pH dependency remain to be elucidated (8).
A three-dimensional (3D) intermediate-resolution structure of hCTR1 was recently determined using cryoEM techniques (9), providing a first glance into the unique fold of this homotrimeric protein. It was proposed that the central pore of the structure is mainly shaped by TM2 [the second transmembrane (TM) helix] from each of the subunits. Members of the CTR family are characterized by two previously identified sequence motifs: an MxxxM motif in TM2 and a GxxxG motif in TM3 (Fig. S1A and ref. 6), both entirely conserved. De Feo and coworkers approximately mapped the residues constituting these motifs onto the cryoEM map (9). Met150 and Met154 of the MxxxM motif, implicated in copper binding, were suggested to reside at the narrow extracellular entrance to the pore, whereas Gly167 and Gly171 of the GxxxG motif were proposed to mediate a tight interface between TM3 and TM1.
Many of the molecular and functional properties of the TM domain have remained obscure, because the positions of the individual residues could not be detected in the intermediate-resolution map. More elaborate structural data can provide additional clues needed to decipher the copper transport mechanism of hCTR1 and to gain insight into regulation and functional motion. Toward this goal, we explore the structure and dynamics of hCTR1 using computational approaches. We generated a Cα-trace model based on the method of Fleishman et al. (10) and subsequently, to provide insight into functional motion, carried out a normal mode analysis using elastic network models (11, 12).
An intermediate-resolution map for producing a TM model structure was first employed by Baldwin and coworkers, who constructed a Cα-trace model of vertebrate rhodopsin (13). The model was generated from a structure at 7 Å resolution in the membrane plane using sequence-based and biochemical constraints. When the crystal structure of higher resolution was later determined, the orientations of TM helices in the model were found to be quite accurate (3.2 Å rmsd) (14). Expanding on this pioneering approach, Fleishman and coworkers developed an automatic method for TM-model prediction from cryoEM maps based essentially on evolutionary conservation (10). This approach was used to model the bacterial multidrug resistance transporter EmrE (15). The model superimposed well on the X-ray structure, which came out later, with core rmsd of 1.4 Å (16).
Currently of renewed interest, normal mode analysis using elastic network models can provide valuable insight into equilibrium dynamics of TM proteins (12). Specifically, the Gaussian network model (GNM) (17, 18) and the anisotropic network model (ANM) (19) provide coarse-grained mode analysis to estimate the magnitudes and directions of thermal fluctuations. These techniques have proven useful in demonstrating long-time or large-scale functional motion of proteins. It is now recognized that structural rearrangements revealed by the most cooperative (or slow) modes of the normal mode analysis often correlate with functional motions in TM proteins; such motions include pore opening, ion gating, and signal transduction (12).
We present herein a study of the structure-function relationship of hCTR1. Our model structure proposes the precise locations and interactions of highly conserved sequence motifs, along with molecular-level interpretation of biochemical data. Our analysis of cooperative dynamics reveals potential functional motions that may offer insight into both the copper transport mechanism and pH regulation.
Results
Computing the Cα Model.
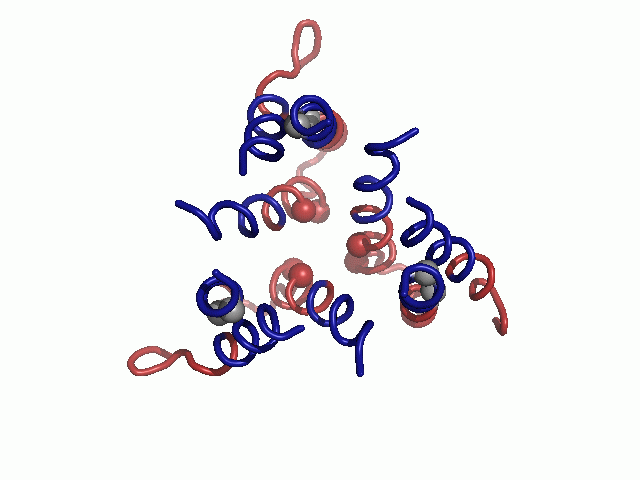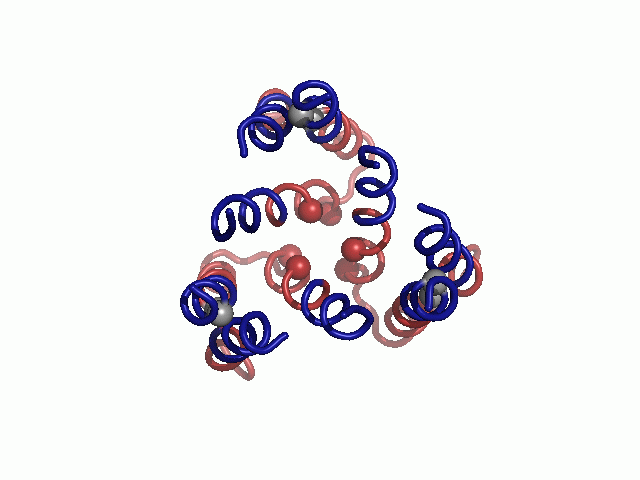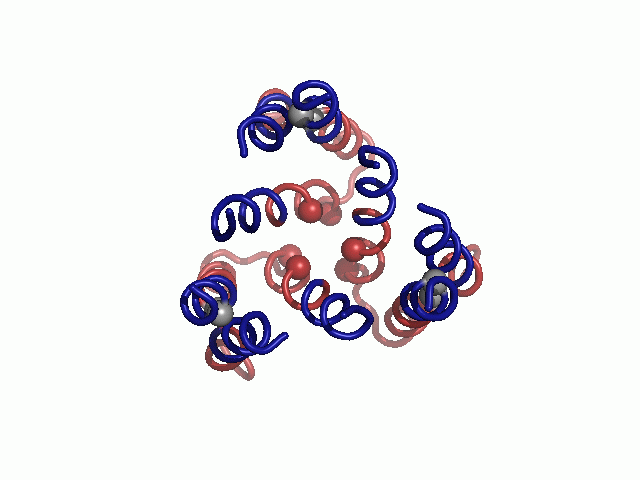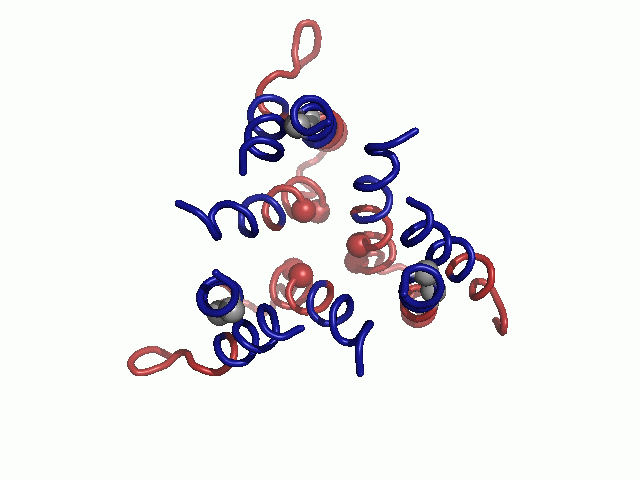
After manually extracting the principal axes of the TM segments from a cryoEM map, we constructed the TM helices, incorporating minor kinks in TM1 and TM3 to better match the contours of these helices in the density map (Fig. 1). We then investigated possible combinations of helix assignments and topological orientation of the helices to the principal axes. Reassuringly, the highest ranked helix assignment and topology were in agreement with those previously presented by De Feo et al. (9) (Table S1). Refining our initial model, we added the short, highly conserved loop between TM2 and TM3 (Fig. 1).
Fig. 1.

Evolutionary conservation profile of the hCTR1 full sequence and TM model. In both panels, the coloring is according to the evolutionary conservation scores calculated via the ConSurf server (34, http://consurf.tau.ac.il), with turquoise-to-maroon signifying variable-to-conserved positions (shown by the color bar). (A) The predicted topology of hCTR1. The three TM segments are marked. (B) Intracellular view of the Cα-trace model of the TM domain hCTR1, fitted onto the cryoEM map (9) (mesh). Residues of the MxxxM and GxxxG motifs are shown as spheres, and the three TM helices are marked on one of the subunits.
In the final model, highly conserved residues populated packed interfaces between helices or lined the central pore (Fig. 1B). Specifically, residues of the TM2 MxxxM and TM3 GxxxG motifs were predicted to reside in strategic locations. Met154 and Met150 formed two sequential threesomes of pore-facing methionine residues, designated herein as the “Met triads.” The first triad was composed of Met154 from each of the three subunits; the three Met154 residues were situated on a single plane at the pore’s extracellular end. Similarly, the second triad, comprising Met150, was located one helix turn deeper into the membrane core. Gly167 and Gly171 resided at the interface of TM3 with neighboring TM1, mediating the most densely packed contact region of the structure. Less conserved residues were situated in the slacker interfaces, whereas variable residues were exposed to the lipids, as anticipated. The physicochemical characteristics of the hCTR1 model are discussed in SI Text (Fig. S1).
Model Is Consistent with Experimental Data.
To investigate the model’s validity, we turned to published site-directed mutagenesis (Table S2), which was not used for model building. In addition to the elaborate mutagenesis data available for residues of the MxxxM and GxxxG motifs (20–23), we used data from Eisses and Kaplan, who performed mutations in several polar and charged residues in the TM domain (20), and from Aller et al., who initiated a Trp scan of the third TM helix of a homologous yeast protein, yCTR3 (23). The various studies, however, employed different experimental systems to assess the effects of mutations on protein structure and function. To simplify the comparison of the diverse biochemical data, we roughly divided the mutations into three groups: deleterious, neutral (nondeleterious), and partial (Table S2). In several positions, different substitutions had conflicting effects. We designated mutations in these positions as deleterious if at least one of the performed substitutions resulted in functional impairment.
Mapped on the model, sensitive-to-mutation positions lined the pore, resided in the tight TM1-3 interface or accommodated the conserved TM2-3 loop at the narrow extracellular entrance (Fig. 2 and Table S3). The majority of the nonsensitive residues, on the other hand, faced the lipids. Whereas some partially sensitive positions resided at wider pore-facing segments, the remaining ones, along with a few nonsensitive positions, were found at relatively slack helix–helix interfaces. As this pattern is anticipated for a TM protein, we conclude that our hCTR1 model is in good agreement with prior experimental results (Fig. 2 and Table S3). The agreement with experimental data implies that the model structure correctly depicts the essence of the elements of the native structure and the interactions between them.
Fig. 2.

Validation of the model structure via mutagenesis data. Mutagenesis data mapped on the hCTR1 model. Three different views are presented. Cα-atoms of residues for which mutational data are available are shown as spheres and colored according to the functional effect of the mutation, with red and green depicting positions sensitive and nonsensitive to substitutions, respectively. Positions in which mutations induced partial function are colored yellow.
Global Movements and Cooperative Dynamics.
Hinge regions governing global movements and the directions of functional motion are often identified through the lowest-frequency GNM and ANM modes, respectively (11, 12, 24). In addition to revealing the relative mobility of each residue, reflected in the GNM-derived mean-square fluctuations, the GNM analysis discloses interresidue positive and negative dynamical cooperativity. In this study, we examined fluctuations and cooperative dynamics of hCTR1 revealed by the five slowest GNM modes, along with directed motions derived from the corresponding symmetric ANM modes.
The two slowest GNM modes were degenerate, with the same contribution to the overall motion (approximately 6.5%, Fig. S2). Indeed, averaging the square displacements in these two modes resulted in a symmetric fluctuations profile for the three subunits. Remarkably, the hinges of this most cooperative mode (hereafter referred to as mode 1,2) precisely coincided with Met150 and Met154 of the TM2 MxxxM motif, along with the highly conserved Tyr147 (Fig. 3A). This is in good agreement with experimental and evolutionary data, which assigned the methionines a pivotal role in function (25, 26). Examining the cooperative motion of mode 1,2, we observed intrasubunit positive correlation among TM1, TM3, and the C-terminal half of TM2, from position 145 (Fig. 3 B and C). Between subunits, however, positive correlation was introduced exclusively between TM1, the C-terminus of TM2 and TM3 of one subunit and the N-terminal part of TM2 of one of the neighboring subunits (Fig. 3C). This designated TM2 as the sole mediator of cooperative dynamics and association of the three subunits in mode 1,2. We specifically note that Glu84 and His139 of a neighboring subunit showed positive correlation (Fig. 3C). Complementary to the cooperative dynamics of its matching GNM mode 1,2, the corresponding ANM mode (Table S4 and Fig. S3A) predicted that TM1, TM3, and the TM2-3 loop of the same subunit moved together with the N-terminal part of TM2 of an adjacent subunit in a rotational movement relative to the symmetry axis (perpendicular to the membrane plane) (Figs. 3C and 4 and Movie S1). This movement resulted in partial closing of the cytoplasmic pore region, mediated mostly by TM2. The pore’s extracellular entrance region, however, remained immobile (Fig. 4 A and C and Movie S1).
Fig. 3.

Square displacements and cooperative dynamics of the most cooperative GNM mode (mode 1,2). (A) Square displacements of mode 1,2. Prominent hinges Met150 and Met154 are marked, as well as the three chains. (B) Cooperative dynamics in mode 1,2. The three TM helices and chains are marked. Red-to-blue (according to the color bar) indicates positive-to-negative correlation of the structural dynamics between each residue pair. TM1, TM3 and the C-terminus of TM2 within each subunit are positively correlated with the N-terminus of TM2 of one of the two neighboring subunits. (C) Mapping of cooperative dynamics on the model. The hCTR1 model is shown as ribbons and colored according to the correlation of TM1 of chain A (TM1A) with the other residues. TM helices that showed positive correlation with TM1A are marked. Cα atoms of residues of the MxxxM and GxxxG motifs, as well as Glu84 and His139, are shown as spheres.
Fig. 4.

The ANM mode matched to GNM mode 1,2 (Table S4 and Movie S1). The conformations predicted by the ANM mode associated with GNM mode 1,2 (Table S4) are colored white, with approximated hinges at the MxxxM motif region in gray. As in GNM mode 1,2, motion was divided into three separate structural elements, each consisting of TM1 and TM3 from the same subunit, along with the N-terminus of TM2 of an adjacent subunit (marked). Conformations of one such structural element are colored according to the direction of motion, ranging from yellow to green and back. (A) Cytoplasmic view. (B) Side view. (C) Extracellular view.
The third slowest GNM mode and the subsequent two modes (the latter were combined into mode 4,5) displayed similar eigenvalues and fluctuations (Fig. S4A) and were aligned to two comparable ANM modes (Table S4 and Fig. S3B). Focusing on the third GNM mode, we detected three hinge regions, roughly in positions 76–77 of TM1, 146–149 of TM2 and 165–168 of TM3 (Fig. 5A); the latter range was, remarkably, associated with the GxxxG motif. Mapped on the model structure, these hinges were positioned together on a plane parallel to the membrane surface, dividing each TM segment into two distinct rigid extra- and intracellular parts approximately at its center. Exploring the cooperative dynamics of this GNM mode, we observed a symmetric behavior of the three subunits, in which the extra- and intracellular parts were of opposite correlations (Fig. 5B). The corresponding ANM mode (Fig. S3B and Table S4) indeed exhibited two distinct movements: At the narrow pore entrance, the extracellular parts of TM2 and TM3, along with the TM2-3 loop, rotated around the symmetry axis, altering the positioning of Met150 and Met154, among other residues (Fig. 5C and Movie S2). At the intracellular end, the intracellular regions of the TM helices approached and moved away from the putative symmetry axis at the pore center point (Fig. 5C and Movie S3).
Fig. 5.

Functional and cooperative dynamics of the third GNM and corresponding ANM modes. (A) Square displacements of the third GNM mode; the hinge positions are indicated on chain A. (B) Cooperative dynamics in GNM mode 3, colored as in Fig. 3B. Interresidue correlations are shown on one subunit only; the same correlations were obtained for the other two. The boundaries of the TM helices are marked. The cytoplasmic and the extracellular regions of the helices show opposite correlations. (C) The corresponding ANM mode (Table S4, Movie S2, and Movie S3). The conformations are colored according to the cooperative dynamics of GNM mode 3 and shown in three different views. The arrows mark the directions of the motions of the cytoplasmic (blue) and extracellular (red) helix regions.
Discussion
We generated a Cα-trace model of hCTR1’s TM domain that is supported by mutagenesis data. Whereas a previous study revealed the general architecture of hCTR1 and approximated the location of sequence motifs, herein we predict the positioning of the Cα-atoms of all residues within the TM domain. Our model reveals specific hydrophilic residues contributing to the pore (Fig. S1B) and others engaged in interhelical contacts, and it also shows the organization of residues of highly conserved sequence motifs (Fig. 1B). As our structural framework provides molecular-level insight into the hCTR1 structure, we believe it can advance future investigations of the structure-function relationship of hCTR1. To gain insight into the transport mechanism, we investigated functional motions associated with this unique structural fold. Our analysis revealed that the detected global movements unravel novel attributes, possibly associated with gating, selectivity, pH regulation, and overall conformational changes occurring during copper transport, as discussed below.
Accuracy of the hCTR1 Model and Its Suitability for Elastic Network Model Studies.
Inaccuracy of the Cα-trace model could result from various factors. First, we approximate that the rotation angles of the native helices may deviate by up to 20° from those proposed in the model owing to inherent limitations of the algorithm’s scoring function (10). Second, the low resolution along the membrane normal does not allow precise determination of the helix registers along the principal axes, which in turn may affect the contacts between tilted TM2 and neighboring helices. It may also affect the pore width. Substantial density, however, connects the density rods of TM2 and TM3 in the cryoEM map, presumably corresponding to the short loop between these segments (Fig. 1B). Consequently, we could determine the registers of TM2 and TM3 with higher confidence. The register of TM1 was then determined by aligning its hydrophobic center in a single plane with those of TM2 and TM3, as performed previously in the modeling of the EmrE transporter (15). This register allowed the Gly residues of the GxxxG motif to be packed against small residues in TM1, as they should be. Nevertheless, deviation in the precise boundaries of TM1 may still alter the position of TM1 relative to the other helices, affecting the predicted interhelical interfaces. Using the comparisons of the EmrE transporter (15) and rhodopsin (13) models to their later-determined structures [(16) and (27), respectively] as blind tests of the approach, we estimate that the hCTR1 model may deviate by up to 3.5 Å rmsd from the native structure.
Considering these limitations of the model’s accuracy, we chose to study motion using normal mode analysis with elastic network models, in which the global modes of motion are robust to the structure resolution and to small variations in topology (12). Whereas the slowest modes of motion were shown to be directly relevant to biological dynamics (11, 12), it should be noted that this approach relies, in essence, on the protein’s structural contacts, and the relation to its chemical properties is considered only implicitly. Hence, we cannot rule out that the actual dynamics may differ somewhat from those proposed by the elastic network model analysis. Additionally, as hCTR1’s extramembrane regions (Fig. 1A) could not be modeled, motion was predicted for the isolated TM domain. We note, however, that the motion of an isolated partial structure can provide insight into the motion of a larger structure: in a separate analysis, the slowest GNM modes of the hCTR1 monomer revealed minima found also for the trimer (Fig. S5). Thus, it is likely that the inherent motion of the trimer is relevant for the complete structure.
Minima detected in the shapes of the most cooperative GNM/ANM modes often represent functional residues (11, 12). Indeed, residues of the MxxxM and GxxxG motifs emerged as hinges in the most cooperative modes (Figs. 3A and 5A), whereas the GxxxG residues were also identified as peaks in the fastest GNM modes (SI Text and Fig. S6B). This could not be established a priori, because the analysis does not account for the residues’ physicochemical nature, level of evolutionary conservation, etc. We therefore deduced that beyond the available biochemical data (Fig. 2), these findings are supporting evidence for the validity of our model structure. They underscore the significance of new putative hinges and highly mobile regions in localized modes; i.e., residues 76–77, 80 and 146–149, as functionally and structurally plausible sites. Additionally, whereas the residues of GxxxG were previously attributed a purely structural role (9, 23), our results suggest that these conserved positions actually possess a dual structural-functional role. As hinge points, the flexible glycines facilitate and govern the global dynamics, which in turn enable the functionality of the protein.
Pore-Related Movements.
Two orthogonal motions affected the pore conformation, especially at its cytoplasmic end (Figs. 4A and 5C and Movie S1, Movie S2, and Movie S3). In one mode of motion, helices from neighboring subunits fluctuated cooperatively in a torsion-like movement around the pore center, and in the second the cytoplasmic portions of all helices approached and moved away from the pore center. The alternative “deformed” conformations sampled by both ANM modes conformed to the expected conservation pattern and hydrophobic nature of a TM protein, just as the original model structure did (Fig. 1B and Fig. S1B). This indicates that such structural dynamics could be accommodated by the unique architecture of hCTR1. In view of the cone-like structure of hCTR1, narrowing of the wide-cytoplasmic pore region depicted by these movements would facilitate a more compact translocation pathway for copper ions, embedded mainly in the hydrophilic residues of TM2 as well as in polar and charged residues at the edges of TM1 and TM3. Such conformation could be more suitable for transport and may disclose the relationship between possible inactive and active states.
Two Met Triads, One Ion?
The strategic positioning of the known copper-binding residues Met150 and Met154 at the extracellular entrance of the pore (Fig. 1B), forming two sequential triads, intuitively suggests that the residues function as the Cu+ selectivity filter; clearly, this proposition must still be supported by atomic-resolution structural data followed by detailed simulations. Indeed, hCTR1 is selective for copper over other ions (8), and three Met residues are capable of specifically stabilizing Cu+ (28). The findings of our dynamics investigation were in line with this notion, designating Met150 and Met154 as the structural hinges in the most cooperative mode (Fig. 3A), whereas the following mode revealed positive correlation in the motion of these positions (Fig. 5 B and C). Interestingly, in another study, the selectivity filter residues of five potassium channels, and in particular the GYG signature motif, emerged as the prominent hinge regions in the most cooperative mode as well (29).
Recently, De Feo et al. showed that the two Met triads evidently bind a single ion at a time, and the researchers indeed suggested that transport could involve conformational change in this region (9). This still raises an obvious question: If one triad is sufficient for a single ion, what is the functional significance of the two sequential triads of the CTR family? We suggest a potential explanation. Cooperative rotational movement of the extracellular end of TM2 alters the structural disposition of the Met triads (Fig. 5C and Movie S2), and a copper ion, initially bound to the (external) Met154 triad, may proceed along the pore to the Met150 triad, whereas the Met154 triad blocks the entrance of additional ions as an external gate (Fig. 6). The ions are transported one at a time, thus avoiding the disproportionation reaction between two colliding Cu+ ions that leads to Cu(s) and Cu+. Moreover, passing a single ion in each cycle seems like an effective way to tightly regulate the transport process, preventing free copper ions from readily entering living cells and generating reactive, toxic radicals (30). The suggested translocation pathway is complementary to previous findings regarding the functional role of the extramembrane domains at the N- and C-terminal regions (9, 20, 22).
Fig. 6.

Transport mechanism. In the proposed mechanism the copper ions are transported one at a time, and the TM region of hCTR alternates between four conformations. The three subunits are shown in red, blue, and gray, with residues of interest depicted in dark gray and the copper ion in yellow. Step 1: In the basal state, ions do not bind to the TM domain, the Met154 triad serves as an external gate and (unprotonated) His139 does not interact with Glu84. Our model structure is suggested to depict the basal conformation, because it was computed from a cryoEM map determined at pH 7.4 (9), a condition in which hCTR1 exhibits low activity according to Lee et al. (8). Step 2: Activation resulting, for example, from pH shift or ion binding, involves conformation change at the cytoplasmic end, along with a rotational movement at the extracellular end. A copper ion binds to the Met154 triad, and (protonated) His139 interacts with Glu84, stabilizing the conformation. Step 3: Following another conformational change at the extracellular end, the copper ion is passed on to the Met150 triad and the Met154 closes the pore entrance, thereby preventing the entry of a second ion. Step 4: The copper ion passes freely through the polar pore. It is yet to be revealed how copper ions are passed through to the C-terminus and from there to the copper chaperones.
Glu84 and His139, Active State and pH Dependence.
As Glu84 and His139 are highly conserved, hydrophilic residues embedded in the TM domain, one might intuitively infer that both possess functional significance. On the basis of mutagenesis findings, Eisses and Kaplan indeed designated these residues as functional (20), although the residues’ specific functional roles are yet to be determined. Building the side-chains of these amino acids in the model structure, we realized that Glu84 of TM1 and His139 of TM2 from adjacent subunits could possibly interact. Moreover, these adjacent positions showed positively correlated dynamics in GNM mode 1,2 (Fig. 3C), suggesting that they move together in the most global mode. Interestingly, hCTR1’s activity is pH-regulated; the transporter is more active in pH 5.5 than in pH 7.5 (8). Considering the above and assuming that His139 maintains its physiological pKa in the protein environment, we suggest that an interaction between Glu84 and His139 might stabilize an active conformation of hCTR1 in low pH, where His139 would probably be protonated. In higher pH, however, His139 would be deprotonated, resulting in a weaker interaction with Glu84 and destabilization of the putative active state. Indeed, this may clarify not only the molecular basis of hCTR1’s pH-regulated activity but also the obscure hyperactivity observed for the H139R mutant (20), which can interact with Glu84 in a higher pH range as well.
Mechanism of Cu+ Transport.
Combining structural data, functional motion, cooperative dynamics, and available mutagenesis, we attempted to provide additional insight into the transport mechanism of hCTR1’s TM region (Fig. 6). The Met150 and Met154 triads, by controlling gating and selectivity, regulate the passing of a single ion in every cycle, possibly via the directed motion presented in Fig. 5C and in Movie S2. Fluctuations at the cytoplasmic ends could modify the pore conformation (Figs. 4A and 5C, Movie S1, and Movie S3), adjusting the hydrophilic environment encountered by the copper ion for optimal transport. The intersubunit contact Glu84-His139 may stabilize such an active conformation, favorable at low-pH conditions. Notably, the positive correlation between the TM2-3 loop, TM1 and TM3 in the slowest mode (Fig. 3C) might hint at vital allosteric interactivity between the cytoplasmic and extracellular parts of the TM domain. This interactivity would be crucial for transducing signals stimulated, for example, by binding of an ion in the extracellular side, or, alternatively, by pH change. These suggestions can be useful to guide future experimental efforts and should be further assessed in light of emerging empirical investigations. Solid empiric validation for these structure-based mechanistic interpretations, however, would be best obtained through at least one, but preferably more, experimentally determined high-resolution conformational states of hCTR1.
Methods
Building the Model Structure.
We approximated the helix boundaries in the hCTR1 sequence and calculated the conservation scores of each amino acid position. We also manually extracted the principal axes of the helices from the hCTR1 cryoEM map, introducing kinks to follow the helix density rods as precisely as possible (9). Because TM1 and TM3 are partly bent in the map, we introduced into the original algorithm (10) a new feature that allowed incorporation of the kinked axes. Using the same scoring function, which favored conformations in which conserved or charged amino acids were buried in the protein core while variable or uncharged amino acids were exposed, we constructed Cα-trace helices and separately rotated each helix around its principal axis using a 5° increment (Fig. S7). The final model was based on the best-scored rotational angle for each of the nine (3 × 3) helices. We then generated the short TM2-3 loop (positions 155–160) using the protein local optimization program (31). Further elaboration regarding the model-building procedure is provided in SI Text.
Normal Mode Analysis with Elastic Network Models.
In this study, we employed the slow modes of motion as detected by the GNM and ANM models (17–19, 32). As the underlying potential function of the GNM is more physically realistic than that of the ANM, the former model is more robust in the prediction of fluctuations (11, 24). We therefore employed the slowest modes from the GNM analysis to detect the magnitudes of fluctuations and to identify cooperative dynamics (11, 17, 18). In accordance with previous definitions (12, 33), we designated regions of lowest mobility (as determined by the GNM); i.e., minima, as “hinges” in cases in which the regions were located between segments of opposite dynamical correlation. The normalized correlations between the residue fluctuations describe the dynamical cooperativity; two positively-correlated residues move in the same direction, whereas two negatively-correlated positions move in opposite directions from each other. Through the analysis of positively and negatively correlated fluctuations in the slowest modes, we identified the dynamic domains and the cooperation between them, implicated in the overall motion of the protein. As the slowest modes often reflect functional motion (11, 12, 24), the dynamically-correlated domains were expected to represent functional units.
GNM is isotropic and cannot account for the direction of the fluctuations. Therefore, we incorporated ANM-derived slow modes (19) for predicting the directions of motion and conformations, in compliance with the fluctuations described by the matching GNM modes (Table S4 and Fig. S3). Additionally, the predicted global modes for symmetric structures may include modes that maintain the structural symmetry with the same type of deformation in all monomers or in a subgroup of monomers (e.g., refs. 29 and 33). Thus, we superimposed the fluctuations of detected degenerative modes, obtaining symmetrically shaped deformations that more accurately described the global motion. More details are available in SI Text.
Supplementary Material
Acknowledgments.
We thank Vinzenz Unger for helpful discussions. This work was supported by Israel Science Foundation Grant 611/07 (N.B.-T.), and by the North Atlantic Treaty Organization traveling Grant CBP.MD.CLG 983009 (T.H. and N.B.-T.). M.S. was supported by the Edmond J. Safra Bioinformatics program at Tel Aviv University. T.H. acknowledges the Turkish Academy of Sciences (TUBA).
Note Added in Proof.
While the manuscript was in review, a full Trp scan of TM1 and TM2 of hCTR1 was published (35). Our model structure is in excellent agreement with this data (Fig. S8).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0914717107/-/DCSupplemental.
References
- 1.Pena MM, Lee J, Thiele DJ. A delicate balance: homeostatic control of copper uptake and distribution. J Nutr. 1999;129(7):1251–1260. doi: 10.1093/jn/129.7.1251. [DOI] [PubMed] [Google Scholar]
- 2.DiDonato M, Sarkar B. Copper transport and its alterations in Menkes and Wilson diseases. Biochim Biophys Acta. 1997;1360(1):3–16. doi: 10.1016/s0925-4439(96)00064-6. [DOI] [PubMed] [Google Scholar]
- 3.Strausak D, Mercer JF, Dieter HH, Stremmel W, Multhaup G. Copper in disorders with neurological symptoms: Alzheimer’s, Menkes, and Wilson diseases. Brain Res Bull. 2001;55(2):175–185. doi: 10.1016/s0361-9230(01)00454-3. [DOI] [PubMed] [Google Scholar]
- 4.Zanusso G, et al. pH-dependent prion protein conformation in classical Creutzfeldt–Jakob disease. J Biol Chem. 2001;276(44):40377–40380. doi: 10.1074/jbc.C100458200. [DOI] [PubMed] [Google Scholar]
- 5.Dumay QC, Debut AJ, Mansour NM, Saier MH., Jr The copper transporter (Ctr) family of Cu+ uptake systems. J Mol Microb Biotech. 2006;11(1–2):10–19. doi: 10.1159/000092815. [DOI] [PubMed] [Google Scholar]
- 6.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4(3):176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 7.Kuo MT, Chen HH, Song IS, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26(1):71–83. doi: 10.1007/s10555-007-9045-3. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Pena MM, Nose Y, Thiele DJ. Biochemical characterization of the human copper transporter Ctr1. J Biol Chem. 2002;277(6):4380–4387. doi: 10.1074/jbc.M104728200. [DOI] [PubMed] [Google Scholar]
- 9.De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Three-dimensional structure of the human copper transporter hCTR1. Proc Natl Acad Sci USA. 2009;106(11):4237–4242. doi: 10.1073/pnas.0810286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleishman SJ, Harrington S, Friesner RA, Honig B, Ben-Tal N. An automatic method for predicting transmembrane protein structures using cryo-EM and evolutionary data. Biophys J. 2004;87(5):3448–3459. doi: 10.1529/biophysj.104.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahar I, Rader AJ. Coarse-grained normal mode analysis in structural biology. Curr Opin Struct Biol. 2005;15(5):586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bahar I, Lezon TR, Bakan A, Shrivastava IH. Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chem Rev. 2010;110(3):1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldwin JM, Schertler GF, Unger VM. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol. 1997;272(1):144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 14.Fleishman SJ, Unger VM, Ben-Tal N. Transmembrane protein structures without X-rays. Trends Biochem Sci. 2006;31(2):106–113. doi: 10.1016/j.tibs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Fleishman SJ, et al. Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J Mol Biol. 2006;364(1):54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 16.Chen YJ, et al. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104(48):18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys Rev Lett. 1997;79(16):3090–3093. [Google Scholar]
- 18.Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2(3):173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 19.Atilgan AR, et al. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80(1):505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eisses JF, Kaplan JH. The mechanism of copper uptake mediated by human CTR1: A mutational analysis. J Biol Chem. 2005;280(44):37159–37168. doi: 10.1074/jbc.M508822200. [DOI] [PubMed] [Google Scholar]
- 21.Liang ZD, Stockton D, Savaraj N, Tien Kuo M. Mechanistic comparison of human high-affinity copper transporter 1-mediated transport between copper ion and cisplatin. Mol Pharmacol. 2009;76(4):843–853. doi: 10.1124/mol.109.056416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puig S, Lee J, Lau M, Thiele DJ. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J Biol Chem. 2002;277(29):26021–26030. doi: 10.1074/jbc.M202547200. [DOI] [PubMed] [Google Scholar]
- 23.Aller SG, Eng ET, De Feo CJ, Unger VM. Eukaryotic CTR copper uptake transporters require two faces of the third transmembrane domain for helix packing, oligomerization, and function. J Biol Chem. 2004;279(51):53435–53441. doi: 10.1074/jbc.M409421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rader AJ, Chennubhotla C, Yang L-W, Bahar I, Cui Q. Normal Mode Analysis—Theory and Applications to Biological and Chemical Systems. London: Chapman & Hall; 2006. The Gaussian network model: Theory and applications; pp. 41–63. [Google Scholar]
- 25.Nose Y, Rees EM, Thiele DJ. Structure of the Ctr1 copper trans‘PORE’ter reveals novel architecture. Trends Biochem Sci. 2006;31(11):604–607. doi: 10.1016/j.tibs.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 26.De Feo CJ, Aller SG, Unger VM. A structural perspective on copper uptake in eukaryotes. Biometals. 2007;20(3–4):705–716. doi: 10.1007/s10534-006-9054-7. [DOI] [PubMed] [Google Scholar]
- 27.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, Nadas IA, Kim MA, Franz KJ. A Mets motif peptide found in copper transport proteins selectively binds Cu(I) with methionine-only coordination. Inorg Chem. 2005;44(26):9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
- 29.Shrivastava IH, Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys J. 2006;90(11):3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59(5):1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson MP, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 32.Bahar I, Kaplan M, Jernigan RL. Short-range conformational energies, secondary structure propensities, and recognition of correct sequence-structure matches. Proteins. 1997;29(3):292–308. doi: 10.1002/(sici)1097-0134(199711)29:3<292::aid-prot4>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 33.Valadie H, Lacapcre JJ, Sanejouand YH, Etchebest C. Dynamical properties of the MscL of Escherichia coli: A normal mode analysis. J Mol Biol. 2003;332(3):657–674. doi: 10.1016/s0022-2836(03)00851-9. [DOI] [PubMed] [Google Scholar]
- 34.Landau M, et al. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Feo CJ, Mootien S, Unger VM. Tryptophan scanning analysis of the membrane domain of CTR-copper transporters. J Membrane Biol. 2010;234(2):113–123. doi: 10.1007/s00232-010-9239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
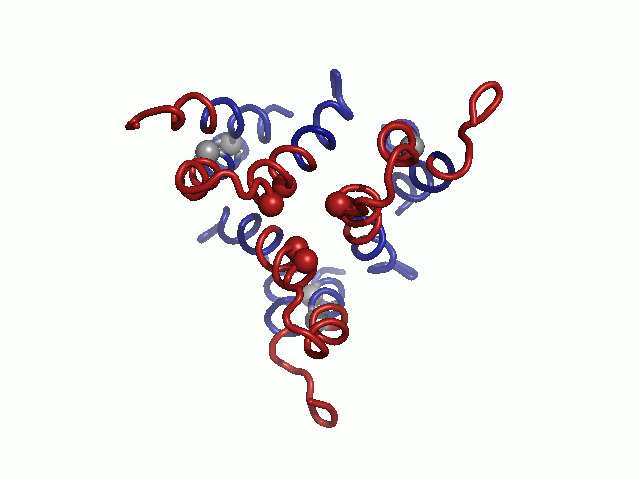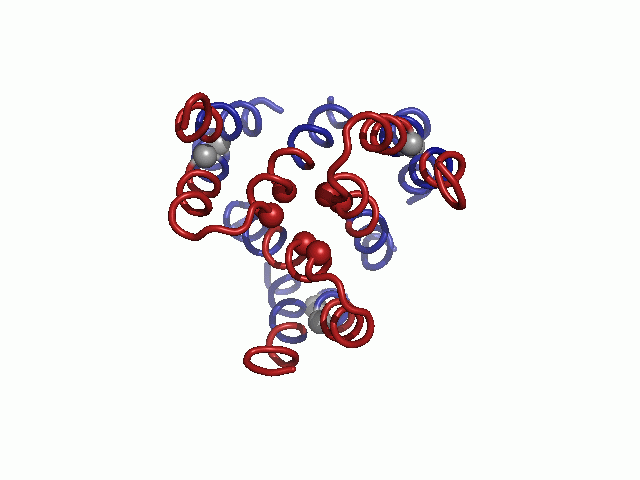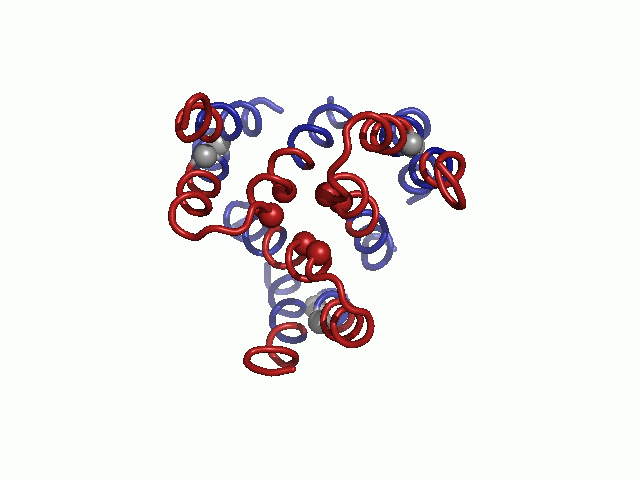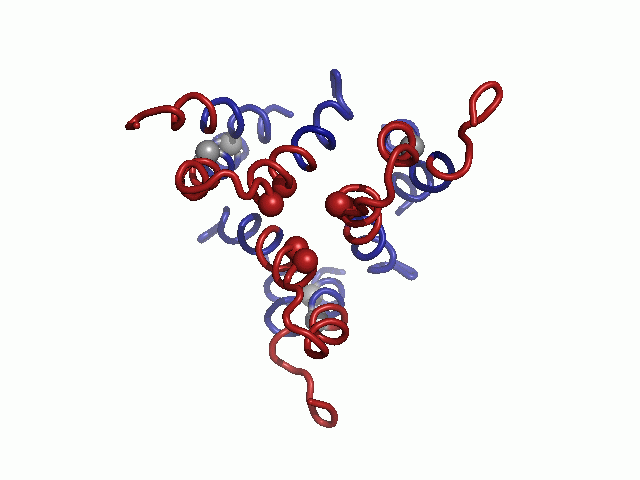{kind=link}
{kind=link}