

Abstract
NMR structural studies of membrane proteins (MP) are hampered by complications in MP expression, technical difficulties associated with the slow process of NMR spectral peak assignment, and limited distance information obtainable for transmembrane (TM) helices. To overcome the inherent challenges in the determination of MP structures, we have developed a rapid and cost-efficient strategy that combines cell-free (CF) protein synthesis, optimized combinatorial dual-isotope labeling for nearly instant resonance assignment, and fast acquisition of long-distance information using paramagnetic probes. Here we report three backbone structures for the TM domains of the three classes of Escherichia coli histidine kinase receptors (HKRs). The ArcB and QseC TM domains are both two-helical motifs, whereas the KdpD TM domain comprises a four-helical bundle with shorter second and third helices. The interhelical distances (up to 12 Å) reveal weak interactions within the TM domains of all three receptors. Determined consecutively within 8 months, these structures offer insight into the abundant and underrepresented in the Protein Data Bank class of 2–4 TM crossers and demonstrate the efficiency of our CF combinatorial dual-labeling strategy, which can be applied to solve MP structures in high numbers and at a high speed. Our results greatly expand the current knowledge of HKR structure, opening the doors to studies on their widespread and pharmaceutically important bacterial signaling mechanism.
Keywords: backbone NMR structure, cell-free synthesis, combinatorial selective labeling
Histidine kinase receptors (HKRs) are part of a two-component system, in which an HKR in the bacterial inner membrane transmits a signal to a response regulator located in the cytoplasm (1). This signaling system constitutes the predominant signal transduction mechanism by which bacteria interact with their environment (1), however, many aspects of its signaling mechanism are unknown. Based on the part of the receptor used to sense environmental stimuli, HKRs are grouped into three structural classes. Class 1, the largest class, is characterized by the presence of an extracytoplasmic sensory domain that senses external stimuli and transmitting the signal across the membrane. Class 2 HKRs lack an apparent extracytoplasmic domain and the stimuli-sensing region is believed to be in the membrane domain itself. Class 3 is characterized by a cytoplasmic sensory domain. For this study, we selected QseC (class 1, quorum sensor), ArcB (class 2, aerobic respiratory control sensor), and KdpD (class 3, K+ sensor) as representatives from each of the three classes. HKRs are multidomain proteins with diverse domain structures and topologies (Fig. 1): QseC has two transmembrane (TM) helices with an intervening 130 amino acid periplasmic sensor domain, ArcB has two TM helices connected by a short periplasmic loop, and KdpD has four TM helices with short interhelical loops. The cytoplasmic regions of HKRs comprise a dimerization domain and a kinase domain, several structures of which have been reported (2–9), including that of ArcB. There are also several reported periplasmic sensor domain structures (10–13). However, due to the highly flexible multidomain nature of HKRs, there is still no structure of a full-length receptor, nor has any structural information on an HKR membrane domain been reported to date. Such a structure is essential to understand the mechanistic aspect of signal transduction.
Fig. 1.

Three classes of HKRs. (A) Schematic representation of TM domains of three classes of HKRs and (B) ribbon representation of 3D structures of the TM domains of E. coli HKRs ArcB, QseC, and KdpD.
Despite impressive progress in the structure determination of membrane proteins (MPs) by X-ray crystallography and NMR spectroscopy in recent years (see reviews in refs. 14 and 15), only about 250 structures of unique integral MPs have been determined, representing less than 1% of known protein structures (16). In addition to problems with expression, solubilization, and purification of integral MPs, X-ray and NMR methods are hampered with inherent technical difficulties. Diffraction quality crystals of integral MPs are very difficult to obtain because solubilized protein-detergent complexes do not usually form ordered crystal lattices. Furthermore, the mobility of TM helices leads to strong broadening of the signals in NMR spectra and presents problems with spectra quality, resonance assignment, and detection of long-range interactions, all of which are necessary to calculate the structure of a protein by NMR. Spin label-based paramagnetic relaxation enhancement (PRE) approaches have been used to address the paucity of long-distance constraints associated with α-helical integral MPs (17, 18). However, the cost of isotope labeling by in vivo heterologous expression in cells of both prokaryotic and eukaryotic origins is often prohibitive for NMR structural studies of even well-expressed MPs.
We addressed the aforementioned technical difficulties, developing a strategy for a cost-efficient structure determination of MPs that has allowed us to quickly solve the backbone structures of three HKR membrane domains by NMR. The chosen structures represent the three classes of this widespread bacterial TM signaling system and belong to the abundant group of 2–4 TM crossers, which is poorly represented in the Protein Data Bank (PDB). By studying the domains in isolation, we present the structural insights into the signal-transducing membrane domains of these receptors. The results demonstrate the efficiency of the combinatorial dual-labeling (CDL) strategy. Thus, based on synergy of fast cell-free (CF) expression and rapid NMR analysis, CDL opens up possibilities for accelerated structural analysis of MPs.
Results
Structures of MPs Produced in CF System are Comparable to Those from Escherichia coli.
To synthesize membrane domains of selected histidine kinases we used the precipitating CF (p-CF) expression mode (19), in which a protein is produced as a precipitate that is subsequently solubilized by a nondenaturing detergent (a comprehensive description is given in SI Materials and Methods). The p-CF mode is extremely useful because it allows NMR studies of MPs without purification (Fig. S1). Because all of the CF reaction components are soluble, they remain in a supernatant and are easily removable after reaction by pellet wash. As a result, the target MP can be expressed without affinity tags that might affect its structure or stability. Although reminiscent of bacterial inclusion bodies, the MP precipitate can be solubilized with a mild lipid-like detergent (19) unlike true inclusion bodies, which require strong denaturing conditions (20). Therefore, it is likely that the CF precipitate has at least a partially folded structure.
To verify this, we investigated the underlying structure of the precipitates by magic angle spinning solid-state NMR (MAS SS-NMR). Uniformly 13C-labeled ArcB(1-115) and KdpD(397-502) precipitates were measured directly after their production in the p-CF reaction. By measuring the deviation of the 13C chemical shifts away from their typical values for a random coil peptide, we could differentiate between helical, beta, and random coil secondary structures (21) in the precipitates. In these two samples, all of the observed 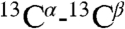 cross-peaks for alanine and valine residues fell within the regions of the 13C-13C correlation spectra typical for α-helices (21) (Fig. 2A and Fig. S2A). The
cross-peaks for alanine and valine residues fell within the regions of the 13C-13C correlation spectra typical for α-helices (21) (Fig. 2A and Fig. S2A). The 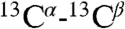 cross-peaks for valine and alanine residues that reside in the loop regions are probably broadened beyond the detectability limits in the MAS SS-NMR spectra. Moreover, the spectrum of the ArcB(1-115) precipitate is very similar to the spectrum of the ArcB(1-115) sample lyophilized after solubilization with a detergent (Fig. 2A, red contours). The presence of secondary structure in the precipitate was confirmed by solution NMR measurements of proton-deuterium (H-D) exchange following detergent solubilization. The results show that, in ArcB(1-115), 11 out of 16 valines and 5 out of 7 alanine are located in protected and therefore likely TM helical regions. Thus MAS SS-NMR analysis of chemical shifts together with solution NMR data on the exchange of the labile backbone protons in the precipitate (Figs. S3 and S4) have unambiguous interpretation: The TM helices of ArcB(1-115) and KdpD(397-502) existed as secondary structure elements before precipitation in a CF reaction.
cross-peaks for valine and alanine residues that reside in the loop regions are probably broadened beyond the detectability limits in the MAS SS-NMR spectra. Moreover, the spectrum of the ArcB(1-115) precipitate is very similar to the spectrum of the ArcB(1-115) sample lyophilized after solubilization with a detergent (Fig. 2A, red contours). The presence of secondary structure in the precipitate was confirmed by solution NMR measurements of proton-deuterium (H-D) exchange following detergent solubilization. The results show that, in ArcB(1-115), 11 out of 16 valines and 5 out of 7 alanine are located in protected and therefore likely TM helical regions. Thus MAS SS-NMR analysis of chemical shifts together with solution NMR data on the exchange of the labile backbone protons in the precipitate (Figs. S3 and S4) have unambiguous interpretation: The TM helices of ArcB(1-115) and KdpD(397-502) existed as secondary structure elements before precipitation in a CF reaction.
Fig. 2.

Verification of fold of ArcB(1-115), produced by CF system. (A) Overlay of 13C DARR-NMR spectra (213.765 MHz) of uniformly 13C-labeled ArcB(1-115) expressed in p-CF reaction. Black contours correspond to the spectra of the washed precipitant of the p-CF reaction. Red contours correspond to the spectra of the ArcB(1-115) sample lyophilized after the precipitant was solubilized in 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LMPG) . The green and cyan lines correspond to random coil 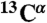 ,
, 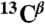 chemical shifts for valine (Val) and alanine (Ala), respectively; arrows show regions of chemical shifts corresponding to the α-helical conformation. (B and C) [1H-15N]-TROSY-HSQC spectra of 15N-labeled ArcB(1-115) expressed (B) by p-CF synthesis and in (C) E coli. (B) The precipitated protein was washed and solubilized in 5% LMPG. (C) The protein was extracted and purified from cell membrane with the FC-12 detergent and the detergent was exchanged to LMPG. Cross-peaks denoted by red arrows correspond to the tag linker residues in the E. coli-expressed protein.
chemical shifts for valine (Val) and alanine (Ala), respectively; arrows show regions of chemical shifts corresponding to the α-helical conformation. (B and C) [1H-15N]-TROSY-HSQC spectra of 15N-labeled ArcB(1-115) expressed (B) by p-CF synthesis and in (C) E coli. (B) The precipitated protein was washed and solubilized in 5% LMPG. (C) The protein was extracted and purified from cell membrane with the FC-12 detergent and the detergent was exchanged to LMPG. Cross-peaks denoted by red arrows correspond to the tag linker residues in the E. coli-expressed protein.
To further validate the p-CF expression system, we compared it with the standard E. coli expression in terms of sample quality and structure, as well as time and cost efficiency (Fig. 3 and Table S1). ArcB(1-115) was expressed and purified using both approaches. The E. coli system required five consecutive days, beginning with the transformation and growth of bacteria in minimal media and then extraction and purification of the protein from the cell membrane, in order to start the first NMR measurement. In contrast, using the p-CF synthesis, the first NMR measurement was possible in under 24 h, following an overnight expression and solubilization of the protein. The comparison of [1H-15N]-TROSY-HSQC spectra obtained from protein produced by both the CF (Fig. 2B) and the E. coli expression systems (Fig. 2C) shows that the positions of all the backbone cross-peaks are nearly identical. The difference in the number of the cross-peaks is a result of the slight difference in the constructs used (the E. coli expression requires an affinity tag). The backbone NOE data and 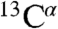 chemical shifts (Fig. S5) are also very similar for both samples. Taken together, these results lead us to conclude that structures of ArcB(1-115) prepared using the CF and the E. coli expression systems are the same.
chemical shifts (Fig. S5) are also very similar for both samples. Taken together, these results lead us to conclude that structures of ArcB(1-115) prepared using the CF and the E. coli expression systems are the same.
Fig. 3.

Comparison of performance of the CF system with the standard E. coli system. SDS-PAGE shows marker (M); CF reaction mixture (RM) before reaction at 0 h (1); CF RM after ArcB(1-115) expression at 15 h (2); precipitate after ArcB(1-115) p-CF (3); E. coli (EC) expressed ArcB(1-115) after extraction, purification, Tag cleavage, size-exclusion chromatography (SEC), detergent exchange on Q-sepharose, and concentration (4); arrow indicates ArcB(1-115).
Sequence-Optimized Isotope Labeling Scheme for Rapid Resonance Assignments.
Structure determination by NMR requires the sequential assignment of backbone resonances, a process that is particularly laborious for α-helical MPs (discussed in ref. 15). An alternative method of backbone resonance assignment utilizing dual selective 15N and 13C labeling and heteronuclear 13C-15N spin coupling was originally proposed by Kainosho and Tsuji (22) and improved later by the high selectivity of isotope labeling in CF synthesis and the efficiency of modern NMR heteronuclear techniques (23). Several approaches were built upon this method to perform site-selective screening (24) and to accelerate the resonance assignment by using dual combinatorial labeling (25–27). Simplified approach utilizing combinatorial selective 15N labeling was proposed for determination of the amino acid types for 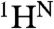 ,
, 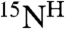 resonances (28). Combinatorial selective labeling approaches (reviewed in refs. 29 and 30) use a different subset of labeled amino acids in each sample in order to reduce the number of samples required to achieve the assignment as compared to one-by-one selective labeling. Most of the existing combinatorial approaches use labeling schemes that are sequence blind and optimized for globular proteins (25, 26, 28). These work for proteins that give good signal dispersion and good overall spectra quality, but are impractical for MPs.
resonances (28). Combinatorial selective labeling approaches (reviewed in refs. 29 and 30) use a different subset of labeled amino acids in each sample in order to reduce the number of samples required to achieve the assignment as compared to one-by-one selective labeling. Most of the existing combinatorial approaches use labeling schemes that are sequence blind and optimized for globular proteins (25, 26, 28). These work for proteins that give good signal dispersion and good overall spectra quality, but are impractical for MPs.
Expanding on the method originally proposed by Trbovic (27) to resolve ambiguities in the NMR assignment of CF-expressed MPs, we created a CDL strategy that is applicable to MPs (details are described in Materials and Methods). At the core of the CDL strategy is the individual optimization of the labeling scheme for each protein sequence and each selected set of amino acids for isotopic labeling. To derive these sequence-dependent schemes, we have developed the software MCCL (Monte Carlo Combinatorial Labeling, available at http://sbl.salk.edu/combipro), which calculates the optimal labeling combination for a given protein sequence with a defined number of samples using the Monte Carlo approach. As a result, the CDL method ensures that a minimal number of samples are needed to assign the type of the first and the second amino acid in every pair in a sequence, meanwhile minimizing the spectral complexity of each sample.
The CDL strategy was used for the design of [15N-13C]-labeling schemes for both KdpD(397-502) (Fig. 4B) and QseC(1-185) (Table S2), consisting of six and seven labeled samples, respectively. Within 1 day after spectra collection, 27% and 22% of 1H-15N cross-peaks for KdpD(397-502) and QseC(1-185), respectively, were unambiguously assigned. Additionally, the amino acid type was defined for 100% and 74% of the 1H-15N cross-peaks for KdpD(397-502) and QseC(1-185), respectively. The incomplete rapid assignment provides evenly distributed multiple starting points for traditional sequential assignment throughout the sequence. Starting from and building upon the results of the CDL-derived assignment, the standard sequential assignment procedure (31, 32) was tremendously accelerated, yielding an assignment for 100% of KdpD(397-502) and 76% of QseC(1-185) backbone resonances in 2–3 weeks. ArcB(1-115) resonances (96% of the backbone, 88% of Cβ, and most of Hα and Hβ) were assigned using the standard sequential assignment protocol. The rapid assignment of backbone resonances enabled us to analyze the secondary structure based on the chemical shift index and the hydrogen exchange data (summarized in Fig. S4) and to proceed with de novo NMR structure determination using the structural constraints collected for assigned backbone atoms.
Fig. 4.

The CDL strategy for the “point-directed assignment” of NMR spectra. (A) The [1H-15N] cross-peak in HSQC will appear only if the second residue in a pair is 15N labeled (second box tagged 1). The [1H-15N] cross-peak in the HSQC spectrum and the [1H-15N-13C] cross-peak in the HNCO spectrum will both appear only if the peptide group is double [13C-15N]-labeled (third box tagged 2). (B) Dual 15N/13C combinatorial selective labeling scheme designed specifically for backbone assignment of KdpD(397-502) (see details in SI Text). (C) Assignment of 1H-15N cross-peaks of KdpD(397-502) using a combinatorial scheme of selective 13C, 15N labeling, presented in panel B. The overlays of [1H-15N]-TROSY-HSQC (red contours) and [1H-15N] projection of TROSY-HNCO (blue contours) spectra are shown for each sample (I–VI). Absence of a cross-peak (tag “0”), a cross-peak present in TROSY only (tag “1”), and cross-peaks present in both the TROSY and the HNCO spectra (tag “2”) in each combinatorially labeled sample determine the code (sequence of the tags) for every cross-peak A, B, and C in a uniformly labeled sample. The scheme-designed code which is identical with the code determined from the recorded spectra defines a pair of amino acids for the 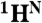 ,
, 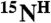 , and
, and 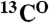 resonances.
resonances.
HKR TM Domain Structures.
Having verified that the p-CF system yields a folded protein and completed the NMR resonance assignments, we collected the NOE and PRE data required to calculate the structures of ArcB(1-115), QseC(1-185), and KdpD(397-502) (see Materials and Methods). The folds depicted in Figs. 1B and 5 show the helical nature of the TM domains from these three proteins whose calculated structures have a crystallographic equivalent resolution (33) in the range of 2.7–3.2 Å (Table 1).
Fig. 5.

Twenty superimposed structures of the TM domains of (A) ArcB(1-115), (B) QseC(1-185), and (C) KdpD(397-502). Backbones are shown for the stable regions: ArcB(1-115), residues 20-83; QseC(1-185), residues 10-38; and 156-185 and KdpD(397-502), residues 397-502. Consecutive TM helices are colored in the following order: green, blue, orange, and coral. Structures on the right are rotated 90° vertically.
Table 1.
Summary of NMR data and statistics for the calculated sets of 20 lowest energy structures of ArcB(1-115), QseC(1-185), and KdpD(397-502)
| ArcB(1-115) | QseC(1-185) | KdpD(397-502) | |
| NMR distance constraints | |||
| NOE* | 72 | — | — |
| PRE | 291 | 295 | 845 |
| Hydrogen bonds | 31 | 28 | 56 |
| NMR dihedral angle constraints | |||
| Phi | 37 | 42 | 72 |
| Psi | 37 | 42 | 72 |
| Structures statistics† | |||
| Violations (mean and SD) | |||
| Distance constraints, Å | 0.80 ± 0.01 | 1.18 ± 0.12 | 0.72 ± 0.02 |
| Dihedral angle constraints, ° | 10.3 ± 1.70 | 10.3 ± 0.40 | 5.70 ± 0.51 |
| Max. distance violation, Å | 0.17 ± 0.01 | 0.27 ± 0.03 | 0.22 ± 0.02 |
| Max. dihedral angle violation, ° | 1.74 ± 0.01 | 2.12 ± 0.40 | 0.81 ± 0.01 |
| Backbone rms deviation, Å | |||
| Average pairwise in the set | 1.45 ± 0.45 | 2.35 ± 0.85 | 1.61 ± 0.48 |
| To the mean structure | 1.41 ± 0.46 | 2.18 ± 0.86 | 1.56 ± 0.49 |
| Equivalent resolution‡ (Å) | 2.9 | 3.2 | 2.7 |
*Interresidue sequential (|i - j| = 1) constraints only.
†Applied in TM helical regions.
‡Calculated by PROCHECK program (33).
The structures of ArcB(1-115) and QseC(1-185) (Figs. 1B and 5 A and B) are both two-helical motifs whose helices have the appropriate length (30–32 Å) to cross the lipid bilayer. The large periplasmic signaling domain of QseC was not included in the structure calculations and so its TM domain is simply composed of two antiparallel weakly interacting α-helices with a crossing angle of 157 ± 4°. Similarly, the connecting loop in ArcB(1-115) is not well defined and its TM domain comprises two antiparallel α-helices with a crossing angle of 142 ± 6.5° and a distance of 11.1 Å at the closest point between the helices (all helical packing parameters are summarized in Table S3). In comparison, the crossing angle between two helices of HTR-II Transducer in complex with sensory Rhodopsin (34) is 169° and the distance between the helices is 10 Å, whereas for two tightly packed helices in dimeric human glycophorin A, the distance is just 6.4 Å (35). Prolines at position 67 of ArcB and positions 166 and 173 in QseC disrupt the helical hydrogen bond patterns (see the1H exchange data for ArcB(1-115) in Fig. S4) and create kinks of 22 ± 2° [ArcB(1-115)], 22 ± 5° and 24 ± 4° [QseC(1-185)] in the second helix, adding local flexibility to the helices and increasing the interhelical distances near the periplasmic side of the membrane, thus additionally weakening the helix–helix interactions.
The TM domain of KdpD comprises a four-helix bundle (Figs. 1B and 5C, and Table S3), in which the second and third helices are relatively short (23–24 Å) and loosely packed with the crossing angle of -165 ± 6° and the interhelical distance of ∼9.4 Å. The second helix interacts with the first and third helices only. The first and the fourth helix show the crossing angle of -157 ± 4°. These two helices weakly interact only near their cytoplasmic ends and this is the only consistent interaction involving the first helix, which causes the whole bundle to be packed rather loosely. It is possible that the loose helical packing observed in all three structures could be an artifact of the detergent solubilization or particular choice of HKR truncation. Nonetheless, the TM structures lead us to surmise that the loose helical packing provides an inherent flexibility in the TM domains and that this is perhaps essential to the mechanism of signal transduction across the membrane. The idea needs further verification including computational dynamics simulation studies.
Discussion
We have taken a deconstructionist approach to tackling the problems of MP structure determination. Because the size and mobility of MPs are limiting factors in NMR analysis, the study of individual domains has obvious advantages. Although domain-wise structure determination is nothing new to crystallography, the impetus is usually to remove offending domains (those that inhibit crystallization), which in the case of MPs is most often the membrane domain itself, or conversely engineering artificial soluble protein domains into MPs in order to obtain diffraction quality MP crystals (36). NMR techniques, on the other hand, are more amenable to the structural study of flexible membrane domains. However, this does not mean that such domains necessarily retain their native states, and the interpretation of the structures needs to be made with this caveat in mind.
The hallmark feature of these three TM domain structures appears to be their loose helical packing. The packing of TM α-helices is related to protein function and can be rigid, as in the case of channel pores like KcsA (37), ionotropic receptors like nAChR (38), the glutamate receptor channel (39), and tightly packed multihelical proteins like membrane respiratory enzymes (40), or flexible, as observed in the case of many metabotropic membrane receptors like G protein-coupled receptors (36) and kinase receptors. The majority of the solved structures of integral MPs (> 97%) represents proteins that actively or passively transport something (a molecule, ion, proton, or electron) across the biological membrane (channels and transporters) and proteins that tightly bind another molecule to catalyze a reaction (oxidases, ATPases, intramembrane proteases, etc.). The metabotropic membrane receptors are still a much underrepresented family in the PDB. Their primary role in a cell is to transmit signals through the membrane. Therefore, they do not require a well-defined conformational state of the TM domain that would be needed, for example, for coordinating transported ions or molecules. On the contrary, in order to transmit a signal, the TM domain must act as a conformational switch, an action which can be supported by its intrinsic mobility (41).
Furthermore, although it is known that HKRs act as dimers in the membrane, the fact that all of the structures are monomers can be explained by the absence of the cytoplasmic dimerization domain in our constructs. The dimerization of HKRs is unlikely mediated by TM domains as reflected by their loose helical packing. Further studies need to be done to address the significance of the loose helical packing of the transmembrane signaling mechanism of HKRs.
Concluding Remarks
The three structures presented in this study offer a glimpse into the abundant class of 2-4 TM crossers, underrepresented in the PDB, and provide an important inroad toward understanding the mechanistic aspects of the conformation-driven signal transduction process. The CDL strategy, founded in the synergy between the CF and NMR methods, opens up new possibilities for fast determination of backbone structures of MPs, especially those recalcitrant to crystallization. It is also complementary with the recently proposed approach by Raman et al. (42) for fast determination of backbone structure of large globular proteins. Backbone structures determined quickly by the CDL strategy would provide excellent starting points for high-throughput modeling of a large number of classes of integral MPs and further structure-function prediction.
Materials and Methods
Protein Expression, Characterization, and NMR Sample Preparation.
ArcB(1-115) was expressed with a thrombin-cleavable N-terminal His9 tag in E. coli BL21 DE3 cells (Invitrogen) and purified as described in SI Materials and Methods. ArcB(1-115), QseC(1-185), and KdpD(397-502) were expressed as p-CF in the absence of detergents (19). Washed precipitate was solubilized in 300 μL 5% (wt/vol) 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (Avanti Polar Lipids; Anatrace), 20 mM Mes-BisTris pH 5.5 [ArcB(1-115)] or pH 6.0 [QseC(1-185) and KdpD(397-502)]. The proteins were characterized by SDS-PAGE, surface-enhanced laser desorption/ionization MS analysis, and light scattering coupled with size-exclusion chromatography and refracting index measurements (Fig. S2). Preparation of the samples for H-D exchange experiments, solid-state NMR, and PRE measurements is described in SI Materials and Methods.
NMR Experiments.
Solid-state NMR, 2D 13C-DARR, experiments (43) were performed on Bruker AVANCE 850 spectrometer (Centre for Biomolecular Magnetic Resonance). High-resolution NMR spectra of ArcB(1-115) expressed in E. coli were recorded at 45 °C on a Bruker AVANCE 900 MHz spectrometer (Korea Basic Science Institute). NMR spectra of TM domains of ArcB, QseC, and KdpD expressed in the CF system were recorded at 45° and 37 °C on a Bruker AVANCE 700 MHz spectrometer (Salk). Details of NMR experiments are described in SI Materials and Methods.
CDL Strategy.
The CDL strategy comprises four steps: (i) designing the combinatorial labeling scheme with the MCCL program; (ii) parallel expression of 6–7 CDL samples using the p-CF expression system (0.5–1.0 mL of reaction mixture for each CDL sample) and solubilization in the same buffer to eliminate any differences in cross-peak positions; (iii) short measurement of [1H-15N]-TROSY-HSQC and 2D-HNCO spectra (about 0.5–1.5 h per spectrum) for each CDL sample, all the samples for the combinatorial assignment of a particular protein were measured in only 1–2 days, depending on the concentration of the protein; (iv) analysis of the spectra and assignment of 1H-15N cross-peaks using the CARA program (44).
Structure Calculation and Analysis.
The 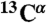 chemical shift deviations from random coil values were used to define backbone torsion angle restraints (45). Sequential distance constraints were derived from the integral intensities of NOE cross-peaks measured in 3D 15N-resolved TROSY-[1H,1H]-NOESY (mixing time 120 ms). The long-range distance constraints were derived by analysis of the PRE effect as described (18). An interactive procedure, which included structure calculation by the CYANA program (46) followed by the distance constraints refinement, was used to calculate the backbone spatial structures of ArcB(1-115), QseC(1-185), and KdpD(397-502). The summary of the constraints used in the calculation of the structures is presented in Table 1. The 20 conformers with the lowest target function of the last CYANA calculation cycle were energy-minimized using the CNS program (47). The helical packing parameters, such as interhelical crossing angles, interhelical distances, and helical kinks, were subsequently analyzed with the Helix Packing Pair program (48).
chemical shift deviations from random coil values were used to define backbone torsion angle restraints (45). Sequential distance constraints were derived from the integral intensities of NOE cross-peaks measured in 3D 15N-resolved TROSY-[1H,1H]-NOESY (mixing time 120 ms). The long-range distance constraints were derived by analysis of the PRE effect as described (18). An interactive procedure, which included structure calculation by the CYANA program (46) followed by the distance constraints refinement, was used to calculate the backbone spatial structures of ArcB(1-115), QseC(1-185), and KdpD(397-502). The summary of the constraints used in the calculation of the structures is presented in Table 1. The 20 conformers with the lowest target function of the last CYANA calculation cycle were energy-minimized using the CNS program (47). The helical packing parameters, such as interhelical crossing angles, interhelical distances, and helical kinks, were subsequently analyzed with the Helix Packing Pair program (48).
Point-Directed Assignment.
The point-directed assignment is based on selective dual-isotope (15N and 13C) labeling of two types of amino acids, located consecutively in a protein sequence. For every pair of residues in which the first amino acid is labeled with 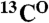 and the second amino acid is labeled with
and the second amino acid is labeled with 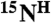 , cross-peaks in both the [15N-1H]-HSQC and the HNCO spectra arise (tag “2” in Fig. 4A). If the second amino acid in a pair is labeled with
, cross-peaks in both the [15N-1H]-HSQC and the HNCO spectra arise (tag “2” in Fig. 4A). If the second amino acid in a pair is labeled with 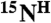 and the first amino acid is not labeled with
and the first amino acid is not labeled with 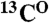 , there will only be a cross-peak in the HSQC spectrum (tag “1” in Fig. 4A). For pairs in which the second amino acid is not labeled with 15N, there will be no cross-peaks in either spectrum (tag “0” in Fig. 4A). Thus, by analyzing the presence and absence of cross-peaks in the [15N-1H]-HSQC and the HNCO spectra recorded from a selectively labeled sample, one can define the type of amino acids for backbone resonances participating in these cross-peaks and bind them to a particular amino acid pair. If a pair is unique in the sequence, an exact assignment of the
, there will only be a cross-peak in the HSQC spectrum (tag “1” in Fig. 4A). For pairs in which the second amino acid is not labeled with 15N, there will be no cross-peaks in either spectrum (tag “0” in Fig. 4A). Thus, by analyzing the presence and absence of cross-peaks in the [15N-1H]-HSQC and the HNCO spectra recorded from a selectively labeled sample, one can define the type of amino acids for backbone resonances participating in these cross-peaks and bind them to a particular amino acid pair. If a pair is unique in the sequence, an exact assignment of the 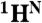 ,
, 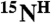 , and
, and 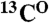 resonances to the residues that comprise that pair is instantly made.
resonances to the residues that comprise that pair is instantly made.
Numerically, each pair of residues in a given sample is specified by a tag depending on its labeling combination, as explained in Figure 4A. Therefore, in a series of combinatorially labeled samples a pair of residues is defined by a series of tags, that is, a code which is directly related to the presence or absence of cross-peaks in both spectra for each sample. The scheme-designed code which is identical with the code derived from the recorded spectra defines a pair of amino acids for the 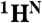 ,
, 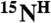 , and
, and 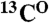 resonances. If the code is unique, i.e., there is only one pair in the sequence it corresponds to, the assignment of the
resonances. If the code is unique, i.e., there is only one pair in the sequence it corresponds to, the assignment of the 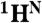 and
and 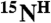 resonances to the second residue, as well as the assignment of the
resonances to the second residue, as well as the assignment of the 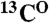 resonance to the first residue of the pair, is made. This simple analysis provides an unambiguous assignment for ∼30–40% of the backbone
resonance to the first residue of the pair, is made. This simple analysis provides an unambiguous assignment for ∼30–40% of the backbone 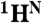 ,
, 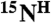 , and
, and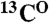 resonances and defines the amino acid type for the rest of the backbone
resonances and defines the amino acid type for the rest of the backbone 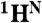 ,
, 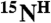 , and
, and 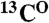 resonances, limiting the number of their possible positions in a sequence to as few as 2–4.
resonances, limiting the number of their possible positions in a sequence to as few as 2–4.
CDL Assignment Process.
The CDL assignment process is demonstrated for three KdpD(397-502) cross-peaks in Figure 4C. Here, an 1H-15N cross-peak for residue C is present in the TROSY spectra of samples I, III, IV, and V and in the HN plane of the HNCO spectrum of sample IV, therefore, its code is 101210 (the digit place corresponds to sample number). This code is unique and corresponds to the Phe481-Ala482 pair in the sequence, which gives us an unambiguous assignment for Ala482. Cross-peak B has the code 021102 and was assigned to three possible Ala-Val pairs in the sequence (Val411/472/483). Cross-peak A has the code 011101 and was assigned to nine possible pairs, with Val as the second amino acid in every pair and Arg, Val, or Thr, which are not labeled by 13C, as the first amino acid.
Supplementary Material
Acknowledgments.
This work was supported by National Institutes of Health (GM074929), Incheon Free Economic Zone, and World-Class University program (S.C.), and Korea Membrane Protein Initiative and 21C Frontier Microbial Genomics program (Y.H.J.). C.K. was partly supported by Deutsche Forschungs Gemeinschaft Research Fellowship.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The NMR-derived data (atomic coordinates, chemical shifts, and restraints) have been deposited in the Protein Data Bank, www.pdb.org, under accession codes 2KSD [ArcB(1-115)], 2KSE [QseC(1-185)], and 2KSF [KdpD(397-502)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1001656107/-/DCSupplemental.
References
- 1.Wolanin PM, Thomason PA, Stock JB. Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 2002;3(10) doi: 10.1186/gb-2002-3-10-reviews3013. REVIEWS3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Etzkorn M, et al. Plasticity of the PAS domain and a potential role for signal transduction in the histidine kinase DcuS. Nat Struct Mol Biol. 2008;15:1031–1039. doi: 10.1038/nsmb.1493. [DOI] [PubMed] [Google Scholar]
- 3.Rogov VV, et al. A new structural domain in the Escherichia coli RcsC hybrid sensor kinase connects histidine kinase and phosphoreceiver domains. J Mol Biol. 2006;364:68–79. doi: 10.1016/j.jmb.2006.07.052. [DOI] [PubMed] [Google Scholar]
- 4.Marina A, Mott C, Auyzenberg A, Hendrickson WA, Waldburger CD. Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J Biol Chem. 2001;276:41182–41190. doi: 10.1074/jbc.M106080200. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, et al. NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature. 1998;396:88–92. doi: 10.1038/23968. [DOI] [PubMed] [Google Scholar]
- 6.Tomomori C, et al. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat Struct Biol. 1999;6:729–734. doi: 10.1038/11495. [DOI] [PubMed] [Google Scholar]
- 7.Ikegami T, et al. Solution structure and dynamic character of the histidine-containing phosphotransfer domain of anaerobic sensor kinase ArcB from Escherichia coli. Biochemistry. 2001;40:375–386. doi: 10.1021/bi001619g. [DOI] [PubMed] [Google Scholar]
- 8.Kato M, Mizuno T, Shimizu T, Hakoshima T. Insights into multistep phosphorelay from the crystal structure of the C-terminal HPt domain of ArcB. Cell. 1997;88:717–723. doi: 10.1016/s0092-8674(00)81914-5. [DOI] [PubMed] [Google Scholar]
- 9.Rogov VV, Bernhard F, Lohr F, Dotsch V. Solution structure of the Escherichia coli YojN histidine-phosphotransferase domain and its interaction with cognate phosphoryl receiver domains. J Mol Biol. 2004;343:1035–1048. doi: 10.1016/j.jmb.2004.08.096. [DOI] [PubMed] [Google Scholar]
- 10.Pappalardo L, et al. The NMR structure of the sensory domain of the membranous two-component fumarate sensor (histidine protein kinase) DcuS of Escherichia coli. J Biol Chem. 2003;278(40):39185–39188. doi: 10.1074/jbc.C300344200. [DOI] [PubMed] [Google Scholar]
- 11.Cheung J, Bingman CA, Reyngold M, Hendrickson WA, Waldburger CD. Crystal structure of a functional dimer of the PhoQ sensor domain. J Biol Chem. 2008;283:13762–13770. doi: 10.1074/jbc.M710592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung J, Hendrickson WA. Structural analysis of ligand stimulation of the histidine kinase NarX. Structure. 2009;17:190–201. doi: 10.1016/j.str.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore JO, Hendrickson WA. Structural analysis of sensor domains from the TMAO-responsive histidine kinase receptor TorS. Structure. 2009;17:1195–1204. doi: 10.1016/j.str.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 14.McLuskey K, Roszak AW, Zhu Y, Isaacs NW. Crystal structures of all-alpha type membrane proteins. Eur Biophys J. 2010;39(5):723–755. doi: 10.1007/s00249-009-0546-6. [DOI] [PubMed] [Google Scholar]
- 15.Kim HK, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Mag Res Sp. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White SH. Biophysical dissection of membrane proteins. Nature. 2009;459(7245):344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- 17.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 18.Roosild TP, et al. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 19.Klammt C, et al. High level cell-free expression and specific labeling of integral membrane proteins. Eur J Biochem. 2004;271:568–580. doi: 10.1111/j.1432-1033.2003.03959.x. [DOI] [PubMed] [Google Scholar]
- 20.Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 21.Wishart DS, Sykes BD. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 22.Kainosho M, Tsuji T. Assignment of the three methionyl carbonyl carbon resonances in Streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution. Biochemistry. 1982;21:6273–6279. doi: 10.1021/bi00267a036. [DOI] [PubMed] [Google Scholar]
- 23.Yabuki T, et al. Dual amino acid-selective and site-directed stable-isotope labeling of the human c-Ha-Ras protein by cell-free synthesis. J Biomol NMR. 1998;11:295–306. doi: 10.1023/a:1008276001545. [DOI] [PubMed] [Google Scholar]
- 24.Weigelt J, van Dongen M, Uppenberg J, Schultz J, Wikström M. Site-selective screening by NMR spectroscopy with labeled amino acid pairs. J Am Chem Soc. 2002;124:2446–2447. doi: 10.1021/ja0178261. [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Pelton JG, Cho HS, Wemmer DE. Protein signal assignments using specific labeling and cell-free synthesis. J Biomol NMR. 2004;28:235–247. doi: 10.1023/B:JNMR.0000013697.10256.74. [DOI] [PubMed] [Google Scholar]
- 26.Parker MJ, Aulton-Jones M, Hounslow AM, Craven CJ. A combinatorial selective labeling method for the assignment of backbone amide NMR resonances. J Am Chem Soc. 2004;126:5020–5021. doi: 10.1021/ja039601r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trbovic N, et al. Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc. 2005;127:13504–13505. doi: 10.1021/ja0540270. [DOI] [PubMed] [Google Scholar]
- 28.Wu PS, et al. Amino-acid type identification in 15N-HSQC spectra by combinatorial selective 15N-labelling. J Biomol NMR. 2006;34:13–21. doi: 10.1007/s10858-005-5021-9. [DOI] [PubMed] [Google Scholar]
- 29.Ozawa K, Wu PS, Dixon NE, Otting G. N-Labelled proteins by cell-free protein synthesis. Strategies for high-throughput NMR studies of proteins and protein-ligand complexes. FEBS J. 2006;273:4154–4159. doi: 10.1111/j.1742-4658.2006.05433.x. [DOI] [PubMed] [Google Scholar]
- 30.Sobhanifar S, et al. Cell-free expression and stable isotope labelling strategies for membrane proteins. J Biomol NMR. 2010;46:33–43. doi: 10.1007/s10858-009-9364-5. [DOI] [PubMed] [Google Scholar]
- 31.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. pp. 130–161. [Google Scholar]
- 32.Clore GM, Gronenborn AM. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods Enzymol. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Gordeliy VI, et al. Molecular basis of transmembrane signalling by sensory rhodopsin II-transducer complex. Nature. 2002;419:484–487. doi: 10.1038/nature01109. [DOI] [PubMed] [Google Scholar]
- 35.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 36.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 38.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 39.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wittig I, Schagger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 41.Hendrickson WA. Transduction of biochemical signals across cell membranes. Q Rev Biophys. 2005;38:321–330. doi: 10.1017/S0033583506004136. [DOI] [PubMed] [Google Scholar]
- 42.Raman S, et al. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 44.Keller R. The Computer Aided Resonance Assignment Tutorial. Goldau, Switzerland: Cantina Verlag; 2004. [Google Scholar]
- 45.Luginbuhl P, Szyperski T, Wuthrich K. Statistical basis for the use of 13Calpha chemical shifts in protein structure determination. J Magn Reson Ser B. 1995;109:229–233. [Google Scholar]
- 46.Guntert P. Automated NMR structure calculation with CYANA. Method Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 47.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Dalton JA, Michalopoulos I, Westhead DR. Calculation of helix packing angles in protein structures. Bioinformatics. 2003;19:1298–1299. doi: 10.1093/bioinformatics/btg141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.