

Abstract
Two million Americans suffer from pulmonary emphysema, costing $2.5 billion/year and contributing to 100,000 deaths/year. Emphysema is thought to result from an imbalance between elastase and endogenous inhibitors of elastase, leading to tissue destruction and a loss of alveoli. Decades of research have still not resulted in an effective treatment other than stopping cigarette smoking, a highly addictive behavior. On the basis of our previous work, we hypothesize that small molecule inhibitors of human neutrophil elastase are ineffective because of rapid clearance from the lungs. To develop a long-acting elastase inhibitor with a lung pharmacodynamic profile that has minimal immunogenicity, we covalently linked an elastase inhibitor, similar to a trifluoro inhibitor that was used in clinical trials, to a 25-amino-acid fragment of human surfactant peptide B. We used this construct to prevent human neutrophil elastase-induced emphysema in a rodent model. The elastase inhibitor alone, although in a 70-fold molar excess to elastase in a mixture with <0.6% residual elastase activity, provided no protection from elastase-induced emphysema. Covalently combining an endogenous peptide from the target organ with a synthetic small molecule inhibitor is a unique way of endowing an active compound with the pharmacodynamic profile needed to create in vivo efficacy.
Keywords: lung cancer, resistant infections, tuberculosis, asthma
Inhalation is an important means of systemically delivering drugs that require immediate onset of action because the lungs provide a facile conduit to the bloodstream (1). Conversely, rapid lung clearance of small molecules causes a significant challenge for the development of pulmonary therapeutics. Rodent models of elastase-induced emphysema, for example, highlight the challenges for the development of effective drugs for pulmonary diseases. Potent in vitro human neutrophil elastase (HNE) inhibitors are cleared from the animals’ lungs in minutes and may even exacerbate HNE-induced emphysema (2), whereas one intratracheally instilled dose of elastase causes maximum lung damage after 4 wk (3). The combination of long-term chronic pulmonary damage that characterizes the progression of emphysema with the rapid lung clearance (4) and potential nephrotoxicity (5) of small molecule drugs indicates that there is a profound need for new ways of producing pulmonary therapeutics with long lung residence time pharmacodynamics.
Because emphysema is a chronic disease, any treatment will likely be administered over the course of the patient's life, and thus long-acting agents may induce an immune response that would negate the therapeutic effect. The ability of the treatment to access all regions of the vast lung surface area must also be considered. One way to slow the clearance of a small molecule inhibitor from the lung is to attach it to a larger polymer (6). For instance, the covalent linkage of a hydrophilic polymer to a peptidyl carbamate inhibitor increases the half-life by 100-fold in the lungs of hamsters and provides protection against HNE-induced emphysema (7). We hypothesized that covalent linkage of a potent small molecule HNE inhibitor to the N-terminal 1–25 residues of human surfactant peptide B (SP-B) would have (i) long lung residence time when administered intratracheally and provide long-term protection against emphysema (7), (ii) minimal immunogenicity because the majority of the construct is composed of a natural protein sequence that is normally present within the alveolar mucus lining, and (iii) distribution throughout the entire lung surface area providing pervasive protection against HNE challenge.
Although pulmonary surfactant is a complex mixture of lipids and peptides, SP-B is the clear choice as a covalent partner for an HNE inhibitor because <1% by weight mixed with lipids dramatically enhances adsorption, reduces surface tension, and restores normal pressure–volume mechanics in rat lungs depleted of surfactant (8). The N-terminal 1–25 (SP-B1-25) peptide fragment can be used because its surface-active properties are comparable to the whole SP-B 79-mer (9, 10). Discovery Labs has shown in phase III clinical studies that one dose of (Lys-Leu-Leu-Leu-Leu)4-Lys (11), a mimetic of SP-B1-25, added to cow lavage dramatically reduces mortality in severely preterm infants (12). These results indicate that SP-B1-25 mimetics have long lung residence time. Thus, the native sequence of this SP-B fragment covalently linked to an HNE inhibitor was evaluated in vitro and in vivo to determine if this approach may provide long-term protection against HNE-mediated emphysema. The peptide-inhibitor construct was significantly more effective at preventing emphysema in mouse lungs subjected to HNE treatment than was the small molecule inhibitor added alone at a higher molar concentration. Hence, the peptide-inhibitor construct appears to satisfy the emphysema therapeutic requirements of (a) coverage of the entire lung surface area, (b) prolonged duration of action, and (c) nonimmunogenicity.
Results
Table 1 shows the structures and molecular masses of the six compounds synthesized for this study. They consist of three small molecule tripeptide inhibitors (X0, X1, and X2), an N-terminal 1–25 peptide fragment of SP-B (SP-B1-25), and two peptide-inhibitor constructs (SP-B1-25-X1 and SP-B1-25-X2). The compounds were initially tested in a series of standard kinetic assays measuring HNE inhibition with synthetic and insoluble elastin sub-strates. The most effective small molecule inhibitor and peptide-inhibitor construct were then evaluated in an elastase-induced emphysema mouse model.
Table 1.
Summary of synthesized compounds
| ID | Classification | Structure | Molecular mass, Da |
| X0 | Small molecule inhibitor | 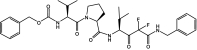 |
614 |
| X1 | Small molecule inhibitor | 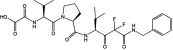 |
552 |
| X2 | Small molecule inhibitor | 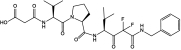 |
566 |
| SP-B1-25 | Peptide | NH2-F-P-I-P-L-P-Y-C-W-L-C-R-A-L-I-K-R-I-Q-A-M-I-P-K-G-COOH | 2927 |
| SP-B1-25-X1 | Peptide-inhibitor construct | 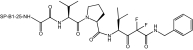 |
3461 |
| SP-B1-25-X2 | Peptide-inhibitor construct | 3475 |
In Vitro Assessment of Elastase Inhibitors.
The main goal of the in vitro assessment was to validate the HNE inhibitory activity of the synthesized compounds, particularly the peptide-inhibitor constructs, before in vivo testing. The elastin solubilization test was chosen for this primary task because it represents a physiologically relevant assay for HNE activity. The product-time curve of this assay is linear over a large period and provides a reasonable comparison of enzymatic reaction rates from a single-time-point sampling. The subsequent evaluation with kinetic assays (concentration-response and substrate-response tests) using a synthetic substrate and multiple-time-point sampling was included to establish some information on the inhibitory mechanisms although it was not the intention of this study to thoroughly characterize these compounds.
Elastin Solubilization Tests.
The ability of the compounds to inhibit the solubilization of elastin by elastase was determined and compared to heparin, a compound previously shown to be a potent elastase inhibitor (12). In Fig. S1A, the effects of the small molecule inhibitors X0, X1, and X2 (synthesized and characterized as described in Figs. S2 and S3) are shown at inhibitor/enzyme (I/E) molar ratios of 1, 5, and 12. Whereas all three exhibited increasing inhibition with increasing concentration, the magnitude of this trend was quite different for each sample. X1 appeared to be barely active, inhibiting the reaction by only 9% (vi/vo = 0.91) at the highest concentration (I/E ratio of 12). X2 was a much better inhibitor than X1; for example, at an I/E ratio of 12, it inhibited the reaction by 61% (vi/vo = 0.39). X0, however, was the most active inhibitor of the group. At the lowest concentration (I/E ratio of 1), this compound inhibited the reaction by 43% (vi/vo = 0.57), and at the higher concentrations, it was as good as or better than heparin (I/E ratio of 2.3), reaching 81% (vi/vo = 0.19) and 86% (vi/vo = 0.14) inhibition at I/E ratios of 5 and 12, respectively. Using 3H-elastin as the substrate and an I/E ratio of 30, inhibition of 99.4% (vi/vo = 0.006) was achieved with X0.
In Fig. S1B, the relative rates of elastin digestion in the presence of SP-B1-25 (characterized as described in Fig. S4), SP-B1-25-X1, and SP-B1–25-X2 are indicated for three I/E ratios of 1, 5, and 12 (14 for SP-B1-25). As expected, the SP-B1-25 peptide had no effect on the reaction at any concentration. However, when linked to the small molecule inhibitors, the peptide-inhibitor constructs showed increasing inhibitory action with increasing concentration, with SP-B1-25-X2 appearing more effective. For example, SP-B1-25-X1 inhibited the reaction by 4% (vi/vo = 0.96) at an I/E ratio of 1 and by 29% (vi/vo = 0.71) at an I/E ratio of 12, whereas SP-B1-25-X2 inhibited it by 10% (vi/vo = 0.90) and by 43% (vi/vo = 0.57) at the same I/E ratios. Even at the highest concentration (I/E ratio of 12), both SP-B1-25-X1 and SP-B1-25-X2 were less effective than heparin at an I/E ratio of 2.3.
Concentration-Response Tests.
Fig. S1 C and D indicates the results for five inhibitors and heparin at I/E ratios of 1, 10, and 100. It is worth noting that heparin performed as expected in these tests. Known for being a tight-binding, hyperbolic inhibitor (13), heparin significantly inhibited HNE hydrolysis by ∼57% at the lowest I/E ratio and then maintained approximately the same activity for the higher concentrations without approaching 100% inhibition.
In Fig. S1C, the small molecule inhibitors X0, X1, and X2 demonstrated an ability to completely suppress the reaction at the highest concentration (I/E ratio of 100); in fact, X0 and X2 showed this behavior even at the lower I/E ratio of 10. Nevertheless, these inhibitors share one attribute with heparin, substantial inhibition at an I/E ratio of 1. Both X0 and X2 per-formed better than heparin at this concentration, inhibiting the reaction by 90% (vi/vo = 0.10) and 64% (vi/vo = 0.36), respectively. X1 gave a weaker performance at this concentration but managed to inhibit by 70% (vi/vo = 0.30) at an I/E ratio of 10. On the basis of these results, X0, X1, and X2 appear to be tight-binding, linear inhibitors (nonclassical inhibitors) of HNE at these experimental conditions. However, even with this classification, the compounds represent a range of action from the very strong tight binding of X0 to the borderline tight binding of X1.
A comparison of the plots in Fig. S1D shows that the peptide-inhibitor constructs SP-B1-25-X1 and SP-B1-25-X2 behave differently from heparin. Both are rather weak inhibitors until they are present in large molar excess (I/E > 10). For example, at an I/E ratio of 1, SP-B1-25-X1 and SP-B1-25-X2 inhibited <10%, and at an I/E ratio of 10, they inhibited 13% (vi/vo = 0.87) and 25% (vi/vo = 0.75), respectively. However, when their concentrations were increased to an I/E ratio of 100, SP-B1-25-X2 almost completely inhibited the reaction (vi/vo = 0.01), and SP-B1-25-X1 was on track to do the same at a higher concentration (vi/vo = 0.15). From these results, SP-B1-25-X1 and SP-B1-25-X2 appear to be classical inhibitors (linear and not tight binding) of HNE under the conditions of this experiment. It is possible that the linking of the 25-amino-acid SP-B peptide alters the various options by which the small molecule inhibitor can access the elastase active site, thus producing a slightly different kinetic mechanism of inhibition than observed with the unlinked compounds. However, a more detailed analysis of the influence of these compounds on elastase kinetics is required to fully define the relative mechanisms of inhibition.
Substrate-Response Tests.
The results of the substrate-response tests for the small molecule inhibitors X0, X1, and X2 are shown in Fig. S1E. The Lineweaver–Burk plot illustrates the inhibitory action of X0 and X2 at 60 nM (I/E ratio of 0.5) and X1 at 240 nM (I/E ratio of 2) on HNE hydrolysis of Suc-Ala3-pNA over a substrate range of 0.6–6.0 mM. The data were fit by nonlinear regression to the Michaelis–Menten equation (Methods), and the best-fit values for the kinetic parameters, Km and Vm (Table S1) were used to plot the lines in Fig. S1E. For X1 and X2, the lines intersect the control at approximately the y axis, suggesting competitive inhibition, whereas for X0, the line intersects the control to the left of the y axis (but not on the x axis), suggesting mixed inhibition. It is suspected that X0 might also be a competitive inhibitor, but experimental measurements at higher substrate concentrations are required to correctly reflect the intersection point of this strong tight-binding inhibitor (14). As a result, X0, together with X1 and X2, was assumed to follow a competitive mechanism of inhibition. Although concentration-response tests at low inhibitor concentrations are necessary to determine the dissociation constant (Ki) of these tight-binding inhibitors, as a first approximation, all three compounds were treated as classical, competitive inhibitors. Values of Ki were estimated to be 12 nM for X0, 630 nM for X1, and 97 nM for X2. These Ki values are likely overestimates of the real values for the small molecule inhibitors. If the equations for classical inhibition and tight-binding inhibition are each solved for Ki, the results show that Ki (classical) is always greater than Ki (tight-binding), regardless of the mechanism (15, 16). Thus, it is not surprising that a smaller value of Ki = 0.4 nM was reported for X0 (17).
The Lineweaver–Burk plot in Fig. S1F displays the inhibitory effects of the peptide-inhibitor constructs SP-B1-25-X1 and SP-B1-25-X2 at a concentration of 1200 nM (I/E ratio of 10) over a substrate range of 0.6–6.0 mM. The best-fit values for Km and Vm (Table S1) were determined and used to generate the lines in Fig. S1F. The intersection of the lines on the y axis at the same value as the control indicates a competitive inhibition mechanism for both SP-B1–25-X1 and SP-B1-25-X2. Because these compounds fit the model for classical inhibitors (Fig. S1D), the values of Ki were calculated to be 2.3 μM for SP-B1-25-X1 and 1.6 μM for SP-B1-25-X2. A check of the constraint [E]o/Ki < 0.1, where [E]o is the initial enzyme concentration, confirmed negligible tight binding between these inhibitors and the enzyme (18).
In Vivo Assessment of Elastase Inhibitors.
The SP-B1–25 peptide covalently linked to a molecule from a tripeptide mimetic family that produced a clinical emphysema candidate (19) is shown in Table 1 (SP-B1–25-X2). The tripeptide mimetic molecule was chosen because a member of this family successfully passed phase I clinical trials (17). In standard kinetic assays, the tripeptide mimetic molecule X0 is an extremely tight-binding inhibitor of HNE with a Ki < 12 nM (Table S1), in agreement with the published results (17). SP-B1-25 possesses no measurable inhibition of HNE enzymatic activity, and the covalent construct of SP-B1–25 with the tripeptide mimetic (SP-B1-25-X2) has a Ki of 1.6 μM (Table S1). Using elastin substrate, a molar ratio of SP-B1-25-X2 to HNE of >12 was required to achieve 50% inhibition of HNE.
The first in vivo study was conducted by intratracheally instilling 50 μg of HNE into three anesthetized mice. One mouse died immediately, so all subsequent studies were done with 40 μg of HNE. Mice were anesthetized and intratracheally instilled with 100 μL of one of the six solutions listed in Table 2. After 2 h, select numbers of mice in four groups were subjected to bronchoalveolar lavage with saline, the lavage was centrifuged, and the red blood cell pellet was separated from the supernatant and weighed. The HNE group (HNE/PBS) showed bleeding, the saline group (PBS) had no bleeding, the tripeptide inhibitor group (X0/HNE) had a 34% reduction in blood cells by weight relative to the HNE group, and the tripeptide covalently linked to SP-B1-25 group (SP-B1–25-X2/HNE) had an 87% reduction in blood cells by weight relative to the HNE group. The results are summarized in Table 2.
Table 2.
Effect of HNE and inhibitors administered intratracheally on induction of hemorrhage at 2 h and airspace enlargement at 4 wk
| Treatment | Red blood cell pellet, g | Mean linear intercept, μm |
| PBS | 0 (3) | 44.40 ± 6.50 (4) |
| HNE/PBS | 0.053 ± 0.028 (4) | 51.45 ± 5.78 (10) |
| X0/PBS | — | 43.27 ± 3.82 (4) |
| X0/HNE | 0.035 ± 0.006 (4) | 53.27 ± 8.27 (8) |
| SP-B1-25-X2/PBS | — | 40.94 ± 1.59 (8) |
| SP-B1-25-X2/HNE | 0.007 ± 0.007 (3) | 45.82 ± 3.30 (8) |
Values are the mean ± SD (number of animals in parentheses). The red blood cell pellet was obtained by saline bronchoalveolar lavage of the lungs 2 h after treatment followed by centrifugation. The HNE/PBS and X0/HNE groups had larger red blood cell pellets than the PBS control, but the SP-B1-25-X2/HNE group did not. The mean linear intercept (MLI), a measure of mean alveolar size, was determined as described and increased as alveolar walls were destroyed. There were no significant differences by ANOVA among the three non-HNE controls (PBS, X0/PBS, SP-B1-25-X2/PBS), and the MLI values were combined. The combined control MLI data were compared with values of the three HNE groups. The MLI values of the HNE/PBS and X0/HNE groups were significantly different from the combined control data, but the data of the SP-B1-25-X2/HNE group were not different. Thus the inhibitor linked to the peptide prevents emphysema, but the free inhibitor does not.
The tripeptide mimetic X0 was used in a 70-fold molar excess to HNE. The elastase activity with elastin substrate of only a 30-fold molar excess had been found to exhibit 0.6% of the original elastase activity. SP-B1-25-X2 was used in 30-fold molar excess to HNE but had much less elastase inhibitory activity than the tripeptide mimetic. These concentrations were chosen on the basis of maximum solubility. Over the next 4 wk the mice gained weight.
After 4 wk, all mice were killed, the lungs were removed and fixed, and mean airspace sizes were determined as described in Methods. Representative tissue samples of two animals from the saline group are shown in Fig. 1 A and B. Tissue samples of two animals from the HNE group are shown in Fig. 1 C and D. The tissue samples from the saline group have the high-density alveolar wall structure characteristic of healthy lungs. The HNE group has many large airspaces that are typically observed in severe cases of emphysema. Fig. 2 A and B shows tissue samples from two animals exposed to HNE mixed with a 70-fold molar excess of small molecule inhibitor X0, and Fig. 2 C and D shows tissue samples from two animals exposed to HNE mixed with a 30-fold molar excess of the SP-B1-25 small molecule inhibitor covalent construct (SP-B1-25-X2). In a blind study, a pathology expert could not distinguish between the animals treated with the small molecule inhibitor X0 combined with HNE and the animals exposed to HNE alone (mean distance between alveolar septa of 53.27 ± 8.27 μm (8) vs. 51.45 ± 5.78 μm (10), respectively) (Table 2). The severe alveolar wall destruction was similar with or without the tripeptide mimetic inhibitor when coadministered with near lethal doses of HNE. The pathology expert also could not distinguish between the saline group and animals given HNE and treated with SP-B1–25 covalently linked to the small molecule inhibitor (SP-B1-25-X2) (mean distance between alveolar septa of 44.40 ± 6.50 μm (4) vs. 45.82 ± 3.30 μm (8), respectively) (Table 2). Thus, the tripeptide mimetic alone provides little or no protection against intratracheal HNE challenge at 2 h or 4 wk after administration, whereas the tripeptide mimetic covalently linked to the SP-B1-25 fragment provided complete protection for 4 wk.
Fig. 1.

Sections of mouse lung tissue from PBS and HNE/PBS groups after 4 wk. At the end of testing, animals were killed, lungs were removed, and tissue samples were taken from the hilar and the central regions of the left lung and from the caudal region of the right lung. Each slide was composed of three separate tissue samples. (A and B) Animals instilled with saline had normal lung structure. (C and D) Animals instilled with HNE had extensive lung damage as seen by the large airspaces.
Fig. 2.

Sections of mouse lung tissue from X0/HNE and SP-B1-25-X2/HNE groups after 4 wk. (A and B) Animals given the potent small molecule inhibitor X0 with HNE developed large airspaces characteristic of emphysema (Fig. 1 C and D). The inhibitor, although having a Ki < 12 nM in vitro, had no efficacy in vivo. (C and D) When the small molecule inhibitor X2 was covalently linked to a surfactant peptide fragment to form the construct SP-B1-25-X2, it lost 50-fold potency in vitro but provided complete protection in vivo even after 4 wk.
Discussion
The hypotheses of this work are motivated by the need to (i) attain a physiologically relevant very long lung residence time for a pulmonary therapeutic because the lungs are extremely adept at clearance of foreign entities, (ii) isolate the active agent to the lungs to minimize or eliminate systemic circulation and thus toxicity, and (iii) achieve a high level of efficacy with a very low dose of active agent because the entire dose will be spread across the lung surface area through the actions of SP-B1-25, resulting in a very high effective dose. SP-B1-25 was chosen as the covalent partner for a synthetic small molecule HNE inhibitor because this peptide possesses a high percentage of the lung active surface properties of the whole SP-B protein. SP-B1-25 has an ideal lung pharmacodynamic profile because its intrinsic function is to spread across the entire lung–air interface to reduce surface tension and facilitate breathing. Delivering the SP-B1-25 tripeptide mimetic inhibitor covalent construct intratracheally and obtaining complete protection from near-lethal HNE challenge demonstrate that no high-pressure inhaler is required. Using the human sequence should minimize the potential for eliciting an immune response. In addition, this first successful study suggests a long lung residence time of a SP-B1-25 tripeptide mimetic construct. Since multiple mechanisms (20–25) exist for harboring elastase in the lungs, pulmonary damage approaches an asymptote 4 wk after the first lung injury.
Traditional drug discovery starts with target identification and validation and then some type of screening to obtain a lead small molecule. Pharmacodynamics are generally not considered until a compound survives a torturous journey through preclinical studies that include in vivo toxicology and pharmacokinetics. This work attempts to create a pulmonary therapeutic by starting with the physiological and pharmacodynamic considerations of the lungs. We hypothesize that the best way to do this is by incorporating an endogenous lung molecule into the therapeutic strategy. Our results provide good evidence that a surfactant-linked approach can be used to develop long-acting pulmonary therapeutics with the potential for enhanced efficacy, reduced toxicity, and simplified patient dosing (26).
Conclusions
Because efficient clearance is an intrinsic function of the lungs and pulmonary isolation of a therapeutic is desirable to lower systemic circulation and toxicity in general, the surfactant-linked approach should be applicable to a wide range of lung ailments. For example, cancer chemotherapy-induced immunodeficiency (27) and cystic fibrosis (28) commonly lead to Gram-negative lung infections. The polymyxin class of antibiotics, discovered from the species Bacillus polymyxa in the 1940s, is effective against many Gram-negative bacteria but has been generally abandoned on account of neurotoxicity (29). There is a resurgence of interest in polymyxins because of the alarming resurgence of resistant Gram-negative lung infections (30). Current research into the pharmacodynamics of aerosolized delivery (31–33) of antibiotics indicates that a covalent SP-B1-25 polymyxin construct (Fig. 3A) may be ideal for treating resistant Gram-negative lung infections. To treat cystic fibrosis (CF), an inhaled adenosine triphosphate (ATP) analog, Denufosol, targeted at lung chloride channel purinergic P2Y receptors with the goal of compensating for dysfunctional CF transmembrane conductance regulator (CFTR) ion flow, is showing excellent promise in clinical trials (34). Covalent linkage of ATP or an analog such as Denufosol to SP-B1-25 (Fig. 3B) may enhance this unique way of treating CF. Interestingly, because immortalized macrophages harboring tuberculosis (TB) overexpress the autophagic purinergic P2X receptor, the SP-B1-25-ATP covalent construct may also be a good treatment for latent TB (35). For lung cancer, there is evidence that enhanced efficacy may be achieved by inhibiting multiple epidermal growth factor (EGF) pathways (36). A promising new strategy toward this goal is to shut down EGF ligand cell-surface shedding from lung cancer cells by inhibiting the sheddase, also known as a disintegrin and metalloproteinase (ADAM) (37, 38). Unfor-tunately, 3 decades of research into matrix metalloproteinase (MMP) small molecule inhibition have still not produced an anticancer agent (39). Covalently linking a zinc metalloproteinase inhibitor such as marimastat to SP-B1-25 (Fig. 3C), with inhaled delivery, may shut down the multiple EGF pathways via ADAM inhibition locally in the lung without the systemic circulation and toxicity that have played a major role in preventing the success of past MMP drug discovery efforts. Similar considerations may result in a long-acting antihistamine (Fig. 3D) (40), b2-agonist for asthma (41), and M3 antagonist for chronic obstructive pulmonary disease (COPD) (42) and hypertensive COPD (43).
Fig. 3.

Proposed SP-B1-25–linked therapeutics for other lung ailments. Polymyxins disrupt Gram-negative bacterial membranes and are highly effective even against multidrug-resistant strains, but they have not been widely used because of toxicity concerns. Covalent linkage of a polymyxin to a surfactant peptide fragment (A) can be used to isolate this compound to the lungs and thus potentially minimize or circumvent systemic toxicity. Activating a purinergic receptor requires ATP or an ATP analog. The purinergic P2Y receptor may be targeted to treat cystic fibrosis, and the P2X receptor may be targeted to treat latent tuberculosis. Covalent linkage of ATP to the surfactant peptide (B) may enhance the efficacy of both protocols. Shutting down the EGF family of pathways by stopping cell-surface EGF ligand shedding is a promising new lung cancer strategy. This can be accomplished by inhibiting the ADAM metalloproteinases, but no inhibitor of this class of enzymes has ever successfully passed clinical trials. A strategy of isolating such an inhibitor to the lungs via surfactant linkage (C) may circumvent the pervasive toxicity problems with these types of compounds. A long-duration, infrequent dosing antihistamine (D) may be created with a surfactant-linked strategy.
Methods
Synthesis of Compounds.
See SI Text.
In Vitro Assessment of Elastase Inhibitors.
See SI Text.
In Vivo Assessment of Elastase Inhibitors.
In vitro testing suggested that X0 was the most effective tripeptide mimetic of the three inhibitors and that when derivatized and linked to SP-B1-25 as SP-B1-25-X2 should be more effective than SP-B1–25-X1. DMSO solutions of X0 (0.14 M) and SP-B1-25-X2 (0.34 M) were prepared. Aliquots of these two solutions were added to PBS or HNE so that the DMSO content was 0.3% in all cases.
Fifty-six 10-week-old female mice (C57Bl6) from Jackson Laboratories were treated. The protocol for treatment of mice was approved by our Institutional Animal Care and Use Committee, and our animal facility is approved by the American Association for the Accreditation of Laboratory Animal Care. Mice were divided into six groups: PBS, HNE in PBS (400 μg/mL, 1.4 × 10−5 M), X0 (1.0 × 10−3 M) in PBS, X0 (1.0 × 10−3 M) combined with HNE (1.4 × 10−5 M) in PBS, SP-B1-25-X2 (4.2 × 10−4 M) in PBS, and SP-B1-25-X2 (4.2 × 10−4 M) combined with HNE (1.4 × 10−5 M) in PBS. All solutions contained 0.3% DMSO.
The mice were anesthetized by inhalation of isoflurane, weighed, and given intratracheal instillation of 0.1 mL of the indicated solution. Two hours later, 12 of the mice were killed for bronchoalveolar lavage (BAL). The animals were anesthetized with pentobarbital sodium, after which the trachea was exposed surgically and a cannula inserted. The cannula was used to introduce 1 mL saline to inflate the lungs and withdraw as much of the BAL fluid as possible. The BAL fluid was noted for color, transferred to Eppendorf tubes, and centrifuged to pellet the red blood cells. The supernatant was removed, and the pellet was weighed.
Four weeks later, the remaining animals were anesthetized with pentobarbital sodium, weighed, and exsanguinated by cutting the abdominal aorta. The trachea was exposed surgically, and a cannula was tied into the trachea. The lungs were inflated with paraformaldehyde fixative and excised. Two transverse slabs of tissue from the left lung and one slab of tissue from the right caudal lung lobe were cut from the lungs, dehydrated, and embedded in paraffin. Four 4-μm sections were cut from each slab for staining and measurement of mean linear intercept (MLI). The quantitative histological measurements were made with an image analysis system consisting of a Nikon E600 microscope, a Spot RT Slider digital camera, and a computer with Image-Pro Plus image analysis software. Two fields from each of the three lung sections were photographed. From each field, five areas of interest, free of airways and muscular blood vessels, were picked for measurement of the number of intersections of virtual lines of known length with alveolar septa. An increase in the mean distance between alveolar septa indicates enlarged airspaces and is consistent with emphysema.
The Kruskal–Wallis test was used to compare grouped data. Probability values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by SolMap Pharmaceuticals, Inc. and by Grant HL088772 from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1001349107/-/DCSupplemental.
References
- 1.Gonda I. Systemic delivery of drugs to humans via inhalation. J Aerosol Med. 2006;19:47–53. doi: 10.1089/jam.2006.19.47. [DOI] [PubMed] [Google Scholar]
- 2.Stone PJ, Lucey EC, Snider GL. Induction and exacerbation of emphysema in hamsters with human neutrophil elastase inactivated reversibly by a peptide boronic acid. Am Rev Respir Dis. 1990;141:47–52. doi: 10.1164/ajrccm/141.1.47. [DOI] [PubMed] [Google Scholar]
- 3.Lucey EC, Stone PJ, Christensen TG, Breuer R, Snider GL. An 18-month study of the effects on hamster lungs of intratracheally administered human neutrophil elastase. Exp Lung Res. 1988;14:671–686. doi: 10.3109/01902148809087836. [DOI] [PubMed] [Google Scholar]
- 4.Ip MP, Kleinerman J, Ranga V, Sorensen J, Powers JC. The effects of small doses of oligopeptide elastase inhibitors on elastase-induced emphysema in hamsters: A dose-response study. Am Rev Respir Dis. 1981;124:714–717. doi: 10.1164/arrd.1981.124.6.714. [DOI] [PubMed] [Google Scholar]
- 5.Ranga V, Kleinerman J, Ip MP, Sorensen J, Powers JC. Effects of oligopeptide chloromethylketone administered after elastase: Renal toxicity and lack of prevention of experimental emphysema. Am Rev Respir Dis. 1981;124:613–618. doi: 10.1164/arrd.1981.124.5.613. [DOI] [PubMed] [Google Scholar]
- 6.Lucey EC, Stone PJ, Digenis GA, Snider GL. A polymer-bound elastase inhibitor is effective in preventing human neutrophil elastase-induced emphysema. Ann N Y Acad Sci. 1991;624:341–342. doi: 10.1111/j.1749-6632.1991.tb17041.x. [DOI] [PubMed] [Google Scholar]
- 7.Stone PJ, Lucey EC, Noskova D, Digenis GA, Snider GL. Covalently linking a peptidyl carbamate elastase inhibitor to a hydrophilic polymer increases its effectiveness in preventing emphysema and secretory cell metaplasia in the hamster. Am Rev Respir Dis. 1992;146:457–461. doi: 10.1164/ajrccm/146.2.457. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Baatz JE, Holm BA, Notter RH. Content-dependent activity of lung surfactant protein B in mixtures with lipids. Am J Physiol Lung Cell Mol Physiol. 2002;283:L897–L906. doi: 10.1152/ajplung.00431.2001. [DOI] [PubMed] [Google Scholar]
- 9.Gupta M, Hernández-Juviel JM, Waring AJ, Bruni R, Walther FJ. Comparison of functional efficacy of surfactant protein B analogues in lavaged rats. Eur Respir J. 2000;16:1129–1133. doi: 10.1034/j.1399-3003.2000.16f19.x. [DOI] [PubMed] [Google Scholar]
- 10.Seurynck-Servoss SL, Dohm MT, Barron AE. Effects of including an N-terminal insertion region and arginine-mimetic side chains in helical peptoid analogues of lung surfactant protein B. Biochemistry. 2006;45:11809–11818. doi: 10.1021/bi060617e. [DOI] [PubMed] [Google Scholar]
- 11.Revak SD, et al. Efficacy of synthetic peptide-containing surfactant in the treatment of respiratory distress syndrome in preterm infant rhesus monkeys. Pediatr Res. 1996;39:715–724. doi: 10.1203/00006450-199604000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Lal MK, Sinha SK. Surfactant respiratory therapy using Surfaxin/sinapultide. Ther Adv Respir Dis. 2008;2:339–344. doi: 10.1177/1753465808097113. [DOI] [PubMed] [Google Scholar]
- 13.Spencer JL, Stone PJ, Nugent MA. New insights into the inhibition of human neutrophil elastase by heparin. Biochemistry. 2006;45:9104–9120. doi: 10.1021/bi060338r. [DOI] [PubMed] [Google Scholar]
- 14.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 15.Henderson PJ. A linear equation that describes the steady-state kinetics of enzymes and subcellular particles interacting with tightly bound inhibitors. Biochem J. 1972;127:321–333. doi: 10.1042/bj1270321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: John Wiley & Sons; 1993. [Google Scholar]
- 17.Edwards PD, Bernstein PR. Synthetic inhibitors of elastase. Med Res Rev. 1994;14:127–194. doi: 10.1002/med.2610140202. [DOI] [PubMed] [Google Scholar]
- 18.Greco WR, Hakala MT. Evaluation of methods for estimating the dissociation constant of tight binding enzyme inhibitors. J Biol Chem. 1979;254:12104–12109. [PubMed] [Google Scholar]
- 19.Williams JC, Stein RL, Giles RE, Krell RD. Biochemistry and pharmacology of ICI 200,880, a synthetic peptide inhibitor of human neutrophil elastase. Ann N Y Acad Sci. 1991;624:230–243. doi: 10.1111/j.1749-6632.1991.tb17022.x. [DOI] [PubMed] [Google Scholar]
- 20.McGowan SE, Stone PJ, Calore JD, Snider GL, Franzblau C. The fate of neutrophil elastase incorporated by human alveolar macrophages. Am Rev Respir Dis. 1983;127:449–455. doi: 10.1164/arrd.1983.127.4.449. [DOI] [PubMed] [Google Scholar]
- 21.Morris SM, Stone PJ, Snider GL, Albright JT, Franzblau C. Ultrastructural changes in hamster lung four hours to twenty-four days after exposure to elastase. Anat Rec. 1981;201:523–535. doi: 10.1002/ar.1092010309. [DOI] [PubMed] [Google Scholar]
- 22.Raub JA, Mercer RR, Miller FJ, Graham JA, O'Neil JJ. Dose response of elastase-induced emphysema in hamsters. Am Rev Respir Dis. 1982;125:432–435. doi: 10.1164/arrd.1982.125.4.432. [DOI] [PubMed] [Google Scholar]
- 23.Stone PJ, Calore JD, Snider GL, Franzblau C. Role of alpha-macroglobulin-elastase complexes in the pathogenesis of elastase-induced emphysema in hamsters. J Clin Invest. 1982;69:920–931. doi: 10.1172/JCI110531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werb Z, Gordon S. Elastase secretion by stimulated macrophages. Characterization and regulation. J Exp Med. 1975;142:361–377. doi: 10.1084/jem.142.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaslow MC, et al. Human neutrophil elastase does not bind to alpha 1-protease inhibitor that has been exposed to activated human neutrophils. Am Rev Respir Dis. 1983;128:434–439. doi: 10.1164/arrd.1983.128.3.434. [DOI] [PubMed] [Google Scholar]
- 26.Cazzola M, Matera MG. Novel long-acting bronchodilators for COPD and asthma. Br J Pharmacol. 2008;155:291–299. doi: 10.1038/bjp.2008.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vento S, Cainelli F, Temesgen Z. Lung infections after cancer chemotherapy. Lancet Oncol. 2008;9:982–992. doi: 10.1016/S1470-2045(08)70255-9. [DOI] [PubMed] [Google Scholar]
- 28.Kidd TJ, et al. Low rates of Pseudomonas aeruginosa misidentification in cystic fibrosis patients. J Clin Microbiol. 2009;47:1503–1509. doi: 10.1128/JCM.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falagas ME, Kasiakou SK. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit Care. 2006;10:R27. doi: 10.1186/cc3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold TM, Forrest GN, Messmer KJ. Polymyxin antibiotics for gram-negative infections. Am J Health Syst Pharm. 2007;64:819–826. doi: 10.2146/ajhp060473. [DOI] [PubMed] [Google Scholar]
- 31.Dudley MN, Loutit J, Griffith DC. Aerosol antibiotics: Considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol. 2008;19:637–643. doi: 10.1016/j.copbio.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Michalopoulos A, Kasiakou SK, Falagas ME. The significance of different formulations of aerosolized colistin. Crit Care. 2005;9:417–418. doi: 10.1186/cc3506. and author reply (2005) 417–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michalopoulos A, et al. Aerosolized colistin for the treatment of nosocomial pneumonia due to multidrug-resistant Gram-negative bacteria in patients without cystic fibrosis. Crit Care. 2005;9:R53–R59. doi: 10.1186/cc3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kellerman D, et al. Denufosol: A review of studies with inhaled P2Y(2) agonists that led to phase 3. Pulm Pharmacol Ther. 2008;21:600–607. doi: 10.1016/j.pupt.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Placido R, et al. P2X(7) purinergic receptors and extracellular ATP mediate apoptosis of human monocytes/macrophages infected with Mycobacterium tuberculosis reducing the intracellular bacterial viability. Cell Immunol. 2006;244:10–18. doi: 10.1016/j.cellimm.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ., 2nd Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol Ther. 2009;122:1–8. doi: 10.1016/j.pharmthera.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou BB, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fridman JS, et al. Selective inhibition of ADAM metalloproteases as a novel approach for modulating ErbB pathways in cancer. Clin Cancer Res. 2007;13:1892–1902. doi: 10.1158/1078-0432.CCR-06-2116. [DOI] [PubMed] [Google Scholar]
- 39.Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006;25:115–136. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- 40.Chen C. Physicochemical, pharmacological and pharmacokinetic properties of the zwitterionic antihistamines cetirizine and levocetirizine. Curr Med Chem. 2008;15:2173–2191. doi: 10.2174/092986708785747625. [DOI] [PubMed] [Google Scholar]
- 41.Walters EH, Gibson PG, Lasserson TJ, Walters JA. Long-acting beta2-agonists for chronic asthma in adults and children where background therapy contains varied or no inhaled corticosteroid. Cochrane Database Syst Rev. 2007;(1):CD001385. doi: 10.1002/14651858.CD001385.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scullion JE. The development of anticholinergics in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:33–40. doi: 10.2147/copd.2007.2.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kario K, Uno H, Shimada K. Sleep-predominant lowering of ambulatory blood pressure by bedtime inhalation of a novel muscarinic M3 receptor antagonist: A new “bronchoantihypertensive” strategy targeting the lung in hypertension with chronic obstructive pulmonary disease. Hypertens Res. 2008;31:817–821. doi: 10.1291/hypres.31.817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.