

Abstract
Developmental events can be monitored at the cellular and molecular levels by using noninvasive imaging techniques. Among the biomolecules that might be targeted for imaging analysis, glycans occupy a privileged position by virtue of their primary location on the cell surface. We previously described a chemical method to image glycans during zebrafish larval development; however, we were unable to detect glycans during the first 24 hours of embryogenesis, a very dynamic period in development. Here we report an approach to the imaging of glycans that enables their visualization in the enveloping layer during the early stages of zebrafish embryogenesis. We microinjected embryos with azidosugars at the one-cell stage, allowed the zebrafish to develop, and detected the metabolically labeled glycans with copper-free click chemistry. Mucin-type O-glycans could be imaged as early as 7 hours postfertilization, during the gastrula stage of development. Additionally, we used a nonmetabolic approach to label sialylated glycans with an independent chemistry, enabling the simultaneous imaging of these two distinct classes of glycans. Imaging analysis of glycan trafficking revealed dramatic reorganization of glycans on the second time scale, including rapid migration to the cleavage furrow of mitotic cells. These studies yield insight into the biosynthesis and dynamics of glycans in the enveloping layer during embryogenesis and provide a platform for imaging other biomolecular targets by microinjection of appropriately functionalized biosynthetic precursors.
Keywords: azide, cyclooctyne, bioorthogonal, oxime, cytokinesis
Embryogenesis is a highly dynamic process characterized by rapid cell division, differentiation, cell migration, and morphogenesis (1). Developmental biologists have sought noninvasive imaging techniques to capture in detail these physiological changes (2–4). Molecular imaging tools such as fluorescent protein fusions and antibody conjugates, which can be used to track specific biomolecules in vivo, have enhanced our understanding of embryogenesis at a molecular level (5). The zebrafish, a popular vertebrate model of embryogenesis, has provided deep insight into the cellular and molecular details of development by virtue of its transparent embryos and external development (6). The zebrafish was thus an ideal model organism to apply chemical tools for imaging glycans, which are key regulators of developmental processes but are difficult to visualize (7).
All cell surfaces are adorned with glycans, which are oligosaccharide structures found as posttranslational modifications of integral membrane proteins and as head groups of membrane-resident lipids (8). In development, glycans have numerous functions, which derive from both their bulk properties as well as their specific molecular structures (9). However, genetic redundancy among biosynthetic genes has frustrated efforts to study certain classes of glycans, notably mucin-type O-glycans and sialylated glycans (9). Critically, glycans cannot be visualized easily by using the standard tools of molecular imaging because of the nongenetically encoded nature of their biosynthesis and the dearth of available glycan-binding antibodies (7, 10).
We have developed a two-step chemical strategy for labeling glycans with imaging agents in vivo. This approach entails (a) metabolic labeling with synthetic azidosugars that hijack glycan biosynthesis, followed by (b) covalent chemical tagging of the azide-labeled glycans with a compound bearing both an azide-reactive group and an imaging probe (11). In previous work, we showed that a membrane-permeable azidosugar, peracetylated N-azidoacetylgalactosamine (Ac4GalNAz), can be used to metabolically label mucin-type O-glycans in developing zebrafish (12). In a second step, the metabolically labeled zebrafish larvae were incubated in solutions containing a fluorophore conjugate of a difluorinated cyclooctyne (DIFO) reagent, enabling visualization of glycans in vivo by using copper-free click chemistry (12, 13).
Those studies revealed dramatic differences in cell-surface expression, intracellular trafficking patterns, and tissue distributions of glycans at different stages of zebrafish larval development. However, we were unable to detect labeled glycans in zebrafish embryos earlier than 24 hours postfertilization (hpf). Because many important developmental events including cell migration, tissue morphogenesis, and cell differentiation occur in the first 24 hours of zebrafish embryogenesis, and glycan biosynthesis is known to occur before 24 hpf (14, 15), we sought to develop methods to image glycans in early embryos.
Here we report that microinjection of azidosugars enables imaging of cell-surface glycans in the enveloping layer of zebrafish embryos as early as 7 hpf, a few hours after expression of zygotic genes is known to occur (16). Additionally, we employed a complementary, nonmetabolic method to target a second class of glycans, those bearing sialic acid. This technological advance enabled the simultaneous imaging of both classes of glycans in vivo. Time-lapse and multicolor imaging experiments highlighted differences between O-glycans and sialylated glycans in the cells of the enveloping layer during the gastrulation and segmentation periods. Our studies also revealed a dramatic and rapid reorganization of cell-surface glycans during mitosis.
Results and Discussion
Microinjection of the Nucleotide Sugar UDP-GalNAz Enables Visualization of Glycans During Gastrulation.
En route to incorporation into mucin-type O-glycans, Ac4GalNAz must first traverse the biosynthetic steps of the N-acetylgalactosamine (GalNAc) salvage pathway, resulting in its conversion to the nucleotide sugar uridine diphosphatidyl N-azidoacetylgalactosamine (UDP-GalNAz) (Fig. 1A). UDP-GalNAz, the azido variant of the natural nucleotide sugar UDP-GalNAc, serves as a substrate for the polypeptide GalNAc transferases (ppGalNAcTs), which transfer GalNAz to Ser and Thr residues of nascent glycoproteins (17). To achieve metabolic labeling earlier in embryogenesis, we sought to bypass the GalNAc salvage pathway by using the downstream intermediate UDP-GalNAz as a labeling substrate. However, because this compound is not cell-permeable, we elected to microinject it directly into the yolk sac of the embryo. At the one-cell stage, cell-impermeable reagents that are microinjected into the yolk gain access to all cells in the developing organism (18).
Fig. 1.

In vivo imaging of glycans in early zebrafish embryos using microinjection of azidosugars and detection with copper-free click chemistry. (A) Metabolic labeling of mucin-type O-glycans with azidosugars via the GalNAc salvage pathway. The enzymatic transformations shown are catalyzed by (i) nonspecific esterases, (ii) GalNAc-1-phosphate kinase, (iii) UDP-GalNAc pyrophosphorylase, and (iv) ppGalNAcTs and other glycosyltransferases. (B) Two-step strategy for imaging glycans in vivo. (C and D) Zebrafish embryos were microinjected with UDP-GalNAz (C, top), GalNAz (D, top), UDP-GalNAc (C, bottom), or no sugar (D, bottom), along with the tracer dye rhodamine-dextran, allowed to develop to 7 hpf, reacted with DIFO-488 (100 μM, 1 h), and imaged by confocal microscopy. Shown are maximum intensity z-projection images. Green, DIFO-488; red, rhodamine-dextran. (Scale bar: 200 μm.)
Following microinjection with 25 pmol of synthetic UDP-GalNAz (17) or the control sugar UDP-GalNAc, the embryos were allowed to develop for several hours and then bathed in a solution of an Alexa Fluor 488 conjugate of DIFO, termed DIFO-488 (13) (SI Appendix, Fig. S1), to detect azide-labeled glycans (Fig. 1B). We performed the copper-free click reaction at various time points, and we were able to detect, by confocal microscopy, labeling of cell-surface glycans in vivo as early as the 65% epiboly stage of gastrulation (∼7 hpf) but not before (Fig. 1C and SI Appendix, Fig. S2). A larger dose of UDP-GalNAz, 125 pmol, also yielded robust labeling of cell-surface glycans with DIFO-488, but this dose caused developmental defects by 24 hpf (SI Appendix, Fig. S3A). Thus, we elected to inject 25 pmol of azidosugar in subsequent experiments.
These important results validated that microinjection of a nucleotide sugar precursor could enable imaging of glycans at much earlier stages of development than was achievable by bathing with Ac4GalNAz. Given that the transcription of zygotic genes starts at 3 hpf (16), 4 hours before our technique yields detectable signal, this microinjection approach enables detection of the early events of glycan biosynthesis. We considered that it might be possible, however, that treatment with a bacterial protease to remove the chorion, which is a standard step in our protocol, might remove some cell-surface glycoproteins and thereby delay the onset of detectable signal using our reagents. To examine this possibility, we performed an analogous imaging experiment in which chorions were removed manually, with forceps, instead. We observed the same results, with earliest detectable labeling at the 65% epiboly stage (SI Appendix, Fig. S4).
Microinjection of GalNAz also Affords Metabolic Labeling During Gastrulation.
Our initial choice of UDP-GalNAz to achieve metabolic labeling at early developmental stages centered on its ability to bypass biosynthetic steps in the GalNAc salvage pathway. To determine whether the GalNAc salvage pathway was responsible for the delay in metabolic labeling that we observed with our bathing protocol, we microinjected the free sugar, GalNAz. Reaction with DIFO-488 at various time points revealed labeling starting at 65% epiboly, the same result as seen with UDP-GalNAz (Fig. 1D and SI Appendix, Fig. S5). Thus, the GalNAc salvage pathway does not appear to be the bottleneck in our labeling technique.
This observation—that microinjecting embryos with GalNAz yields signal significantly before bathing them in Ac4GalNAz does—may be explained by two potential factors. First, microinjection could result in a higher intracellular concentration of azidosugar at earlier time points than bathing. Alternatively, the conversion of Ac4GalNAz to GalNAz by promiscuous cytosolic esterases may be a bottleneck step in the labeling method. To test this second possibility, we microinjected embryos with Ac4GalNAz and reacted them with DIFO-488 at 12 hpf. These Ac4GalNAz-injected embryos exhibited DIFO-derived fluorescence that was comparable in intensity to that from similarly labeled embryos injected instead with free GalNAz (SI Appendix, Fig. S6). These results indicate that cytosolic esterases are able to efficiently convert Ac4GalNAz to GalNAz during the first 12 hours of embryogenesis.
At these early stages of development, we noticed that the cells labeled with DIFO-488 during gastrulation appeared to be members of the enveloping layer, the embryo’s outermost layer of cells (SI Appendix, Fig. S7 A and B). To analyze the labeling of all the cells in the organism, we turned to flow cytometry. GalNAz-injected embryos were reacted with DIFO-488 at 10 hpf to label azide-containing glycans on the enveloping layer. The embryos were subsequently dissociated, and the resulting cell suspension was labeled with an Alexa Fluor 647 conjugate of DIFO (DIFO-647, SI Appendix, Fig. S1) (SI Appendix, Fig. S7C). We found that most of the dissociated cells displayed DIFO-647-derived signal, indicating that most cells of the embryo were metabolically labeled with GalNAz (SI Appendix, Fig. S7D). By contrast, approximately 0.5% of the cells were labeled with DIFO-488, indicating that they were accessible to this reagent while part of the intact embryo. Interestingly, the enveloping layer cells labeled by DIFO-488 when the embryo was intact also displayed the highest DIFO-647 signal, suggesting that these cells may possess higher levels of mucin glycoproteins than other cells in the embryo.
We next extended our labeling time course out to 96 hpf, when the zebrafish are at the larval stage, to evaluate the duration of time that a single bolus of GalNAz or UDP-GalNAz could still yield detectable azide-dependent signal with the DIFO reagents. In this experiment, zebrafish injected with the appropriate azidosugar were allowed to develop normally and reacted at a single time point between 12 and 96 hpf with DIFO-488 (SI Appendix, Fig. S8). We found that zebrafish injected with either GalNAz or UDP-GalNAz exhibited azide-dependent fluorescence throughout the four-day experiment. Further, when embryos were treated at 85 hpf with tris(2-carboxyethyl)phosphine to reduce any surface-exposed azides present at that time (12), then allowed to develop to 96 hpf and reacted with an Alexa Fluor 555 conjugate of DIFO (DIFO-555, SI Appendix, Fig. S1), they showed robust labeling, indicating that new azide-containing glycans were presented at the cell surface during this later period (85–96 hpf) (SI Appendix, Fig. S9).
These results prompted us to continue our experiments with GalNAz, which is more readily synthesized in the laboratory and is not prone to hydrolysis in vitro. Importantly, we observed no toxicity or developmental abnormalities during the duration of these four-day experiments, suggesting that microinjection of 25 pmol of GalNAz or UDP-GalNAz followed by detection with copper-free click chemistry is not harmful to the organism.
Two-Color, Time-Resolved Labeling Enables Visualization of O-Glycan Trafficking.
To image two temporally distinct populations of O-glycans, we adopted a two-color labeling strategy involving successive reactions with different DIFO-fluorophore conjugates. In these experiments, embryos were injected with GalNAz at the one-cell stage, allowed to develop to 90% epibody (9 hpf), and then reacted with DIFO-488. The embryos were rinsed and allowed to develop further for 2 or 12 hours, at which point they were reacted with DIFO-555. In this manner, “old” and “new” glycans within the same embryo could be distinguished by using confocal microscopy.
In the experiment with a two-hour window for development and metabolic labeling between the two copper-free click reactions, we observed substantial differences in spatial distribution between the two populations of glycans (Fig. 2A and SI Appendix, Fig. S10). The newer glycans, shown in red, were spread evenly across the cell surface, including close to the edge of the cell. Older glycans, shown in green, had migrated away from this region and were likely undergoing internalization.
Fig. 2.

Two-color, time-resolved labeling enables visualization of O-glycan trafficking. Zebrafish embryos were microinjected with GalNAz, allowed to develop to 9 hpf, and reacted with DIFO-488 (100 μM, 1 h). The embryos were then allowed to further develop for 2 h (A) or 12 h (B), at which point they were reacted with DIFO-555 (100 μM, 1 h) and then imaged by confocal microscopy. Shown are maximum intensity z-projection images of superficial enveloping layer cells. Green, DIFO-488; red, DIFO-555. (Scale bar: 10 μm.)
The two-color experiment with a 12-hour delay between copper-free click reactions demonstrated very little overlap between old and new glycan populations, indicating that substantial glycan biosynthesis and turnover had occurred during that time period (Fig. 2B and SI Appendix, Fig. S10). Interestingly, the DIFO-555 signal from 21 hpf embryos was much more evenly distributed across the surface of the cells than the DIFO-555 signal from embryos reacted at 12 hpf. These results suggest that internalization of glycans may be faster at 12 hpf than at 21 hpf; alternatively, the data may reflect differences in enveloping layer cell morphologies at the two developmental time points.
Collectively, these two-color experiments using GalNAz injections and labeling with DIFO-fluorophore conjugates highlight the dynamics of O-glycan biosynthesis in surface epithelial cells during the early stages of embryogenesis. At this point, we were interested in evaluating whether other sectors of the glycome also exhibited this behavior and whether we might discern differences in the generation, trafficking, and degradation of distinct classes of glycans during embryogenesis.
Imaging of Sialylated Glycans Using a Nonmetabolic Approach.
We turned our attention to sialylated glycans, which are known to perform numerous functions in development (19). Sialic acid, a nine-carbon sugar frequently found as a terminal modification of both N- and O-glycans, can be metabolically labeled by treating cells or organisms with an azide-derivatized precursor, peracetylated N-azidoacetylmannosamine (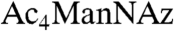 ) (20–22). We have used this approach to image sialylated glycans in developing zebrafish by bathing embryos in solutions of
) (20–22). We have used this approach to image sialylated glycans in developing zebrafish by bathing embryos in solutions of 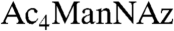 prior to detection using copper-free click chemistry (12). In those experiments, we did not observe any labeling prior to 24 hpf, similar to our results with Ac4GalNAz.
prior to detection using copper-free click chemistry (12). In those experiments, we did not observe any labeling prior to 24 hpf, similar to our results with Ac4GalNAz.
In light of our success at labeling O-glycans during early embryogenesis by microinjection of UDP-GalNAz and GalNAz, we first attempted to image sialylated glycans by using an analogous approach. Here, we microinjected embryos at the one-cell stage with 25 or 250 pmol of ManNAz, allowed them to develop to late gastrula, and reacted them with DIFO-488 to detect sialylated glycans in vivo (SI Appendix, Fig. S11 A and B). Although we did detect labeling of cell-surface glycans above background fluorescence at both doses, the 250 pmol dose caused developmental defects (SI Appendix, Fig. S3B), and the level of labeling with 25 pmol was very modest, suggesting the requirement of a more sensitive labeling technique.
We thus elected to use a nonmetabolic approach for imaging sialylated glycans. It has long been known that chemical treatment of cells or cell lysates with low concentrations of sodium periodate (NaIO4) selectively oxidizes sialic acid residues (23–27), exposing an aldehyde that can be subsequently targeted by reductive amination or oxime/hydrazone-forming reactions (28). Recently, Paulson and coworkers utilized this approach to label sialylated glycans on the surfaces of live cells (29). We endeavored to extend this strategy to a whole-animal context by treating zebrafish embryos with NaIO4 at various stages of embryogenesis and then reacting them with an aminooxy-derivatized Alexa Fluor 488 reagent (AO-488) (Fig. 3A).
Fig. 3.

Imaging of sialylated glycans using a nonmetabolic approach. (A) Schematic for chemical labeling of sialylated glycans by treatment with NaIO4 to expose aldehydes on sialic acids, followed by detection using aminooxy-fluorophore conjugates. (B and C) Zebrafish embryos (10 hpf) were bathed in NaIO4 (500 μM, 30 min, top row) or no reagent (bottom row), reacted with aminooxy-Alexa Fluor 488 (AO-488, 100 μM, 1 h, pH 6.7), and imaged by confocal microscopy. (B, left panel) Brightfield; (B, right panel) and (C), maximum intensity z-projection images of AO-488 fluorescence. [Scale bars: 200 μm (B), 20 μm (C).]
We observed modest labeling of cell surfaces beginning at 65% epiboly, or 7 hpf (SI Appendix, Fig. S11C), and more robust labeling beginning at 10 hpf (Fig. 3B). These results suggest that substantial de novo production of sialic acid-containing glycans begins as gastrulation ends and somitogenesis commences. We confirmed that ManNAz-labeled glycans colocalize with those labeled by NaIO4 and AO-488 (SI Appendix, Fig. S12), which is consistent with literature reports that this method is selective for sialic acids. This chemical approach for tagging sialylated glycans did not result in any noticeable toxicity or developmental defects, as embryos that were treated with NaIO4 and aminooxy fluorophores and then were allowed to develop to 120 hpf were indistinguishable from untreated zebrafish larvae. However, embryos treated with NaIO4 and reacted with AO-488 prior to bud stage demonstrated substantial fluorophore labeling in the yolk cell (SI Appendix, Fig. S11C). Therefore, in all subsequent experiments we employed embryos that had completed epiboly. Furthermore, we reacted embryos with AO-488 and an aniline additive (29, 30) to probe whether this nucleophilic catalyst might improve the labeling efficiency of periodate-oxidized glycans. In our system, the addition of aniline resulted in only a modest increase in cell-surface fluorescence (SI Appendix, Fig. S13), and because of the fragile nature of the embryo, we chose to omit this reagent from our labeling reactions.
Whereas the images of cell-surface sialylated glycans revealed by the periodate method (Fig. 3C) appeared similar to those of O-glycans using the GalNAz method (Fig. 1D), we were eager to determine whether we could distinguish subtle differences between O-glycan and sialylated glycan production. Because the two detection methods that we employed, oxime formation and copper-free click chemistry, are orthogonal to one another, we were able to address this question by performing both labeling reactions on the same population of embryos.
Simultaneous Visualization of O-Glycans and Sialylated Glycans Using Independent Bioorthogonal Chemistries.
In order to ascertain the similarities and differences between O-glycan labeling, using GalNAz and DIFO-fluorophores, and sialic acid labeling, using NaIO4 and aminooxy fluorophores, we performed a dual labeling experiment within the same embryo. Although we have shown previously that azides and ketones can be targeted simultaneously for imaging in cultured cells (31), analogous experiments in vivo have not been reported. Embryos were microinjected with GalNAz or no sugar at the one-cell stage, allowed to develop to 10 or 24 hpf, and then incubated with NaIO4 or no reagent. In all cases, the embryos were subsequently reacted simultaneously with DIFO-555 to label azides and AO-488 to label aldehydes.
We observed substantial labeling with each fluorescent probe, and appropriate negative controls displayed minimal background fluorescence (Fig. 4 and SI Appendix, Figs. S14 and S15). The resulting images revealed considerable colocalization of the GalNAz-labeled and NaIO4-reacted species. This result is to be expected, because many sialic acids appear within mucin-type O-glycans, and many mucin glycoproteins also contain N-glycans, which often display terminal sialic acid residues. These experiments validate the ability of two independent bioorthogonal chemistries to be employed for simultaneous visualization of two classes of glycans within a single living organism.
Fig. 4.

Simultaneous visualization of O-glycans and sialylated glycans using two independent bioorthogonal chemistries. (A) Schematic depicting dual labeling of O-glycans and sialylated glycans. (B and C) Zebrafish embryos were microinjected with GalNAz or no sugar, allowed to develop to 10 hpf, and then bathed in NaIO4 (500 μM, 30 min) or no reagent. Embryos were then reacted in a mixture of DIFO-555 (100 μM) and AO-488 (100 μM) in PBS (pH 6.7) for 1 h, rinsed, and imaged by confocal microscopy. Green, AO-488; red, DIFO-555. [Scale bars: 200 μm (B), 20 μm (C).]
Time-Lapse Monitoring of Mitotic Cells Reveals Dramatic Glycan Reorganization During Cell Division.
During the course of our studies, we noticed an intense staining pattern derived from our reagents at what appeared to be the cleavage furrow of dividing cells (Fig. 5A, Arrowheads and Inset, for GalNAz labeling, and Fig. 3C for NaIO4 treatment). To evaluate whether these cells were indeed undergoing mitosis, we utilized a transgenic zebrafish ubiquitously expressing a GFP-fused histone protein (H2A-GFP), enabling visualization of cell nuclei by confocal microscopy (32). In these experiments, GalNAz was microinjected into embryos at the one-cell stage, and the zebrafish were allowed to develop until the end of epiboly (10 hpf), at which point they were reacted with DIFO-647.
Fig. 5.

Time-lapse monitoring of mitotic cells reveals dramatic glycan reorganization during cell division. (A) Wild-type zebrafish embryos were microinjected with GalNAz, allowed to develop to 10 hpf, reacted with DIFO-488 (100 μM, 1 h), and imaged by confocal microscopy. Arrowheads indicate intense staining at the cleavage furrow of dividing cells. Maximum intensity z-projection images are shown. (B) H2A-GFP transgenic zebrafish embryos were microinjected with GalNAz and allowed to develop to 10 hpf. The embryos were reacted with DIFO-647 (100 μM, 1 h) and imaged by confocal microscopy. Shown are single z-plane frames from a time-lapse movie (Movie S1). Indicated in the top right corner of each image is time (h:min:s). Green, H2A-GFP; red, DIFO-647. (C) H2A-GFP zebrafish were microinjected at the one-cell stage with GalNAz and memCherry mRNA and allowed to develop to 10 hpf. The embryos were reacted with DIFO-647 (100 μM, 1 h) and imaged by confocal microscopy. Blue, H2A-GFP; green, memCherry; red, DIFO-647. Arrowheads indicate location of new membrane between daughter cells. Maximum intensity z-projection images are shown. [Scale bars: 100 μm (A), 20 μm (B, C).]
Time-lapse imaging of the labeled embryos revealed that during metaphase, the DIFO-derived signal was uniform around the cell membrane (Fig. 5B). However, moments after the beginning of anaphase, which is marked by the rapid separation of the sister chromatids, we observed an intensification of the fluorescent signal at the cleavage furrow (Fig. 5B and Movie S1). The DIFO-derived signal continued to concentrate as the two patches of membrane that constitute the furrow began to invaginate during cytokinesis, but surprisingly, we did not observe the two labeled glycan populations touch one another. Instead, the intense signal dissipated over time. We allowed the labeled embryos to continue to develop over subsequent days and observed no developmental abnormalities, suggesting that cell division was indeed proceeding normally.
Labeling of Cell-Surface Glycans Differs Substantially from a Plasma Membrane Marker During Mitosis.
To visualize the plasma membrane during cell division, we coinjected H2A-GFP embryos at the one-cell stage with a mixture of GalNAz and the mRNA for membrane-mCherry (memCherry), a fusion of the membrane-localized domain of Lyn with the fluorescent protein mCherry (33, 34). The embryos were allowed to develop to 10 hpf, at which point they were reacted with DIFO-647 and imaged (Fig. 5C; SI Appendix, Fig. S16; and Movie S2). This experiment revealed two important pieces of information. First, the memCherry signal appeared between the two daughter cells within seconds of the formation of the cleavage furrow, suggesting that cytokinesis was indeed occurring normally. Second, we noticed no intensification of the memCherry signal at the cleavage furrow, in contrast to the DIFO signal.
These results suggest that the substantial recruitment of glycans to the cleavage furrow cannot be explained by a simple reorganization of the plasma membrane. Further, the lack of DIFO-derived signal between daughter cells (Fig. 5C, arrowheads)suggests that glycans within that region of plasma membrane had not migrated from other regions of plasma membrane during the mitotic event. Therefore, the glycans within this patch of membrane are likely biosynthesized de novo, consistent with previous studies of membrane addition during cytokinesis in different organisms (35–37). We attempted to label the putative newly synthesized glycans in the new membrane between daughter cells by reacting GalNAz-injected embryos sequentially with DIFO-555 and, immediately afterward, DIFO-488. In all cases, we observed colocalization of the two labels (SI Appendix, Fig. S17). This result suggests either that the process of new membrane addition at the furrow and subsequent glycan reorganization take place on a faster time scale than that of our labeling protocol or that there are relatively few azide-labeled glycans in this segment of membrane.
Conclusion
We have demonstrated that microinjection of simple and nucleotide azidosugars followed by labeling using copper-free click chemistry can enable visualization of glycan biosynthesis in the enveloping layer of zebrafish embryos as early as 4 hours after the onset of zygotic gene expression. We have also shown that a nonmetabolic approach can be used to image sialylated glycans in the enveloping layer of zebrafish embryos. Simultaneous visualization of O-glycans and sialylated glycans using these independent targeting strategies exposed subtle differences in the distributions of these two partially overlapping sectors of the glycome during embryogenesis.
Through these studies, we also found that cell-surface O-glycans are recruited to the cleavage furrow of dividing enveloping layer cells on second-to-minute time scales. An interesting direction for future study involves the identification of the protein scaffolds to which the glycans are anchored. These mucin-type glycoproteins may play a role in cell division that has not yet been characterized. More generally, this work sets the stage for future studies of other sectors of the glycome, as well as other biomolecules such as proteins, lipids, and cofactors that can be labeled by microinjection of functionalized biosynthetic precursors.
Materials and Methods
Metabolic Labeling of Zebrafish by Microinjection of Embryos with Azidosugars and Detection by Copper-Free Click Chemistry.
Zebrafish embryos at the one-cell, two-cell, or four-cell stage were microinjected into the yolk cell with 1–5 nL of a 5–125 mM solution of the appropriate sugar (UDP-GalNAz, UDP-GalNAc, GalNAz, Ac4GalNAz, or ManNAz) and either rhodamine-dextran (5% wt/vol) or phenol red (0.1% wt/vol) in 0.2 M KCl. For experiments using memCherry, mRNA encoding memCherry (32) was added to the 25 mM GalNAz solution (containing 0.1% wt/vol phenol red and 0.2 M KCl) at a concentration of 20 μg/mL; in this case, 1 nL of the solution was microinjected directly into the blastomere cell at the one-cell stage. Following microinjection, embryos were allowed to develop to 3 hpf, at which point they were manually cleaned. With the exception of the experiment shown in SI Appendix, Fig. S4, the embryos were then enzymatically dechorionated by incubation in a 1 mg/mL solution of pronase in embryo medium (150 mM NaCl, 0.5 mM KCl, 1.0 mM CaCl2, 0.37 mM KH2PO4, 0.05 mM Na2HPO4, 2.0 mM MgSO4, 0.71 mM NaHCO3 in deionized H2O, pH 7.4) at ∼28 °C for approximately 5 min, and rinsed five times. Following dechorionation, the embryos were transferred using a fire-polished wide-bore Pasteur pipet into 1% agarose-coated Petri dishes containing embryo medium. All embryos younger than 12 hpf were stored in Petri dishes or 96-well plates coated with a 1% solution of agarose in embryo medium. When embryos were to be used for imaging beyond 24 hpf, 131 μM N-phenylthiourea (PTU) was included in all media starting at 24 hpf to inhibit melanin production. To detect cell-surface azide-labeled glycans, embryos were transferred to a flat-bottom 96-well plate containing DIFO-488, DIFO-555, or DIFO-647 (100 μL of a 100 μM solution in embryo medium) and allowed to react for 1 h at 28.5 ºC.
Chemical Labeling of Sialylated Glycans Using Sodium Periodate and Aminooxy Reagents.
Zebrafish embryos were dechorionated at 3 hpf using pronase and allowed to further develop. At various time points, the embryos were transferred to a Petri dish containing a freshly made solution of NaIO4 (500 μM) or no reagent in embryo medium and incubated for 30 min at 28.5 ºC. The embryos were then transferred to a Petri dish containing 10 mM glycerol for 2 min to quench any excess NaIO4, followed by another transfer to a dish containing 1× PBS, pH 6.7. Embryos were then transferred to a flat-bottom 96-well plate containing a solution of AO-488 (100 μL of a 100 μM solution in PBS, pH 6.7) and allowed to react for 1 h at 28.5 ºC.
Imaging of Glycans by Confocal Microscopy.
After the labeling reactions, embryos were rinsed by sequential transferring through six 15-cm Petri dishes filled with 25 mL of embryo medium. Embryos were then mounted between two cover slips, stabilized by vacuum grease in each corner, in a solution of 0.6% low melting point agarose in embryo medium. For embryos 24 hpf and older, 2.6 μM tricaine (ethyl 3-aminobenzoate methanesulfonate) was added to the agarose solution prior to mounting and imaging by confocal microscopy. For details on image acquisition and analysis, see SI Appendix.
Supplementary Material
Acknowledgments.
We thank Emilie Delaune for providing memCherry mRNA and much advice; Anjali Ganguli for providing UDP-GalNAz; Holly Aaron (UC Berkeley Molecular Imaging Center), Julian Codelli, Jen St. Hilaire, and Deborah Weinman for technical assistance; and David Halpin and Blake Riggs for helpful discussions. This work was funded by grants to C.R.B. (GM058867) and S.L.A. (GM061952) from the National Institutes of Health. J.M.B. was supported by National Science Foundation and National Defense Science and Engineering predoctoral fellowships, and K.W.D. was supported by a National Science Foundation predoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0912081107/-/DCSupplemental.
References
- 1.Gilbert SF, Singer SR, Tyler MS, Kozlowski RN. Developmental Biology. Sunderland, Mass: Sinauer Associates; 2006. [Google Scholar]
- 2.Yaniv K, et al. Live imaging of lymphatic development in the zebrafish. Nat Med. 2006;12:711–716. doi: 10.1038/nm1427. [DOI] [PubMed] [Google Scholar]
- 3.McMahon A, Supatto W, Fraser SE, Stathopoulos A. Dynamic analyses of Drosophila gastrulation provide insights into collective cell migration. Science. 2008;322:1546–1550. doi: 10.1126/science.1167094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- 5.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 6.Beis D, Stainier DY. In vivo cell biology: Following the zebrafish trend. Trends Cell Biol. 2006;16:105–112. doi: 10.1016/j.tcb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Laughlin ST, Bertozzi CR. Imaging the glycome. Proc Natl Acad Sci USA. 2009;106:12–17. doi: 10.1073/pnas.0811481106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varki A. Essentials of Glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 9.Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 10.Prescher JA, Bertozzi CR. Chemical technologies for probing glycans. Cell. 2006;126:851–854. doi: 10.1016/j.cell.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Prescher JA, Bertozzi CR. Chemistry in living systems. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 12.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. In vivo imaging of membrane-associated glycans in developing zebrafish. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baskin JM, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerardel Y, Chang LY, Maes E, Huang CJ, Khoo KH. Glycomic survey mapping of zebrafish identifies unique sialylation pattern. Glycobiology. 2006;16:244–257. doi: 10.1093/glycob/cwj062. [DOI] [PubMed] [Google Scholar]
- 15.Chang LY, et al. Developmental regulation of oligosialylation in zebrafish. Glycoconj J. 2009;26:247–261. doi: 10.1007/s10719-008-9161-5. [DOI] [PubMed] [Google Scholar]
- 16.Kane DA, Kimmel CB. The zebrafish midblastula transition. Development. 1993;119:447–456. doi: 10.1242/dev.119.2.447. [DOI] [PubMed] [Google Scholar]
- 17.Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Natl Acad Sci USA. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimmel CB, Law RD. Cell lineage of zebrafish blastomeres. I. Cleavage pattern and cytoplasmic bridges between cells. Dev Biol. 1985;108:78–85. doi: 10.1016/0012-1606(85)90010-7. [DOI] [PubMed] [Google Scholar]
- 19.Varki A. Sialic acids in human health and disease. Trends Mol Med. 2008;14:351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 21.Saxon E, et al. Investigating cellular metabolism of synthetic azidosugars with the Staudinger ligation. J Am Chem Soc. 2002;124:14893–14902. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
- 22.Luchansky SJ, Argade S, Hayes BK, Bertozzi CR. Metabolic functionalization of recombinant glycoproteins. Biochemistry. 2004;43:12358–12366. doi: 10.1021/bi049274f. [DOI] [PubMed] [Google Scholar]
- 23.Gahmberg CG, Andersson LC. Selective radioactive labeling of cell surface sialoglycoproteins by periodate-tritiated borohydride. J Biol Chem. 1977;252:5888–5894. [PubMed] [Google Scholar]
- 24.De Bank PA, Kellam B, Kendall DA, Shakesheff KM. Surface engineering of living myoblasts via selective periodate oxidation. Biotechnol Bioeng. 2003;81:800–808. doi: 10.1002/bit.10525. [DOI] [PubMed] [Google Scholar]
- 25.Van Lenten L, Ashwell G. Studies on the chemical and enzymatic modification of glycoproteins. A general method for the tritiation of sialic acid-containing glycoproteins. J Biol Chem. 1971;246:1889–1894. [PubMed] [Google Scholar]
- 26.Spiro RG. Periodate oxidation of the glycoprotein fetuin. J Biol Chem. 1964;239:567–573. [PubMed] [Google Scholar]
- 27.Karkas JD, Chargaff E. Studies on stability of simple derivatives of sialic acid. J Biol Chem. 1964;239:949–957. [PubMed] [Google Scholar]
- 28.Hang HC, Bertozzi CR. Chemoselective approaches to glycoprotein assembly. Acc Chem Res. 2001;34:727–736. doi: 10.1021/ar9901570. [DOI] [PubMed] [Google Scholar]
- 29.Zeng Y, Ramya TN, Dirksen A, Dawson PE, Paulson JC. High-efficiency labeling of sialylated glycoproteins on living cells. Nat Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dirksen A, Hackeng TM, Dawson PE. Nucleophilic catalysis of oxime ligation. Angew Chem Int Ed. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 31.Chang PV, Prescher JA, Hangauer MJ, Bertozzi CR. Imaging cell surface glycans with bioorthogonal chemical reporters. J Am Chem Soc. 2007;129:8400–8401. doi: 10.1021/ja070238o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pauls S, Geldmacher-Voss B, Campos-Ortega JA. A zebrafish histone variant H2A.F/Z and a transgenic H2A.F/Z:GFP fusion protein for in vivo studies of embryonic development. Dev Genes Evol. 2001;211:603–610. doi: 10.1007/s00427-001-0196-x. [DOI] [PubMed] [Google Scholar]
- 33.Lowell CA. Src-family kinases: Rheostats of immune cell signaling. Mol Immunol. 2004;41:631–643. doi: 10.1016/j.molimm.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Megason SG. In toto imaging of embryogenesis with confocal time-lapse microscopy. Methods Mol Biol. 2009;546:317–332. doi: 10.1007/978-1-60327-977-2_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albertson R, Riggs B, Sullivan W. Membrane traffic: A driving force in cytokinesis. Trends Cell Biol. 2005;15:92–101. doi: 10.1016/j.tcb.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Shuster CB, Burgess DR. Targeted new membrane addition in the cleavage furrow is a late, separate event in cytokinesis. Proc Natl Acad Sci USA. 2002;99:3633–3638. doi: 10.1073/pnas.052342699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gromley A, et al. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.