

Abstract
Hepcidin is a negative regulator of iron absorption produced mainly by the liver in response to changes in iron stores and inflammation, and its levels have been shown to regulate the intestinal basolateral iron transporter ferroportin1 (Fp1). Hereditary hemochromatosis patients and Hfe-deficient mice show inappropriate expression of hepcidin but, in apparent contradiction, still retain the ability to regulate iron absorption in response to alterations of iron metabolism. To further understand the molecular relationships among Hfe, hepcidin, and Fp1, we investigated hepcidin and Fp1 regulation in Hfe-deficient mice (Hfe−/− and β2m−/−) in response to iron deprivation, iron loading, and acute inflammation. We found that whereas basal hepcidin levels were manifestly dependent on the presence of Hfe and on the mouse background, all Hfe-deficient mice were still able to regulate hepcidin in situations of altered iron homeostasis. In the liver, Fp1 was modulated in opposite directions by iron and LPS, and its regulation in Hfe-deficient mice was similar to that observed in wild-type mice. In addition, we found that iron-deprived mice were able to mount a robust response after LPS challenge and that Toll-like receptor 4 (TLR-4)-deficient mice fail to regulate hepcidin expression in response to LPS. In conclusion, these results suggest that although Hfe is necessary for the establishment of hepcidin basal levels, it is dispensable for hepcidin regulation through both the iron-sensing and inflammatory pathways, and hepatic Fp1 regulation is largely independent of hepcidin and Hfe. The inflammatory pathway overrides the iron-sensing pathway and is TLR-4 dependent.
Keywords: ferroportin 1, Toll-like receptor 4, β2m, hereditary hemochromatosis, lipopolysaccharide
The maintenance of body iron balance is imposed by the fact that total iron deficiency is incompatible with life, whereas iron excess is dangerously toxic. In mammals, iron metabolism is characterized by a limited external exchange and by efficient reutilization of iron from internal sources. Because the capacity to excrete iron is very limited, total body iron is regulated mainly by controlling the level of absorption from the diet, via intestinal cells (3), such that sufficient iron is provided to keep stores replete, but not excessive, and erythroid demands are met. When the regulatory function of intestinal iron absorption is altered and the amount of iron entering the body exceeds the amount lost over a sustained period of time, iron overload arises (41). Progressive iron deposition eventually leads to tissue damage and fibrosis and ultimately to organ failure.
Hereditary hemochromatosis (HH), a common genetic disorder in Caucasian populations, is characterized by high levels of iron absorption from the diet, leading to iron accumulation in the liver and other organs (41). The gene associated with HH type 1 has been identified as HFE, coding for a major histo-compatibility complex class I (MHC-I)-like molecule (11). As with other MHC-I molecules, HFE needs to associate with β2-microglobulin (β2m) for appropriate expression at the cell surface (12). Accordingly, mice lacking HFE or β2m recapitulate HH in humans and develop iron overload primarily in the liver (25, 45, 49) due to hyperabsorption of iron in the duodenum (4, 45).
Recently, a small, cysteine-rich peptide called hepcidin emerged as a major regulator of iron absorption (20, 40, 42). Hepcidin is cleaved from a larger precursor expressed predominantly in the liver and its mRNA levels are regulated by iron, anemia, hypoxia, and inflammation (38). Mice lacking hepcidin develop iron overload (36), and conversely, mice overexpressing hepcidin incur severe anemia (37). In addition, hepcidin injection in mice has been shown to downregulate iron absorption (21). Collectively, these data offer strong support for the role of hepcidin as a negative regulator of intestinal iron absorption.
The mechanisms by which hepcidin exerts its effects on intestinal iron absorption include transcriptional and posttranslational regulation of ferroportin 1 (Fp1), the only iron exporter in mammals (9). In fact, Fp1 mRNA expression in the gut has been shown to be downregulated by high hepcidin levels (14, 48), and in the human embryonic kidney cell line model HEK-293, hepcidin was shown to bind to Fp1, resulting in internalization and degradation of Fp1 and leading to decreased export of cellular iron (35). The regulation of Fp1 by hepcidin is proposed to complete a homeostatic loop through which iron regulates the secretion of hepcidin by liver cells, which in turn controls the expression and the concentration of Fp1 on the cell surface of gut enterocytes (35).
In iron-loaded HH patients and in Hfe-deficient mice due to the lack of Hfe or β2m, basal hepatic hepcidin mRNA levels have been reported to be either lower than (2, 5, 28, 31) or not significantly different (15, 17, 28, 32) from those in the controls. Because hepcidin levels in normal mice rise in response to iron overload (38, 42), this has been interpreted as the lack of iron sensing and the inability to regulate hepcidin expression in the absence of Hfe, which is in line with its proposed role as a negative regulator of intestinal iron absorption (16). This, however, contradicts other studies that demonstrated that HH patients (29) and both β2m−/− (44) and Hfe−/− (4, 22) mice, in fact, retain the ability to regulate iron absorption levels in response to altered iron metabolism. These observations raise the question of whether Hfe is absolutely required for the regulation of hepcidin and downstream Fp1 levels in response to altered iron metabolism.
In this study, we set out to evaluate the capacity of Hfe-deficient mice (Hfe−/− and β2m−/−) to regulate hepcidin expression and subsequent downstream responses, namely, Fp1 expression, via the iron-sensing and inflammation pathways. Because hepcidin levels in mice have been shown to depend on their genetic background (8, 10, 13), we investigated Hfe-knockout mice from two distinct backgrounds, C57BL/6 (B6.Hfe−/−) and 129/SvEvTac (129Sv.Hfe−/−), by comparing their responses to corresponding wild-type (Wt) mice, B6.Wt and 129Sv.Wt. In addition, we further studied the involvement of upstream elements, namely, Toll-like receptor 4 (TLR-4), in the iron-sensing and inflammatory pathways.
MATERIALS AND METHODS
Animals
All procedures were performed in accordance with Canadian Council on Animal Care guidelines after approval by the institutional animal care committee of the Centre Hospitalier de l’Université de Montréal (CHUM). Hfe−/− mice were kindly provided by Dr. N. C. Andrews [Howard Hughes Medical Institute and Harvard Medical School, Children’s Hospital, Boston, MA (24)] in the 129/SvEvTac background and were backcrossed onto the C57BL/6 (B6) background for 10 generations. B6, β2m−/−, C3H/HeJ, and C3H/HeOuJ mice were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). All animals were permanently housed under specific pathogen-free conditions.
Animal treatments
Control mice were given a commercial diet (Teklad Global 18% protein rodent diet; Harlan Teklad, Madison, WI). Dietary iron overload was produced by giving 8-wk-old mice the same commercial diet supplemented with 2.5% (wt/wt) carbonyl iron (Sigma-Aldrich, St. Louis, MO) for 2 wk. Iron deprivation was induced by feeding mice the same commercial diet deficient in iron for 2 wk.
Acute inflammation was produced by a single dose of LPS (Escherichia coli serotype 055:B5, 5 mg/kg ip, Sigma-Aldrich). Control mice were similarly injected with an equivalent volume of sterile saline solution (0.09% NaCl). The animals were killed 6 h after the injection.
Quantitative RT-PCR
Total RNA was isolated with TRIzol reagent (Invitrogen, Burlington, ON, Canada), and RT was performed with the Thermoscript RT-PCR system (Invitrogen). Hepcidin, Fp1, and β-actin mRNA levels were measured by real-time PCR in a Rotor Gene 3000 real-time DNA detection system (Montreal Biotech, Kirkland, QC, Canada) with the QuantiTect SYBRGreen I PCR kit (Qiagen, Mississauga, ON, Canada), as described (28). The primers employed were: β-actin, 5′-TGTTACCAACTGGGACGACA-3′ and 5′-GGTGTTGAAGGTCTCAAA-3′; hepcidin, 5′AGAGCTGCAGC-CTTTGCAC3′ and 5′GAAGATGCAGATGGGGAAGT3′; and Fp1, 5′CCCATCCCCATAGTCTCTGT3′ and 5′CTTGCAGCAACTGT-GTCACC3′. Expression levels were normalized to the housekeeping gene β-actin.
Hematological measurements and transferrin saturation
Red blood cell (RBC) count, Hb, hematocrit, and mean corpuscular volume were measured with an automated cell counter calibrated for murine samples (ABC vet counter; ABX hématologie, Montpellier, France). Serum iron, total iron-binding capacity, and transferrin saturation were assessed by colorimetric assay (30) with the Kodak Ektachem DT60 system (Johnson & Johnson, Ortho Clinical Diagnostics, Mississauga, ON, Canada).
Measurement of tissue iron concentration
Liver and spleen iron concentrations were assessed by acid digestion of tissue samples, followed by iron quantification with atomic absorption spectroscopy (30).
Hepatocyte isolation and treatments
Hepatocytes were isolated from adult mouse livers according to a procedure already described (34). Isolated cells were resuspended in Williams’ E media with 5% FBS and seeded onto collagen-coated plates at a density of 2.4 × 105 cells per well. After 3 h, unattached cells were removed, and the medium was replaced by fresh medium. Twenty-two hours after isolation, the medium was changed and replaced by fresh medium alone or medium containing IL-6 (20 ng/ml), TNF-α (20 ng/ml) for 24 h, or ferric ammonium citrate (FAC; 10 μM, Sigma-Aldrich) for 16 h.
Statistical analysis
All values in the figures are expressed as means ± SD. Multiple comparisons were statistically evaluated by one-way ANOVA.
RESULTS
Iron restriction
To evaluate the ability of Hfe-deficient mice to regulate hepcidin mRNA levels in response to lowered iron stores, mice were placed for 2 wk on an iron-deficient diet. As reported previously (28), iron parameters, including serum iron, transferrin saturation, and liver iron concentrations in mice kept on the standard diet, were significantly higher in Hfe-deficient animals compared with Wt controls from the same background, and liver iron stores were more elevated in the 129Sv background (322 ± 50 in 129Sv.Wt vs. 1,675 ± 249 μg iron/g dry weight in 129.Hfe−/−; P < 0.0001) compared with B6 (221 ± 20 in B6.Wt vs. 772 ± 202 in B6.Hfe−/− and 671 ± 67 μg iron/g dry weight in B6.β2m−/−; P < 0.001). Iron restriction resulted in a significant decrease of liver iron stores in all mouse strains, whereas erythroid parameters such as Hb and RBC count remained unaffected, indicating that the treatment did not lead to anemia (data not shown).
As seen in Figure 1A, there were no significant differences in hepcidin mRNA basal levels among B6.Wt, B6.Hfe−/− and B6.β2m−/− mice fed a standard diet. After an iron-deficient diet, all strains responded by significantly decreasing hepcidin mRNA levels.
Fig. 1.

Hepcidin is downregulated in the liver of Hfe-deficient mice in response to iron-deprivation. Hepcidin gene expression in the liver of mice fed a standard diet (SD; light gray) or an iron-deficient diet (−Fe; dark gray) from the C57BL/6 (A) and 129/SvEvTac background (B). Hepcidin mRNA expression was quantified by quantitative RT-PCR (qRT-PCR) and normalized to β-actin. The hepcidin/β-actin ratio is shown. The results are presented as means ± SD (n = 3–6 mice/group). Statistical analysis was performed by 1-way ANOVA; **P < 0.001 for comparison with appropriate control mice (SD diet).
A similar response was observed in the 129Sv background (Fig. 1B), with five- and fourfold decreases in hepcidin levels in 129Sv.Wt and 129.Hfe−/− mice fed the iron-deficient diet, respectively. However, and as reported previously (28), in this mouse background, basal hepcidin levels were already significantly lower in 129Sv.Hfe−/− mice fed the standard diet compared with 129Sv.Wt mice (P < 0.001) on the same diet.
These results indicate that Hfe-deficient mice retain the ability to lower hepatic hepcidin levels in situations of iron deprivation.
Iron loading
To evaluate hepcidin regulation in response to increased iron stores, mice were placed for 2 wk on a carbonyl iron-supplemented diet (2.5% wt/wt). This treatment resulted in a two- to threefold increase of liver iron stores in all mouse strains tested (772 ± 202 in B6.Wt, 1,571 ± 251 in B6.Hfe−/−, and 1,317 ± 305 μg iron/g dry weight in B6. β2m−/−; 1,230 ± 227 in 129Sv.Wt and 3,686 ± 989 μg iron/g dry weight in 129.Hfe−/−; P < 0.001 compared with mice on the standard diet). We found a 2.4-fold elevation of hepcidin mRNA levels in B6.Wt mice and a 1.9-fold increment in B6.Hfe−/− and B6. β2m−/− mice fed the iron-enriched diet compared with corresponding controls kept on the standard diet (Fig. 2A). When comparing dietary iron-loaded B6.Wt mice with B6.Hfe−/− and B6. β2m−/− mice, hepcidin levels were significantly higher (P < 0.005) in B6.Wt mice.
Fig. 2.

Hepcidin is up-regulated in the liver of Hfe-deficient mice in response to iron-loading. Hepcidin gene expression in the liver of mice fed a standard diet (SD; light gray) or an iron-enriched diet (+Fe; dark gray) from the C57BL/6 (A) and 129/SvEvTac background (B). Hepcidin mRNA expression was quantified by qRT-PCR and normalized to β-actin. The hepcidin/β-actin ratio is shown. The results are presented as means ± SD (n = 5–7 mice/group). Statistical analysis was performed by 1-way ANOVA; *P < 0.01, and **P < 0.001 for comparison with relevant saline-treated mice (SD diet).
In the 129Sv background, 129Sv.Wt and 129Sv.Hfe−/− mice showed 14- and 15-fold increases in hepcidin levels, respectively, compared with controls fed the standard diet. Although the response observed was similar in Wt and Hfe-deficient mice in terms of fold increase, because basal levels were lower in 129Sv.Hfe−/−, hepcidin remained significantly lower than in 129Sv.Wt (P < 0.001).
Thus Hfe-deficient mice from the two different backgrounds tested were still able to increase hepcidin in response to iron loading, although the absolute levels reached were lower than in iron-loaded Wt mice.
Acute systemic inflammation
Besides iron stores, hepcidin has been shown to also be modulated by inflammation (38). To evaluate changes in iron metabolism and hepatic hepcidin expression by inflammation, we challenged mice with LPS and measured iron parameters as well as hepcidin mRNA levels in the liver 6 h after treatment. The results for the B6 background are presented in Fig. 3. Hepcidin levels rose significantly in all mouse strains (Fig. 3A) and were even higher in LPS-treated B6.Hfe−/− and B6. β2m−/− (3.4- and 3.6-fold increase, respectively) compared with B6.Wt (2-fold increment; P < 0.05). Hypoferremia, evidenced by a two- to fourfold lowering of serum iron and transferrin saturation, was noted in all mouse strains tested (Fig. 3, B and C). In addition, we found a small but significant elevation of liver iron concentrations in all mice challenged by LPS (Fig. 3D), possibly as a result of reduced iron export from liver cells.
Fig. 3.

Hypoferremia and hepcidin induction by inflammation in Hfe-deficient mice from the C57BL/6 background. Hepcidin gene expression (A), serum iron (B), transferrin saturation (C), and iron concentration in the liver (D) in saline-treated control mice (CTL; light gray) and LPS-treated mice (dark gray). Hepcidin mRNA expression was quantified by qRT-PCR and normalized to β-actin. The hepcidin/β-actin ratio is shown. The results are presented as means ± SD (n = 6 mice/group). Statistical analysis was performed by 1-way ANOVA; *P < 0.05, **P < 0.01, and ***P < 0.001 for comparison with relevant control mice.
In the 129Sv background, hepcidin levels were also elevated by LPS in 129Sv.Wt (2-fold increase) as well as in 129Sv.Hfe−/− (8.7-fold increase; Fig. 4A). However, in this mouse background, because basal hepcidin levels in Hfe−/− mice were considerably lower than in Wt controls, hepcidin remained significantly lower than in Wt (P < 0.01) after LPS treatment, as it occurred with the response to iron loading. In addition, hypoferremia also occurred in both LPS-treated mouse strains, with a two- to threefold lowering of serum iron and transferrin saturation (Fig. 4, B and C), but no changes took place in liver iron concentrations (Fig. 4D).
Fig. 4.

Hypoferremia and hepcidin induction by inflammation in Hfe-deficient mice from the 129/SvEvTac background. Hepcidin gene expression (A), serum iron (B), transferrin saturation (C), and iron concentration in the liver (D) in saline-treated control mice (CTL; light gray) and LPS-treated mice (dark gray). Hepcidin mRNA expression was quantified by qRT-PCR and normalized to β-actin. The hepcidin/β-actin ratio is shown. The results are presented as means ± SD (n = 6 mice/group). Statistical analysis was performed by 1-way ANOVA; **P < 0.001 for comparison with respective control mice.
These results show that Hfe-deficient mice, irrespective of the background, can still mount a robust response by elevating hepcidin expression and developing hypoferremia in the systemic inflammation setting induced by LPS treatment.
Fp1 mRNA regulation
Next, to assess whether changes in Fp1 mRNA expression associated with upregulation of hepcidin expression are appropriately elicited in Hfe-deficient mice, we measured Fp1 mRNA expression in two distinct situations leading to augmented hepcidin levels: iron loading and inflammation. We found that, despite the fact that both challenges induced hepcidin mRNA levels, Fp1 mRNA expression in the liver was distinctly regulated. It increased significantly in mice fed the iron-enriched diet but declined in mice treated with LPS (Fig. 5A). The response to iron loading in the liver was similar in all mouse strains tested, i.e., Wt and Hfe-deficient strains. Importantly, basal Fp1 mRNA levels in the liver of Hfe−/− and β2m−/− mice were significantly higher than those in Wt controls (P < 0.001). Furthermore, the difference persisted in mice fed the iron-enriched diet as well as in mice treated with LPS (P < 0.001). This shows that the capacity to regulate Fp1 in the liver in response to disrupted iron homeostasis remains intact in Hfe-deficient mice.
Fig. 5.

Hepatic Fp1 regulation by iron is independent of hepcidin levels. Fp1 mRNA levels in the liver (A) and duodenum (B) in control (CTL; light gray), iron-loaded (+Fe; dark gray), and LPS-treated mice (black). The results are presented as means ± SD (n = 6 mice/group). Statistical analysis was performed by 1-way ANOVA; *P < 0.01, and **P < 0.001 for comparison with relevant control mice. Fp1 mRNA expression was quantified by qRT-PCR and normalized to β-actin. The Fp1/β-actin ratio is shown. C: effect of iron, LPS, IL-6, and TNF-α on Fp1 expression by primary murine hepatocytes. Each symbol represents 1 experiment in which 3 samples of primary murine hepatocytes were incubated with culture medium, FAC, LPS, or the cytokines indicated. The results are expressed as fold changes of Fp1 mRNA normalized to β-actin of treatments compared with CTL.
The regulation of Fp1 mRNA levels was different in the duodenum. First, the distinct regulation observed in the liver of iron-loaded mice compared with LPS-treated mice did not occur, because both treatments resulted in reduced duodenal Fp1 mRNA levels in B6.Wt controls (Fig. 5B). Second, we also found that, as reported by others (31), basal duodenal Fp1 mRNA levels were significantly reduced in both B6.Hfe−/− and B6. β2m−/− mice compared with B6.Wt mice (P < 0.001), which may explain why no further changes were seen in Hfe-deficient mice after the treatments.
To further investigate whether Fp1 can be directly regulated by iron and cytokines in hepatocytes, isolated hepatocytes from B6 mice were treated with FAC, LPS, IL-6, and TNF-α and Fp1 expression was measured. As illustrated in Fig. 5C, we found that iron treatment induced Fp1 mRNA expression in hepatocytes, whereas LPS, TNF-α, and IL-6 treatments suppressed it.
Taken together, these results show that 1) Fp1 mRNA is oppositely regulated in the liver by iron and inflammation, and 2) Hfe-deficient mice retain the ability to regulate hepatic Fp1 mRNA levels through both the iron-sensing and inflammatory pathways.
Iron-sensing versus inflammatory pathway
Because iron deficiency and inflammation elicit opposite responses in hepcidin regulation, we questioned how hepcidin would be modulated by inflammation in iron-deficient mice. B6.Wt mice were placed on the iron-deficient diet and challenged after 2 wk by LPS. Compared with untreated controls, hypoferremia revealed by lower serum iron levels and transferrin saturation was evident both in mice treated only with LPS and in iron-deficient mice challenged with LPS (Fig. 6, A and B). Liver iron levels were decreased slightly in iron-deficient mice and augmented in LPS-treated mice. Combined treatment resulted in decreased liver iron concentrations (Fig. 6C).
Fig. 6.

The inflammatory pathway overrides the iron-sensing pathway. Serum iron (A), transferrin saturation (B), and iron concentration in the liver (C) in control saline-treated mice (CTL), iron-deficient (Fe-def), LPS-treated, and mice with combined treatments (Fe-def + LPS). Hepatic hepcidin (D) and Fp1 gene expression in the liver (E) and duodenum (F). mRNA expression was quantified by qRT-PCR and normalized to β-actin. The hepcidin/β-actin and Fp1/β-actin ratios are shown. The results are presented as means ± SD (n = 6 mice/group). Statistical analysis was performed by 1-way ANOVA; *P < 0.01, and **P < 0.001 for comparison with control mice.
We found that hepcidin mRNA levels in the liver rose significantly with combined treatment (Fig. 6D). Conversely, Fp1 mRNA levels decreased in these mice in both the liver and duodenum (Fig. 6, E and F), as observed in mice treated only with LPS, whereas in iron-deficient mice, Fp1 mRNA levels did not change in the liver but were augmented in the duodenum.
Taken together, these results indicate that the inflammatory pathway overcomes hepcidin and Fp1 regulation by iron stores in both the liver and duodenum.
TLR-4 pathway
Transduction of the LPS signal across the cell membrane is mediated by TLR-4, which, in turn, activates transcription factor NF-kB, resulting in the production of various proinflammatory cytokines, including IL-6 and TNF-α (18). To gain further insight into the possible upstream pathways involved in hepcidin regulation by inflammation, we tested mice deficient in TLR-4 (C3H/HeJ) and Wt mice from the same background (C3H/HeOuJ). As shown in Fig. 7, we found that hepcidin activation by iron levels remained intact in TLR-4-deficient mice but was suppressed when the animals were challenged by LPS. Thus the pathway involved in hepcidin regulation during inflammation is TLR-4 dependent, whereas hepcidin regulation by iron is not dependent on TLR-4.
Fig. 7.
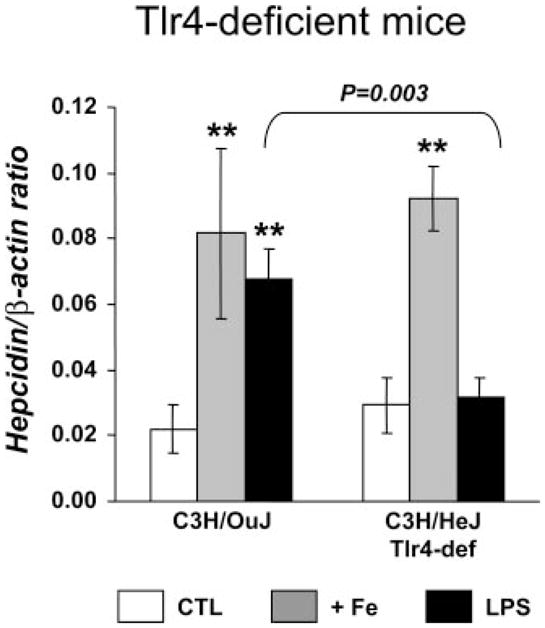
The inflammatory pathway for hepcidin regulation is Toll-like receptor 4 (TLR-4) dependent. Hepatic hepcidin mRNA expression was quantified by qRT-PCR and normalized to β-actin. The hepcidin/β-actin ratio is shown. The results are presented as means ± SD (n = 4–6 mice/group). Statistical analysis was performed by 1-way ANOVA; **P < 0.001 for comparison with relevant control mice.
DISCUSSION
Hfe-independent regulation of hepcidin levels by iron and inflammation
In this study, we showed that Hfe-deficient mice from two different backgrounds retain the ability to regulate hepcidin levels in response to altered iron stores. In fact, Hfe-deficient mice were similar to Wt mice in terms of fold change of hepcidin regulation. However, because basal hepcidin levels, which are considerably affected by background, are inappropriately low in these mice, hepcidin mRNA levels in iron-loaded, Hfe-deficient mice did not reach the values in Wt animals. Similar responses were observed in LPS-treated mice, with the development of hypoferremia in all strains. This suggests that total hepcidin expression levels on iron loading and inflammation are additive to baseline levels. The changes in hepcidin levels after these treatments are presumably sufficient to significantly affect iron absorption levels and thus reconcile the observation that Hfe-deficient mice can regulate iron absorption in response to alterations of iron metabolism (4, 22, 44) with the proposed role for hepcidin as a negative regulator of iron absorption (16). Possible explanations for discrepancies between studies reporting the absence of hepcidin regulation in Hfe−/− mice after iron loading or acute inflammation (2, 43) may include differences in experimental design, i.e., animal background (mixed, C57BL/6 × 129SvTavEv background), different treatments (parenteral vs. dietary iron loading), doses, and treatment schedules. Previous investigations of the response to acute inflammation were performed 1.5 h after LPS injection (43), earlier than studies reporting hepcidin regulation, 6 h in our experiments and in those of Lee et al. (23) and 16 h after Freund’s complete adjuvant injection (15). It is possible that Hfe is required for the initial response and is dispensable later during the inflammatory response. In terms of relevance to the anemia of chronic disease (ACD), long-term responses may be more pertinent, because ACD is associated with chronic inflammation and diseases.
Taken together, these observations suggest that Hfe has an essential role in setting basal hepcidin levels and, hence, basal iron absorption levels but that further regulation of hepcidin by iron stores and inflammation is largely Hfe independent.
The cumulative effect of background in setting hepcidin basal levels may be related to differences in MHC-I molecules. Several lines of evidence indicate that MHC-I molecules other than Hfe may influence the degree of iron loading. For example, Hfeβ2m−/− compound mutants develop more severe iron loading than single knockout mice (24), and MHC-deficient mice incur iron overload (6).
Regulation of Fp1 mRNA in the liver is largely independent of hepcidin levels and Hfe
We showed that the regulation of Fp1 mRNA in the liver was in the opposite direction during acute inflammation and in iron overload. Because both treatments lead to increase hepcidin production, this indicates that the regulation of Fp1 mRNA levels in the liver is independent of hepcidin levels. In contrast, in the duodenum, both treatments lead to the same response, i.e., the suppression of Fp1 mRNA expression. Possibly, Fp1 in the liver is regulated locally by iron, most likely via the iron responsive elements/iron regulatory proteins (IRE/IRP)-dependent system (26, 27), in both macrophages (19) and hepatocytes (current study). However, direct regulation of Fp1 by iron via the IRE/IRP system seems to be suppressed in the duodenum, because Fp1 mRNA is downregulated in the duodenum of iron-loaded Wt mice. The opposite direction of regulation for Fp1 in the gut is also found in sex-linked anemia mice, where Fp1 mRNA and Fp1 protein levels remain elevated, even though enterocyte iron and ferritin levels are significantly higher than in iron-deficient Wt mice (7). Presumably, in the gut, Fp1 mRNA and Fp1 protein levels (1) are regulated by systemic factors, such as hepcidin. Ultimately, depending on which pathway is activated, the iron-sensing or the inflammatory pathways, there seem to be multiple mechanisms for Fp1 regulation in the liver and the gut.
We found that basal hepatic Fp1 mRNA levels were higher in Hfe-deficient mice than in Wt mice. Similarly, hepatic Fp1 mRNA expression is increased in mice that develop iron overloading due to a deficiency in hemojuvelin (Hjv), an upstream regulator of hepcidin expression in the iron-sensing pathway (39), as well as in hepcidin-deficient mice (Usf2−/−) (47). Solely on the basis of the response of Fp1 to iron loading in the gut, this could be interpreted as an inappropriate response to downregulate Fp1 in the liver due to the lack of basal hepcidin expression. However, the fact that the normal response to iron loading in the liver is an increase of Fp1 mRNA and Fp1 protein (1) expression suggests that Hfe-deficient, Hjv−/− and Usf2−/− mice may be appropriately responding to higher iron levels in the liver and thus further indicating that local regulation of Fp1 mRNA by iron in the liver is independent of hepcidin levels.
Inflammatory pathway overrides the iron-sensing pathway for hepcidin and Fp1 regulation
We observed that iron-deprived mice, despite significantly reduced iron stores, still mount a robust response to acute inflammation by increasing hepatic hepcidin mRNA and reducing Fp1 levels in both the liver and duodenum. This suggests that the iron-sensing pathway for hepcidin regulation is overruled during inflammation, a finding that may have significant implications in ACD. In fact, sustained hepcidin expression and Fp1 reduction in the duodenum may result in decreased iron absorption and thus in the development of true iron deficiency in patients with chronic inflammation and cancer. Iron deficiency in these situations would be insufficient to trigger an appropriate response to restore body iron stores and would thus aggravate and perpetuate the anemic state. This is particularly pertinent in cancer settings, where anemia is associated with reductions in the quality of life and survival in cancer patients (46). In fact, anemia causes tissue hypoxia, which has been shown to promote tumor growth and to enhance the resistance of tumor cells to radiotherapy and chemotherapy (46).
Inflammatory pathway for hepcidin regulation is TLR-4 dependent
TLRs are an essential component of signaling receptors expressed on the cell surface that recognize pathogen-associated molecular patterns (PAMP) produced by micro-organisms. LPS is a PAMP that is recognized by TLR-4 and the CD14 coreceptor, leading to subsequent activation of NF-κB and resulting in the production of various proinflammatory cytokines, including IL-6 and TNF-α (33). We found that hepcidin regulation in TLR-4-deficient mice was completely abrogated after LPS treatment but not by iron loading. These results suggest that the inflammatory pathway, but not the iron-sensing pathway of hepcidin regulation, is TLR-4 dependent.
In conclusion, we showed that Hfe is necessary for the establishment of basal hepcidin levels but dispensable for hepcidin regulation through both the iron-sensing and inflammatory pathways. In addition, Fp1 regulation in the liver is independent of hepcidin levels and of Hfe. Finally, the inflammatory pathway overrides the iron-sensing pathway and is TLR-4 dependent.
Acknowledgments
We thank C. Dallaire for work with atomic absorption spectroscopy and O. Da Silva (Research Support Office, Research Centre, CHUM) for editing this manuscript.
GRANTS
This work was supported by the Canadian Institutes of Health Research (CIHR; Grant No. MOP44045). M. M. Santos is the recipient of a CIHR New Investigator Award.
References
- 1.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad KA, Ahmann JR, Migas MC, Waheed A, Britton RS, Bacon BR, Sly WS, Fleming RE. Decreased liver hepcidin expression in the hfe knockout mouse. Blood Cells Mol Dis. 2002;29:361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 3.Aisen P, Wessling-Resnick M, Leibold EA. Iron metabolism. Curr Opin Chem Biol. 1999;3:200–206. doi: 10.1016/S1367-5931(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 4.Ajioka RS, Levy JE, Andrews NC, Kushner JP. Regulation of iron absorption in Hfe mutant mice. Blood. 2002;100:1465–1469. doi: 10.1182/blood-2001-11-0037. [DOI] [PubMed] [Google Scholar]
- 5.Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 6.Cardoso EM, Macedo MG, Rohrlich P, Ribeiro E, Silva MT, Lemonnier FA, de Sousa M. Increased hepatic iron in mice lacking classical MHC class I molecules. Blood. 2002;100:4239–4241. doi: 10.1182/blood-2002-05-1565. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Su T, Attieh ZK, Fox TC, McKie AT, Anderson GJ, Vulpe CD. Systemic regulation of Hephaestin and Ireg1 revealed in studies of genetic and nutritional iron deficiency. Blood. 2003;102:1893–1899. doi: 10.1182/blood-2003-02-0347. [DOI] [PubMed] [Google Scholar]
- 8.Courselaud B, Troadec MB, Fruchon S, Ilyin G, Borot N, Leroyer P, Coppin H, Brissot P, Roth MP, Loreal O. Strain and gender modulate hepatic hepcidin 1 and 2 mRNA expression in mice. Blood Cells Mol Dis. 2004;32:283–289. doi: 10.1016/j.bcmd.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Dupic F, Fruchon S, Bensaid M, Borot N, Radosavljevic M, Loreal O, Brissot P, Gilfillan S, Bahram S, Coppin H, Roth MP. Inactivation of the hemochromatosis gene differentially regulates duodenal expression of iron-related mRNAs between mouse strains. Gastroenterology. 2002;122:745–751. doi: 10.1053/gast.2002.31877. [DOI] [PubMed] [Google Scholar]
- 11.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 12.Feder JN, Tsuchihashi Z, Irrinki A, Lee VK, Mapa FA, Morikang E, Prass CE, Starnes SM, Wolff RK, Parkkila S, Sly WS, Schatzman RC. The hemochromatosis founder mutation in HLA-H disrupts beta 2-microglobulin interaction and cell surface expression. J Biol Chem. 1997;272:14025–14028. doi: 10.1074/jbc.272.22.14025. [DOI] [PubMed] [Google Scholar]
- 13.Fleming RE, Holden CC, Tomatsu S, Waheed A, Brunt EM, Britton RS, Bacon BR, Roopenian DC, Sly WS. Mouse strain differences determine severity of iron accumulation in Hfe knockout model of hereditary hemochromatosis. Proc Natl Acad Sci USA. 2001;98:2707–2711. doi: 10.1073/pnas.051630898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frazer D, Wilkins S, Becker E, Vulpe C, McKie A, Trinder D, Anderson G. Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology. 2002;123:835–844. doi: 10.1053/gast.2002.35353. [DOI] [PubMed] [Google Scholar]
- 15.Frazer DM, Wilkins SJ, Millard KN, McKie AT, Vulpe CD, Anderson GJ. Increased hepcidin expression and hypoferraemia associated with an acute phase response are not affected by inactivation of HFE. Br J Haematol. 2004;126:434–436. doi: 10.1111/j.1365-2141.2004.05044.x. [DOI] [PubMed] [Google Scholar]
- 16.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann T, Muckenthaler M, van der Hoeven F, Brennan K, Gehrke SG, Hubert N, Sergi C, Grone HJ, Kaiser I, Gosch I, Volkmann M, Riedel HD, Hentze MW, Stewart AF, Stremmel W. Iron overload in adult Hfe-deficient mice independent of changes in the steady-state expression of the duodenal iron transporters DMT1 and Ireg1/ferroportin. J Mol Med. 2004;82:39–48. doi: 10.1007/s00109-003-0508-x. [DOI] [PubMed] [Google Scholar]
- 18.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR-4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR-4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 19.Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102:4191–4197. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 20.Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 21.Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schumann K, Debnam ES, Srai SKS. Effect of hepcidin on intestinal iron absorption in mice. Blood. 2004;103:3940–3944. doi: 10.1182/blood-2003-03-0953. [DOI] [PubMed] [Google Scholar]
- 22.Laftah AH, Simpson RJ, Beaumont N, Bahram S, Schumann K, Srai SK. Hypoxic response of iron absorption is not affected by the Hfe gene knock-out in mice. Br J Haematol. 2003;123:170–172. doi: 10.1046/j.1365-2141.2003.04559.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee P, Peng H, Gelbart T, Beutler E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA. 2004;101:9263–9265. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy JE, Montross LK, Andrews NC. Genes that modify the hemochromatosis phenotype in mice. J Clin Invest. 2000;105:1209–1216. doi: 10.1172/JCI9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 26.Liu Xb, Hill P, Haile DJ. Role of the ferroportin iron-responsive element in iron and nitric oxide dependent gene regulation. Blood Cells Mol Dis. 2002;29:315–326. doi: 10.1006/bcmd.2002.0572. [DOI] [PubMed] [Google Scholar]
- 27.Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39:710–715. doi: 10.1016/s0168-8278(03)00408-2. [DOI] [PubMed] [Google Scholar]
- 28.Makui H, Soares RJ, Jiang W, Constante M, Santos MM. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood. 2005;106:2189–2195. doi: 10.1182/blood-2005-02-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaren GD, Nathanson MH, Jacobs A, Trevett D, Thomson W. Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. J Lab Clin Med. 1991;117:390–401. [PubMed] [Google Scholar]
- 30.Miranda CJ, Makui H, Andrews NC, Santos MM. Contributions of beta2-microglobulin-dependent molecules and lymphocytes to iron regulation: insights from HfeRag1(−/−) and β2mRag1(−/−) double knock-out mice. Blood. 2004;103:2847–2849. doi: 10.1182/blood-2003-09-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muckenthaler M, Roy CN, Custodio AO, Minana B, deGraaf J, Montross LK, Andrews NC, Hentze MW. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34:102–107. doi: 10.1038/ng1152. [DOI] [PubMed] [Google Scholar]
- 32.Muckenthaler MU, Rodrigues P, Macedo MG, Minana B, Brennan K, Cardoso EM, Hentze MW, de Sousa M. Molecular analysis of iron overload in [beta]2-microglobulin-deficient mice. Blood Cells Mol Dis. 2004;33:125–131. doi: 10.1016/j.bcmd.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112:521–530. doi: 10.1111/j.1365-2567.2004.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musallam L, Ethier C, Haddad PS, Bilodeau M. Role of EGF receptor tyrosine kinase activity in antiapoptotic effect of EGF on mouse hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1360 –G1369. doi: 10.1152/ajpgi.2001.280.6.G1360. [DOI] [PubMed] [Google Scholar]
- 35.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 36.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grand-champ B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–2186. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 41.Pietrangelo A. Hereditary hemochromatosis–a new look at an old disease. N Engl J Med. 2004;350:2383–2397. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 42.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 43.Roy CN, Custodio AO, de Graaf J, Schneider S, Akpan I, Montross LK, Sanchez M, Gaudino A, Hentze MW, Andrews NC, Muckenthaler MU. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet. 2004;36:481–485. doi: 10.1038/ng1350. [DOI] [PubMed] [Google Scholar]
- 44.Santos M, Clevers H, de Sousa M, Marx JJ. Adaptive response of iron absorption to anemia, increased erythropoiesis, iron deficiency, and iron loading in beta2-microglobulin knockout mice. Blood. 1998;91:3059–3065. [PubMed] [Google Scholar]
- 45.Santos M, Schilham MW, Rademakers LH, Marx JJ, de Sousa M, Clevers H. Defective iron homeostasis in beta 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med. 1996;184:1975–1985. doi: 10.1084/jem.184.5.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spivak JL. The anaemia of cancer: death by a thousand cuts. Nature Rev Cancer. 2005;5:543–555. doi: 10.1038/nrc1648. [DOI] [PubMed] [Google Scholar]
- 47.Viatte L, Lesbordes-Brion JC, Lou DQ, Bennoun M, Nicolas G, Kahn A, Canonne-Hergaux F, Vaulont S. Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood. 2005;105:4861–4864. doi: 10.1182/blood-2004-12-4608. [DOI] [PubMed] [Google Scholar]
- 48.Yeh Ky Yeh M, Glass J. Hepcidin regulation of ferroportin 1 expression in the liver and intestine of the rat. Am J Physiol Gastrointest Liver Physiol. 2004;286:G385–G394. doi: 10.1152/ajpgi.00246.2003. [DOI] [PubMed] [Google Scholar]
- 49.Zhou XY, Tomatsu S, Fleming RE, Parkkila S, Waheed A, Jiang J, Fei Y, Brunt EM, Ruddy DA, Prass CE, Schatzman RC, O’Neill R, Britton RS, Bacon BR, Sly WS. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci USA. 1998;95:2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]