

Abstract
The objective was to assess outcomes of IFNγ-priming upon macrophage activation by the synovial macromolecule high-molecular-weight hyaluronan [HMW-HA] in the context of rheumatoid arthritis inflammation. Human macrophages primed by IFNγ and activated by HMW-HA were evaluated for cytokine secretion by ELISA and Milliplex assay and activation profiles by nuclear transcription factor EIA. IFNγ-primed, HMW-HA-activated macrophages produced elevated levels of TNF and secreted the TH1 cytokine IL-12p70, while IFNγ suppressed HMW-HA-induced secretion of the regulatory cytokine IL-10 and activation of the transcription factor c-Jun. IFNγ modulates the HMW-HA-induced cytokine response profile promoting macrophage activation and inflammatory TH1 cytokine secretion.
Keywords: Rheumatoid arthritis, inflammation, hyaluronan, macrophages, interferon gamma, interleukin-10, interleukin-12, tumor necrosis factor, c-Jun
Introduction
Hyaluronan [HA] is a glycosaminoglycan found in its high molecular weight form [HWM-HA] as repeating D-glucuronic acid, β1-3 linked to N-acetyl-D-glucosamine-β1-4 as a major component of the synovium in vivo[1; 2]. HMW-HA binds cells and initiates intracellular signaling via multiple receptors including CD44 and toll like receptor 4 [TLR4][2; 3]. HMW-HA mediates anti-inflammatory effects on monocytes, osteoblasts and osteoclasts[2; 4], and contributes to healthy joint tissue homeostasis. Injection of HMW-HA into afflicted joints of patients with osteoarthritis has been implemented as an effective therapy, although the mechanism of action remains unclear[5].
Rheumatoid arthritis [RA] is a heterogeneous group of inflammatory disorders characterized by mononuclear synovitis orchestrated by a complex network of immune cells and soluble factors[6; 7]. Joints of RA patients present a challenge to incoming leukocytes due to inflammatory T cell cytokines within the synovium where polarization of CD4 T cells toward a TH1 IFNγ-secreting phenotype is typical[6; 7; 8; 9; 10]. Effects of HMW-HA in the presence of immune altering cytokines, such as interferons, are unknown in humans, and perturbations in the normal anti-inflammatory properties of HMW-HA are likely.
Macrophages are key mediators of pathogenesis in RA[7] through secretion of pro-inflammatory cytokines TNF, IL-6, IL-12p70 and IL-1β[6; 7; 8]. IFNγ alters macrophage biology by induction of “IFNγ-priming”, characterized by suppression of the regulatory cytokine IL-10 and enhancement of the inflammatory cytokines TNF and IL-12p70 in response to activation[11]. IFNγ-induced suppression of IL-10 secretion and enhancement IL-12p70 secretion constitutes an environment that promotes Th1 polarization of CD4 T cells. We hypothesized that IFNγ-priming of macrophages overrides the homeostatic properties of HMWHA resulting in aberrant macrophage activation that could model T cell-dependent RA inflammation in vivo.
Materials and Methods
Reagents
LPS from E. coli, IFNγ and M-CSF were obtained from Sigma (St. Louis, MO). HMW-HA from human umbilical cord was obtained from Calbiochem (San Diego, CA). ELISA kits for human TNF and IL-10 were obtained from BD Biosciences (San Jose, CA). Milliplex 22-plex cytokine/chemokine kit was obtained from Millipore Corp. (St. Charles, MO). A Nuclear extraction kit and TransAM NFκB, MAPK and STAT EIA kits were obtained from Active Motif (Carlsbad, CA). Polymyxin B was obtained from Invivogen (SanDiego, CA). Limulus amebocyte lysate [LAL] assay kit was obtained from Lonza (Allendale, NJ).
Macrophage culture
Human monocytes were provided by H.E. Gendelman at the University of Nebraska. Monocytes were isolated from healthy donors by counter current centrifugal elutriation [12] and were cultured 7 days in the presence of 10% human serum and 1 ng/ml M-CSF to differentiate macrophages. Use of human tissues was approved by Institutional Review Board of the University of Florida.
Cytokine analysis
Macrophages were cultured 18 hours with media (mock) or 10 ng/ml IFNγ (primed). LPS or HMW-HA were added for an additional 6 hours. Supernatants were collected to measure 1.) TNF and IL-10 by ELISA or 2.) the concentration of 22 cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-15, Eotaxin, GM-CSF, IFNγ, IP10, MCP-1, MIP1α, MIP1β, TNFα and VEGF) using Milliplex® technology. To inactivate endotoxin, LPS or HMW-HA were boiled for 15 minutes or treated with 100 ug/ml polymyxin B before adding to macrophage cultures.
LAL assay
Stock solutions of LPS or HMW-HA were serially diluted and endotoxin activity was detected by LAL assay. Where indicated, LPS or HMW-HA samples were pre-treated with 100 ug/ml polymyxin B. LAL assay was stopped with 25% acetic acid and color development was determined by scanning optical density at 405 nm.
Transcription factor analysis
Macrophages were mock- or IFNγ-primed for 18 hours followed by activation with LPS or HA for 2 additional hours. Nuclear lysates were prepared according to the manufacturer’s protocol. Nuclear proteins were quantified by BCA assay (Thermo Fisher, Rockford, IL). TransAM EIA assays for NFκB (p50, p65, p52, c-Rel and RelB), STAT (STAT1, STAT3, STAT5a, STAT5b) and MAPK (ATF-2, c-Myc, c-Jun and MEF-2) were performed according to manufacturer’s protocol with 1 ug nuclear protein per test. Briefly, TransAM EIAs allow quantitation of transcriptionally active proteins by capture with transcription factor family-specific consensus DNA oligonucleotides on plastic wells. Individual transcription factor proteins were then quantified by probing with monoclonal antibodies and TMB substrate (BD Biosciences).
Statistics
Data analysis and statistics were performed with GraphPad Prism version 5 software (La Jolla, CA). Results were considered statistically significant when P<0.05.
Results
IFNγ modulates HMW-HA-induced TNF and IL-10 secretion by macrophages
HMW-HA normally mediates anti-inflammatory effects on mononuclear cells[2; 4]. The activational outcome of HMW-HA on macrophages in the absence or presence of IFNγ was compared with LPS by measuring the cytokines TNF (inflammatory) and IL-10 (regulatory). HMW-HA was as effective as LPS in mediating TNF production by macrophages in the absence of IFNγ priming and as potent as LPS when macrophages were primed with IFNγ (Figure 1, A& B). Macrophages secreted IL-10 in response to HMW-HA or LPS (Figure 1, C & D), although IFNγ priming inhibited IL-10 secretion by either HMW-HA or LPS treated macrophages.
Figure 1. IFNγ priming of macrophages enhances HMW-HA-induced TNF secretion and suppresses HA-induced IL-10 secretion.

Macrophages were mock treated or cultured with 10 ng/ml IFNγ for 18 hrs before activation with LPS (A & C) or HMW-HA (B & D) for 6 hrs (open bars = Mock and shaded bars = IFNγ). LPS-induced TNF secretion (A) and IL-10 secretion (C) were determined by ELISA of culture supernatants. HMW-HA-induced TNF secretion (B) and IL-10 secretion (D) were determined by ELISA of culture supernatants. Data represent mean of 4 independent donors (Error bars = 1 SD. Paired t test, *P<0.05 or †P<0.01). (E) Macrophages were treated with either LPS or HMW-HA, which were untreated [U] or boiled for 15 minutes [B] and TNF was detected by ELISA. (F) Macrophages were treated with either 100 ng/ml LPS (shaded circles) or 100 ug/ml HMW-HA (black circles) in the presence of media alone (Mock) or polymyxin B. LPS (G) or HMW-HA (H) were serially diluted and endotoxin activity was detected by LAL assay without polymyxin B (circles) or with 100 ug/ml polymyxin B (squares).
To rule out that activation of macrophages by HMW-HA was caused by endotoxin in the HMW-HA preparation, three approaches were implemented. First, boiling reduced by almost 100% TNF secretion induced by 100 ng/ml LPS [maximum concentration used in panels A and B], but produced no reduction in TNF secretion by 100 ug/ml or 1 mg/ml HMW-HA (Figure 1E). Next, polymyxin B had a potent dose-dependent inhibitory effect on TNF secretion induced by 100 ng/ml LPS, but polymyxin B failed to significantly reduce HMW-HA-induced TNF secretion (Figure 1F). Finally, endotoxin activity detected by LAL assay in LPS at concentrations ≥ 1 ng/ml was completely abolished when LPS was treated with 100 ug/ml polymyxin B (Figure 1G). In contrast, polymyxin B failed to neutralize the activity detected by LAL assay with HMW-HA (Figure 1H). Non-endotoxin LAL assay activity of biological products is not uncommon, but require LPS inhibitors, such as polymyxin B, to validate LAL assay results[13] Taken together, results from this series of studies show that LAL activity and TNF induction by HMW-HA are endotoxin-independent..
IFNγ-primed macrophages secrete a profile of pro-inflammatory cytokines in response to HMW-HA activation
The synovial cytokine milieu is highly variable among RA patients; however several factors (IL-1α, IL-1β, IL-6 and IL-12p70) are hallmarks of pathogenesis[6; 8]. Macrophages treated ex vivo by HMW-HA or LPS in the presence or absence of IFNγ priming were assessed for secretion of 22 cytokines/chemokines. Eleven were not detected (Eotaxin, IFNγ, IL-13, IL-15, IL-2, IL-4, IL-5, IL-7 and VEGF) or produced at similar levels (IL-8 and MCP-1) regardless of treatment (data not shown). Nine cytokines/chemokines, in addition to TNF and IL-10, were altered by LPS or HMW-HA activation and classified depending on their independence of, or responsiveness to IFNγ-priming (Group I or II, respectively) (Figure 2). LPS and HMW-HA induced secretion of group I cytokine/chemokines GM-CSF, IL-12p40, IL-1α, IL-6, MIP-1α and MIP-1β. IFNγ-priming enhanced LPS induced secretion of group II cytokines IL-12p70 and IL-1β when compared to mock-primed macrophages, which secreted no IL12p70 and little IL-1β. Similarly, IFNγ-priming enhanced HMW-HA induced secretion of IL12p70 with a trend toward enhanced IL-1β secretion (P = 0.077, paired t test). HMW-HA induced IL-12p70 secretion by IFNγ-primed macrophages at levels approximately 8-fold greater than LPS (P=0.029, Mann-Whitney U test). Conversely, HMW-HA induced secretion of IL-1β that was only 63% of levels induced by LPS. IP-10 was induced by both LPS and HMW-HA activation and enhanced by IFNγ-primed macrophages.
Figure 2. IFNγ-primed macrophages respond to LPS or HMW-HA with similar pro-inflammatory cytokine secretion.

Macrophages were mock treated or cultured with 10 ng/ml IFNγ for 18 hrs before activation with media control (open bars), 1 ng/ml LPS (shaded bars) or 1 mg/ml HMW-HA [HA] (black bars) for 6 hrs and supernatants were analyzed by Milliplex Human 22-plex cytokine/chemokine panel. Data represent mean of 4 independent donors (Error bars = 1 SD. Paired t test, *P<0.05, §P=0.077. n.d. = not detected in assay, limits of detection (pg/ml): GM-CSF = 0.23, IL-12p40 = 20.9, IL-6 = 0.79 and IL-12p70 = 0.23).
HMW-HA induces c-Jun nuclear translocation that is inhibited by IFNγ
To assess the molecular pathways that lead to macrophage activation and cytokine secretion in response to LPS or HMW-HA, nuclear trans-activated proteins from the STAT, NFκB and MAPK pathways were assayed. Multiple STAT proteins (STAT3, STAT5a and STAT5b), NFκB proteins (c-Rel, p52 and RelB) and MAPK proteins (ATF-2 and c-Myc) were unaffected by activation with LPS or HMW-HA or IFNγ-priming. STAT1 was expressed at the same level in nuclear lysates from all IFNγ-primed macrophages regardless of activation method (data not shown). Activation of macrophages with either LPS or HMW-HA in the absence of IFN? priming resulted in trans-activation of both NFκB p50 and NFκB p65 which comprise the major NFκB heterodimer complex (Figure 3A-B). Priming of macrophages with IFNγ produced no change in levels of nuclear NFκB p50 or p65 (Figure 3A-B). Either LPS or HMW-HA in the absence of IFN? priming induced trans-activation of the MAPK protein c-Jun, although HMW-HA activation resulted in 3-fold higher levels of c-Jun compared to LPS (Figure 3C). IFNγ-priming enhanced LPS-induced c-Jun activation, but in contrast significantly inhibited HWM-HA-induced c-Jun activation (P<0.05) (Figure 3C). While either LPS or HMW-HA inhibited MEF-2 nuclear trans-activation, with or without IFNγ priming, only HMW-HA-induced inhibition reached statistical significance (Figure 3D).
Figure 3. HMW-HA mediates unique molecular activation of MDM.
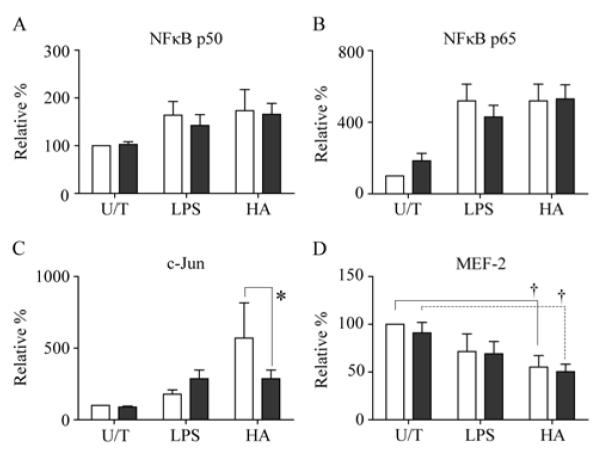
The effects of 1 ng/ml LPS- or 1 mg/ml HMW-HA on activation of mock-primed (open bars) or IFNγ-primed (shaded bars) MDM were determined by nuclear transcription factor EIA. Activation of NFκB (A) p50 and (B) p65 subunits were determined. Activation of MAPK (C) c-Jun and (D) MEF2 were determined. Data are represented as relative protein concentration normalized to mock-primed, untreated [U/T] samples representing 100%. Data represent mean of 4 independent MDM donors (Error bars = 1 S.D. Paired t test, *P=<0.05, †P<0.01).
Discussion
HMW-HA is anti-inflammatory in healthy joints and is utilized in osteoarthritis therapies[5]. Our study demonstrates that, under the influence of IFNγ, HMW-HA can contribute to inflammation of human macrophages, key mediators of RA pathology. Because the local inflammatory environment of RA joints includes Th1 IFNγ-secreting CD4 T cells, our study was modeled after healthy or RA-inflamed synovia where incoming macrophages encounter either HMW-HA alone or HMW-HA in the presence of the Th1 cytokine IFNγ, respectively.
In the absence of IFNγ, HMW-HA induces a distinct cytokine secretion profile characterized by IL-10, but lacking IL-12p70. In contrast, HMW-HA mediates inflammatory activation of IFNγ-primed macrophages, characterized by a cytokine profile that closely resembles in vivo cytokine milieu associated with RA pathogenesis [8; 10]. IFNγ priming enhances HMW-HA-induced secretion of the pro-RA cytokines TNF, IL-12p70 and IL-1β, while secretion of the anti-RA cytokine IL-10 is suppressed.
Secretion of IL-10 by macrophages in response to HMW-HA likely mediates homeostatic effects in vivo where IL-10 is a regulator of inflammation in the joints. IL-10 is present in the joints of RA patients, and neutralization of IL-10 in RA synovial cultures enhances TNF and IL-1β secretion[14]. Likewise, enrichment of RA synovial cultures by exogenous IL-10 inhibits TNF and IL-1β secretion[14]. Inhibition of IL-10 secretion from IFNγ-primed macrophages could be a critical checkpoint in development of RA inflammation.
IL-12p70 was secreted by macrophages only when primed with IFNγ. A novel finding of our study is that IFNγ-primed macrophages activated with HMW-HA secreted IL-12p70 at levels more than 8-fold higher than levels induced by the potent inflammatory molecule LPS. This is a key finding, because IL-12p70 is involved in early RA pathogenesis, likely through a “feed-forward” activation loop whereby IL-12p70 induces local CD4 T cells to develop a TH1 IFNγ-secretion phenotype instead of a Th2 phenotype [8]. Our results provide evidence suggesting an in vivo mechanism of RA inflammation in which IFNγ directs activation of macrophages by endogenous HMW-HA through the suppression of IL-10 and induction of IL-12p70.
The cytokine profiles observed in HMW-HA and LPS activated MDM cultures point to similar, but not identical mechanisms of activation by these two molecules – not entirely surprising considering that HMW-HA and LPS share a common receptor in TLR4 [2]. Yet, the in vivo effects of HA and LPS are quite dissimilar: LPS is pyrogenic and causes pathogenic inflammation while HMW-HA is found within healthy non-inflamed tissues including the synovium. [15]. The NFκB and STAT transcription factor families responded similarly to HMWHA and LPS activation. Only c-Jun and MEF-2 showed marked differences where HMW-HA induced greater activation of c-Jun and greater suppression of MEF-2. Activation of NFκB is implicated in inflammatory cytokine production in RA synovial fibroblasts, whereas c-Jun is unnecessary for IL-6 and IL-8 secretion[16]. Perhaps potent induction of the AP-1 transcription factor subunit c-Jun by HMW-HA competes for promoter sites in inflammatory cytokine genes, limiting the effect of NFκB leading to homeostatic regulation. Activation of the MAPK protein MEF-2 is associated with macrophage[17] and osteoclast[18] differentiation, however the role of this transcription factor remains relatively unknown for macrophage function. HMW-HA might suppress MEF-2 trans-activation as a mechanism of halting differentiation and maintaining tissue homeostasis. If so, this mechanism is unaltered by IFNγ-priming and is unlikely to be relevant to RA-associated inflammation. Overall, the distinct signaling and cytokine secretion profiles displayed by macrophages activated with LPS versus HMW-HA point to unique effects of these molecules.
TLR4 signaling has been implicated in RA pathogenesis, and macrophages from the joints of patients with RA are more sensitive to activation by TLR4 ligands[19]. Increased sensitivity to TLR4 ligands in vivo is consistent with our in vitro model of IFNγ priming; however the actual mechanism is probably more complex than increased sensitivity to TLR4 ligation alone. IFNγ has a unique capability to re-direct intracellular signaling pathways – a phenomenon that is evident in the finding that IFNγ-primed cells treated with IL-10 are shunted away from typical IL-10-induced STAT3 signaling. Rather, IL-10 receptor signaling is redirected through the canonical IFNγ-responsive molecule STAT1[20]. Similar molecular hijacking may quantitatively or qualitatively alter TLR4 and/or CD44 signaling contributing to unique and complex macrophage activation in RA afflicted joints.
The exact mechanism of joint homeostasis induced by HMW-HA remains unclear although IL-10 likely plays a major role. IL-10 not only inhibits Th1 CD4 T cell polarization, but also mediates general immunoregulatory effects on antigen presenting cells [APC]. Alternatively, APC/CD4 T cell interactions in the absence of IL-12p70 may induce Th2 polarization typified by secretion of IL-4. Th2 polarization as a means of immune tolerance in joints is supported by evidence that elevated Th1:Th2 ratio is associated with RA severity[9]. Based on our findings, IFNγ-priming induces global alterations to macrophage function such that normally homeostatic HMW-HA induces pro-RA inflammatory activation, marked by increased TNF, IL-1β and IL-12p70 and suppression of IL-10. In our model, accumulation of Th1 IFNγ-secreting CD4 T cells at synovial tissue (checkpoint 1) sets the stage for pathogenic activation of recruited macrophages by HMW-HA (checkpoint 2). Production of IL-12p70 in the absence of IL-10 by IFNγ-primed HMW-HA-activated macrophages feeds forward to promote further Th1 polarization (checkpoint 3). This self-sustaining loop progresses until chronic exposure to elevated TNF and IL-1β mediates tissue injury through activation of resident osteoclasts (checkpoint 4). Interruption of this cycle presents a potential therapeutic target to prevent and/or reverse the inflammation-induced tissue damage characteristic of RA. Therefore, a thorough systems biology approach to study of IFNγ-priming and HMW-HA activation of macrophages is warranted to identify new soluble and/or intracellular signaling factors that contribute to RA development and progression. Moreover, the molecular profiles discovered here, particularly suppression by IFNγ priming of c-Jun trans-activation induced by HMW-HA, should be evaluated by studies of synovial macrophages from arthritic and healthy joints.
Acknowledgements
We would like to thank Myhanh Che and Howard Gendelman at the University of Nebraska for providing elutriated monocytes. We thank Matthew J. Delano at the University of Florida for critical reading of this manuscript.
GRANT SUPPORT: M.A.W., NIH T32AR007603 S.M.W., NIH U24DE1650901 M.M.G., NIH R01HD032259 and NIH R01AI65265
Footnotes
Author contributions.
MAW – Conceived study, performed most experiments, analyzed data and wrote manuscript.
SMW – Performed Luminex assays and analyzed Luminex data.
GG – Performed TNF ELISAs and some LPS inactivation experiments.
JWS – Oversaw project and contributed to writing manuscript.
MMG – Oversaw project and contributed to writing manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bastow E, Byers S, Golub S, Clarkin C, Pitsillides A, Fosang A. Hyaluronan synthesis and degradation in cartilage and bone. Cell Mol Life Sci. 2008;65:395–413. doi: 10.1007/s00018-007-7360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chang E, Kim H, Ha J, Ryu J, Park K, Kim U, Lee Z, Kim H, Fisher D, Kim H. Hyaluronan inhibits osteoclast differentiation via Toll-like receptor 4. J Cell Sci. 2007;120:166–76. doi: 10.1242/jcs.03310. [DOI] [PubMed] [Google Scholar]
- [3].Muto J, Yamasaki K, Taylor K, Gallo R. Engagement of CD44 by hyaluronan suppresses TLR4 signaling and the septic response to LPS. Mol Immunol. 2009;47:449–56. doi: 10.1016/j.molimm.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lajeunesse D, Delalandre A, Martel-Pelletier J, Pelletier J. Hyaluronic acid reverses the abnormal synthetic activity of human osteoarthritic subchondral bone osteoblasts. Bone. 2003;33:703–10. doi: 10.1016/s8756-3282(03)00206-0. [DOI] [PubMed] [Google Scholar]
- [5].Altman R. Status of hyaluronan supplementation therapy in osteoarthritis. Curr Rheumatol Rep. 2003;5:7–14. doi: 10.1007/s11926-003-0077-6. [DOI] [PubMed] [Google Scholar]
- [6].Brennan F, Beech J. Update on cytokines in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19:296–301. doi: 10.1097/BOR.0b013e32805e87f1. [DOI] [PubMed] [Google Scholar]
- [7].Hamilton J, Tak P. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009;60:1210–21. doi: 10.1002/art.24505. [DOI] [PubMed] [Google Scholar]
- [8].McInnes I, Liew F. Cytokine networks--towards new therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:31–9. doi: 10.1038/ncprheum0020. [DOI] [PubMed] [Google Scholar]
- [9].Schulze-Koops H, Kalden J. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2001;15:677–91. doi: 10.1053/berh.2001.0187. [DOI] [PubMed] [Google Scholar]
- [10].Duke O, Panayi G, Janossy G, Poulter L. An immunohistological analysis of lymphocyte subpopulations and their microenvironment in the synovial membranes of patients with rheumatoid arthritis using monoclonal antibodies. Clin Exp Immunol. 1982;49:22–30. [PMC free article] [PubMed] [Google Scholar]
- [11].Hu X, Chakravarty S, Ivashkiv L. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gendelman H, Orenstein J, Martin M, Ferrua C, Mitra R, Phipps T, Wahl L, Lane H, Fauci A, Burke D. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–41. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ochiai M, Tamura H, Yamamoto A, Aizawa M, Kataoka M, Toyoizumi H, Hroiuchi Y. A limulus amoebocyte lysate activating activity (LAL activity) that lacks biological activities of endotoxin found in biological products. Microbiol. Immunol. 2002;46:527–33. doi: 10.1111/j.1348-0421.2002.tb02730.x. [DOI] [PubMed] [Google Scholar]
- [14].Katsikis P, Chu C, Brennan F, Maini R, Feldmann M. Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J Exp Med. 1994;179:1517–27. doi: 10.1084/jem.179.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pitsillides A, Worrall J, Wilkinson L, Bayliss M, Edwards J. Hyaluronan concentration in non-inflamed and rheumatoid synovium. Br J Rheumatol. 1994;33:5–10. doi: 10.1093/rheumatology/33.1.5. [DOI] [PubMed] [Google Scholar]
- [16].Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope R. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol. 2000;165:7199–206. doi: 10.4049/jimmunol.165.12.7199. [DOI] [PubMed] [Google Scholar]
- [17].Baek Y, Haas S, Hackstein H, Bein G, Hernandez-Santana M, Lehrach H, Sauer S, Seitz H. Identification of novel transcriptional regulators involved in macrophage differentiation and activation in U937 cells. BMC Immunol. 2009;10:18. doi: 10.1186/1471-2172-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Feng H, Cheng T, Steer J, Joyce D, Pavlos N, Leong C, Kular J, Liu J, Feng X, Zheng M, Xu J. Myocyte enhancer factor 2 and microphthalmia-associated transcription factor cooperate with NFATc1 to transactivate the V-ATPase d2 promoter during RANKL-induced osteoclastogenesis. J Biol Chem. 2009;284:14667–76. doi: 10.1074/jbc.M901670200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang Q, Ma Y, Adebayo A, Pope R. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56:2192–201. doi: 10.1002/art.22707. [DOI] [PubMed] [Google Scholar]
- [20].Herrero C, Hu X, Li W, Samuels S, Sharif M, Kotenko S, Ivashkiv L. Reprogramming of IL-10 activity and signaling by IFN-gamma. J Immunol. 2003;171:5034–41. doi: 10.4049/jimmunol.171.10.5034. [DOI] [PubMed] [Google Scholar]