

Abstract
Bluetongue virus (BTV) is the cause of bluetongue, an emerging, arthropod-transmitted disease of ungulates. Bluetongue is characterized by vascular injury with hemorrhage, tissue infarction and widespread edema, lesions that are consistent with those of the so-called viral hemorrhagic fevers. To further investigate the pathogenesis of vascular injury in bluetongue, we utilized an electrical impedance assay and immunofluorescence staining to compare the effects of BTV infection on cultured bovine endothelial cells (bPAEC) with those of inducers of cell death (Triton X-100) and interendothelial gap formation (tissue necrosis factor [TNF]). The data confirm that the adherens junctions of BTV-infected bPAECs remained intact until 24 hours post-infection, and that loss of monolayer impedance precisely coincided with onset of virus-induced cell death. In contrast, recombinant bovine TNF-α caused rapid loss of bPAEC monolayer impedance that was associated with interendothelial gap formation and redistribution of VE-cadherin, but without early cell death. The data from these in vitro studies are consistent with a pathogenesis of bluetongue that involves virus-induced vascular injury leading to thrombosis, hemorrhage and tissue necrosis. However, the contribution of cytokine-induced interendothelial gap formation with subsequent edema and hypovolemic shock contributes to the pathogenesis of bluetongue remains to be fully characterized.
Keywords: Bluetongue, Virus, Endothelium
1. Introduction
Bluetongue virus ([BTV]; genus Orbivirus, family Reoviridae) and related orbiviruses such as African horse sickness virus are the cause of important and apparently emerging arboviral diseases of livestock (Backx et al., 2007; Burrage and Laegreid, 1994; Coetzer and Guthrie, 2004; Darpel et al., 2007; MacLachlan and Guthrie, 2010; Mellor and Hamblin, 2004; Toussaint et al., 2007; Verwoerd and Erasmus, 2004). BTV infection of ruminants occurs throughout tropical and temperate regions of the world, coincident with the distribution of competent Culicoides insect vectors (Gibbs and Greiner, 1994; Pritchard et al., 2004; Tabachnick, 2004). However, there has been a remarkable recent northern expansion of the virus’ global range that has been attributed in part to the impact of climate change (Purse et al., 2008; Purse et al., 2005; Wilson and Mellor, 2008). Bluetongue disease is most common in certain breeds of sheep and species of wildlife, but some BTV strains also cause severe disease in cattle, goats, South American camelids and other wild and domestic ungulates, and even carnivores (Darpel et al., 2007; Jauniaux et al., 2008; Meyer et al., 2009; Verwoerd and Erasmus, 2004).
Bluetongue is characterized by hemorrhage and ulceration of the oral cavity and upper gastrointestinal tract; facial, intermuscular and pulmonary edema; peritoneal, pleural and pericardial effusion; coronitis; necrosis of skeletal and cardiac muscle; and subintimal hemorrhage in the pulmonary artery (MacLachlan et al., 2009). The gross and histopathologic lesions of bluetongue are consistent with a pathogenesis that involves injury to small caliber blood vessels, which in turn leads to increased permeability of affected vessels as well as hemorrhage, thrombosis, and ischemic tissue necrosis (infarction) (Erasmus, 1975; MacLachlan et al., 2008; Mahrt and Osburn, 1986; Moulton, 1961; Spreull, 1905; Verwoerd and Erasmus, 2004). Vascular injury in bluetongue has been attributed to direct virus-mediated injury to endothelial cells (EC), but recent findings suggest that the process of EC injury and dysfunction may be more complex and be attributable, at least in part, to the activities of virus-induced, host-derived inflammatory and vasoactive mediators (DeMaula et al., 2001; DeMaula et al., 2002a; DeMaula et al., 2002b; Drew et al., 2010). These in vitro studies also clearly demonstrated that the EC response, as measured by activation and various mechanisms of cell death, is dependent on and modulated by the presence of proinflammatory mediators. Similarly, lysates of BTV-infected ECs caused an immediate increase in the permeability of monolayers of human ECs, as measured by a decrease in monolayer resistance and confirmed by immunofluorescence labeling that showed redistribution of VE-cadherin, an adherens junction protein of ECs that is important in maintaining monolayer integrity (Chiang et al., 2006). However, crude lysates containing virus and soluble, cell derived mediators were used in these latter experiments, which clearly confuses distinction of the relative roles of virus infection versus mediator activity in modulating monolayer permeability.
The objective of this study was to further investigate the pathogenesis of bluetongue, specifically the mechanisms by which BTV infection alters vascular permeability in susceptible ruminants. An electrical impedance assay and fluorescence staining were used to characterize the effect of BTV infection on cultured bovine ECs. The data show that the decrease in impedance of BTV-infected bovine ECs occurs relatively late in the course of infection and coincides with virus-induced cell death. In contrast, there was little change in monolayer impedance prior to the onset of virus-induced cytopathology, whereas paracellular permeability, as assessed by monolayer impedance and immunofluorescence labeling of VE-cadherin, was altered rapidly by exposure to cytokine mediators such as tissue necrosis factor (TNF).
2. Materials and Methods
2.1. Cells
Baby hamster kidney (BHK-21) cells were obtained from ATCC (CCL-10). The isolation and purification of the bovine pulmonary artery ECs (bPAEC) were described previously (DeMaula et al., 2001). Cells were maintained in complete medium Dulbeco’s minimal essential medium [(DMEM), Gibco, 10313-021], 10 % fetal bovine serum, MEM vitamins, non essential amino acids, L- glutamine, sodium pyruvate (Gibco, 11360), and confluent monolayers of bPAECs of similar passage number (9 or 10) were used in all experiments. The bPAEC were grown on surfaces coated with 10 μg mL−1 human fibronectin (Gibco, 33016-015).
2.2. Antibodies and cytokines
A murine monoclonal antibody (MAb) specific for BTV core protein VP7 (MAb 290 ) has been described previously (Whetter et al., 1989). Goat anti-VE-cadherin was obtained from Santa Cruz Biotech (SCBT, SC-6458). AlexaFluor® 594 donkey anti-mouse IgG (Invitrogen, A21203) and AlexaFluor® 488 donkey anti-goat IgG (Invitrogen, A11055) were used as secondary antibodies, and AlexaFluor® 594 phalloidin (Invitrogen, A12381) was used to label actin. Recombinant bovine TNF – α (rbTNF-α) (Thermo Scientific, RBOTNFAI) was reconstituted and stored according to the manufacturer’s recommendations.
2.3. Virus and recombinant BTV proteins
The United States’ prototype strain of BTV serotype 10 (ATCC, VR-1231) was used for all experiments. The virus inoculum was prepared by infecting BHK-21 cells until the appearance of complete cytopathic effect. The cells were pelleted and sonicated for 2 minutes to release cell associated virus, and the virus containing supernatants and cell lysates were centrifuged at 400 g to remove the cellular debris from the virus suspension. The virus suspension was then ultracentrifuged at 69,000 g through a 5 % sucrose cushion to separate BTV from soluble, cell-derived mediators and the partially purified virus preparation was re-suspended in phenol red - free DMEM (Gibco, 30153), aliquoted and frozen at −80 °C. Virus titers (TCID50) were determined as previously described (Barratt-Boyes et al., 1992; MacLachlan et al., 1984).
Baculovirus-expressed individual BTV structural (VP) and nonstructural (NS) proteins (VP1, VP2, VP4, VP5, VP6, VP7, NS1, and NS2) as well as core-like particles (co-expressed VP3/VP7) were produced as previously described (French and Roy, 1990; Inumaru and Roy, 1987; Kar and Roy, 2003; Loudon and Roy, 1991; Oldfield et al., 1990; Stauber et al., 1997).
2.4. Kinetics of BTV infection of bPAEC
The kinetics of BTV infection of bPAECs were determined using both one-step growth curve analysis and immunofluorescence staining of VP7 at 6-8 hour intervals after infection. Titers of BTV (TCID50) were determined after infection of confluent monolayers of bPAECs in T25 flasks; virus (m.o.i. 1) was absorbed for 1 hour when the monolayer was washed with MEM and complete medium was added. At each time point, cells were scraped from the flask and the cell suspensions were sonicated for 2 minutes. The cell debris was removed by centrifugation (400 g) and the titer (TCID50) of the virus suspension was determined by end point dilution as described previously (DeMaula et al., 2001). bPAEC monolayers for immunofluorescence staining were grown on sterile 12 mm coverslips, and infected with BTV at an m.o.i. of 1. Monolayers were fixed at 6-8 hour intervals after infection with 4 % paraformaldehyde, permeabilized with 0.01 % Triton X-100 (Fischer Scientific, BP151), then incubated for 1 hour with MAb 290 diluted in PBS containing 1.5 % bovine serum albumin (BSA), washed and then incubated with labeled donkey anti-mouse IgG for one hour, washed and mounted in Prolong gold with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, P36935).
2.5. Impedance of bPAEC monolayers after cytokine treatment or BTV infection
The impedance across confluent monolayers of bPAECs was determined using a real-time cell electronic sensor (RT-CES) system (Roche) that measures impedance of individual monolayers over time in a 96-well plate format, where any decrease in impedance is equated to a proportionate increase in monolayer permeability (Atienza et al., 2006; Kirstein et al., 2006; Li et al., 2006). The measurement is reported as a Cell Index (CI). Impedance was monitored in individual wells after addition of known inducers of either inter-endothelial gap formation [rbTNF-α (0.1 μg/mL)] or cell death [Triton X-100 (0.1 % v/v)]. BTV was added to confluent monolayers (m.o.i. 5), and individual recombinant BTV proteins at 125 ng mL−1. The impedance of individual monolayers was monitored in 6 replicates every 30 minutes after addition of TNF or Triton X-100, BTV, or individual recombinant BTV proteins until completion of the experiment at 36 - 48 hours after exposure.
2.6. Cell cytotoxicity
The appearance of cell death in BTV-infected monolayers was evaluated by phase microscopy at 0, 6, 12, 18, 24, 36, and 48 hours after BTV infection at an m.o.i. of 5. Cytotoxicity was also objectively quantitated in a 96 – well format with a protease release assay (Promega, CytoTox-Fluor, G9261), as directed by the manufacturer. Cytotoxicity was determined at 6 hours after exposure of bPAEC to vehicle control, rbTNF – α, or Triton X-100, and at 0, 6, 12, 18, 24, and 36 hours after BTV infection at an m.o.i. of 5.
2.7 Dual labeling of VE-cadherin and actin or BTV
Confluent monolayers of bPAECs were grown in 24 – well culture plates containing sterilized 12 mm, round glass coverslips. Individual EC monolayers were treated for 6 hours with rbTNF-α (0.1 μl mL−1) or vehicle control (complete EC medium), or were infected with BTV (m.o.i. of 5). Post treatment or infection, monolayers were fixed with fresh 4 % paraformaldehyde, permeabilized with 0.01 % Triton X-100, blocked with 10% BSA prior to immunofluorescence staining for VE-cadherin and actin, or VE-cadherin and BTV core protein VP7. Individual monolayers were incubated with goat anti - VE cadherin antibody and MAb 290 specific for BTV core protein VP7 diluted in 1.5% BSA/PBS, followed by AlexaFluor® 488 donkey anti-goat IgG and AlexaFluor® 594 donkey anti-mouse IgG. Actin was labeled with AlexaFluor®594 rhodamine according to the manufacturer’s directions. The coverslips were mounted on Fisher brand Superfrost plus slides with Prolong gold with DAPI. Fluorescence was visualized with an Olympus BX61 equipped with a xenon lamp and appropriate fluorescent filters.
2.8 Statistical analysis
Results are presented as mean ± SD. Treated samples were compared to controls by paired t-tests. For multiple-group comparisons, ANOVA, followed by the Fisher’s LSD post hoc, was used. For analysis of differences over time, repeated measures ANOVA, followed by the Fisher’s LSD post hoc, was used. A p-value of < 0.05 was considered significant. For correlation analysis, Spearman’s correlation was used to compare impedance and cytotoxicity.
3. Results
3.1. Virus growth in bPAECs
There was a substantial increase in virus titer following BTV infection of bPAECs, with a marked increase by 12 hours after infection (Drew et al., 2010). Similarly, there was progressively increased intensity of immunofluorescence staining of BTV core protein VP7 (Fig. 1), with abundant cytoplasmic staining of VP7 in all cells by 12 hours after infection. Prominent holes in the bPAEC monolayer were present by 24 hours after infection, consistent with the obvious cytolysis and nuclear pyknosis and karyorrhexis that were concurrently evident by phase contrast light microscopy (data not shown).
Fig. 1.
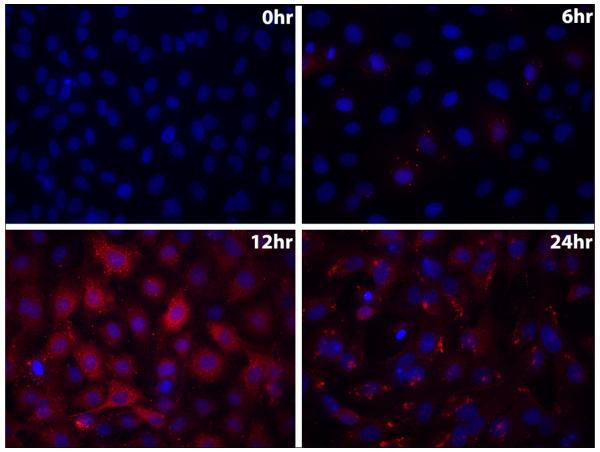
Replication of BTV in bPAECs. Immunofluorescence staining of VP7 (red) over time (200x). Note diffuse cytoplasmic staining of VP7 throughout the monolayer by 12 hours after infection, and that patchy areas of the monolayer are devoid of cells by 24 hours (cytopathic effect).
3.2. Impedance and cytotoxicity of bPAECs treated with inducers of cell death (Triton X-100) and interendothelial gap formation (TNF – α)
The impedance of bPAEC monolayers was abolished immediately after treatment with Triton X-100, and persisted thereafter (Fig. 2A). Similarly, cytotoxicity as assessed by protease release was markedly and significantly increased following Triton X-100 treatment (Fig. 2C). Thus, the decrease in monolayer impedance following Triton X-100 treatment is attributed to cell death (Fig. 2D). The impedance of bPAEC monolayers treated with rbTNF – α was also reduced for the duration of the experiment (Fig. 2B). Although the impedance of the rbTNF – α treated monolayers was significantly decreased at 6 hours after exposure, there was no difference at this time in the viability of the TNF-treated bPAEC monolayers, as determined by protease release assay (Fig. 2C). It is concluded, therefore, that the early decrease in impedance of rbTNF-treated bPAEC monolayers is attributable to interendothelial gap formation and not cell death, as demonstrated by the redistribution of VE – cadherin with formation of microscopically visible gaps between treated bPAECs (Fig. 2E).
Fig. 2.

Effects of recombinant bovine TNF-α (rbTNF-α) and Triton X-100 on bPAEC monolayer impedance and cell viability. (A) Immediate and sustained loss of bPAEC monolayer impedance following Triton X-100 treatment, as compared to vehicle control. (B) Decreasing and sustained decrease in bPAEC monolayer impedance beginning at 2 hours following treatment with rbTNF – α. Individual treatments were each done 4 times in 6 replicate wells. The data are presented as the mean of replicate wells +/− the SD of one representative experiment. * designates significant (p < 0.05) difference between the treated and vehicle control well at 6 hours after treatment. (C) Assessment of cell viability in bPAEC monolayers at 6 hours treatment with either rbTNF-α, Triton X-100, or vehicle control. There was no statistical difference in the ratios of cell death between the vehicle and TNF – α treated monolayers, whereas the ratio of cell death of the Triton X-100 treated bPAECs was significantly increased. Each treatment was carried out 5 times in 3 replicate wells. The data are reported as the mean of the combined ratios of cell death of treated as compared to control monolayers, +/− the SD of one experiment. * represents significant (p < 0.05) differences between the treatment group. (D) Effect of Triton X-100 on bPAEC at 6 hours after treatment. Phase contrast image (200X) demonstrating widespread cytopathic effect and loss of monolayer integrity. (E) Immunofluorescence staining of VE – cadherin (green) and actin (red) in bPAEC at 6 hours after TNF – α treatment (1000X). The TNF-treated bPAECs have prominent intercellular gaps (white arrowheads) and redistribution of VE – cadherin (white arrows) as compared to the vehicle-treated control.
3.3. The onset of reduced impedance of BTV-infected bPAEC monolayers coincides with cell death
The impedance of BTV-infected bPAECs monolayers (m.o.i. 5) was significantly decreased at 12 hours and thereafter (Fig. 3A). The incidence of cell death increased over time in the BTV infected bPAEC monolayers as determined by both protease release assay and visual observation using phase contrast microscopy. Increased cell death as determined by protease release assay was first detected at 12 hours after BTV infection (m.o.i. 5), and was significantly increased thereafter (Fig. 3B). Phase contrast microscopy confirmed that cell death began at 12 hours after infection and progressed over the next 36 hours to complete cytopathic effect (Fig. 3C).
Fig. 3.

Effect of BTV infection (m.o.i. 5) on impedance and viability of bPAEC monolayers. (A) Impedance of BTV-infected monolayers is significantly decreased as compared to uninfected monolayers beginning at 12 hours after infection, and continuing thereafter. Each treatment was done 4 times in 6 replicate wells. The data are presented as the mean of replicate wells +/− the SD. * designates significant (p < 0.05) difference between the time point and the 0hr and 6hr time points. (B) Cell death ratio of BTV-infected monolayers increases over time, beginning at 12 hours after infection. Each treatment was done 5 times in 3 replicate wells, and the data are presented as the means of the ratios of infected to control +/− SD. * indicates significant difference (p < 0.05) (C) Phase contrast microscopy (200 X) illustrates cytopathic effect in the BTV-infected bPAEC monolayers. Substantial holes are present in the infected monolayers by 18 hours after infection.
The correlation between impedance (pooled difference) and cytotoxicity (pooled ratios) at 0, 6, 12, 24, and 36 hours after infection was 0.917 (p < 0.01), confirming that the change in impedance is strongly associated with an increased incidence of cell death within the infected monolayers.
3.4. Adherens junctions remain intact in BTV- infected bPAECs
The effect of BTV infection on the adherens junctions of bPAEC monolayers was evaluated over time by fluorescent labeling of VE-cadherin and BTV core protein VP7. Despite the presence of abundant intracellular BTV antigen at 6 hours after infection and thereafter, there was continuous staining of the adherens junctions of bPAECs in areas of intact monolayer at 12-18 hours after infection (Fig. 4), confirming that interendothelial junctions remained intact even in virus infected bPAECs.
Fig. 4.

Effect of BTV infection (m.o.i 5) on bPAEC intercellular junctions. Immunofluorescence staining for VE – cadherin (green) and BTV core protein VP7 (red) (1000X). The intercellular junctions of infected ECs remain intact at 6, 12, and 18 hours after infection of the bPAEC monolayers (white arrow heads). At 24 hours cytopathic effect is advanced, however intercellular junctions are both intact (white arrow head) in surviving ECs and have gaps (white arrow) formed between adjacent cells. There is a substantial defect in the monolayer where dead cells have sloughed (white asterisks).
3.5. Impedance of bPAEC monolayers is unaltered by exposure to recombinant BTV proteins
The effect of individual recombinant BTV proteins on bPAEC monolayers was assessed by adding each recombinant protein to the culture medium so that it would contact the luminal surface of the bPAEC monolayer. None of the proteins had any significant effect on the impedance of the treated monolayers, nor on the microscopic appearance of the monolayers (data not shown), confirming luminal application of these proteins caused neither cell death or had any obvious paracrine effect on EC barrier integrity.
3. Discussion
We evaluated the effect of BTV infection on monolayers of bPAECs. The data confirm that BTV-infected bPAECs exhibit a delayed decrease in monolayer impedance, and this decrease is highly correlated with the onset of cell death. The data further confirm that the adherens junctions of BTV-infected ECs remain intact throughout the course of infection, and that individual recombinant BTV proteins did not cause cell death or alter bPAEC monolayer impedance. We conclude, therefore, that the change in BTV-infected bPAEC monolayer impedance, and thus permeability, is due largely to cell death. The precise mechanism of virus-induced cell death, whether apoptosis or necrosis, was not determined. Our conclusions differ from those of another recent investigation wherein the authors concluded that BTV infection caused an immediate (after 2 hours) loss of resistance of human EC monolayers (Chiang et al., 2006). The authors of this study further concluded that loss of monolayer resistance and increased permeability were due to the redistribution of EC adherens junctions within the infected monolayers, which in turn would lead to increased paracellular permeability. A potential explanation for the different outcomes of the two studies is the different inocula that were used; specifically, we used partially purified virus that was separated from soluble, cell-derived mediators whereas an unpurified virus inoculum was used in the prior studies. The presence or absence of inflammatory mediators in the virus inoculum clearly can influence both the onset and mechanism of EC death (DeMaula et al., 2001), thus the use of a purified virus inoculum is essential to accurate characterization of the effects of virus infection alone on EC monolayer permeability and integrity.
The data obtained in the current study are consistent with the proposed pathogenesis of bluetongue in ruminants (MacLachlan et al., 2009). Specifically, it is hypothesized that direct virus-mediated injury results in EC destruction and subsequent vascular thrombosis and tissue infarction during the acute phase of BTV infection of susceptible animals, whereas the widespread edema that occurs in the terminal phases of bluetongue, African horse sickness and other virus-induced hemorrhagic fevers is likely the result of EC contraction and increased paracellular permeability induced by host-derived proinflammatory mediators such as TNF (Gowen and Holbrook, 2008; MacLachlan et al., 2009). This conclusion is consistent with the tropism of these viruses for ECs, macrophages and dendritic cells (Barratt-Boyes et al., 1992; Brewer and MacLachlan, 1994; Drew et al., 2010; Ellis et al., 1993; Hemati et al., 2009; Whetter et al., 1989), which are all potent sources of pro-inflammatory cytokine mediators such as TNF, and by ultrastructural studies that have clearly shown restricted virus infection of ECs but more widespread EC injury with accompanying edema in the tissues of horses with African horse sickness and white-tailed deer with bluetongue (Gomez-Villamandos et al., 1999; Howerth and Tyler, 1988; Laegreid et al., 1992). However, the pathogenesis of EC injury and dysfunction in bluetongue and other orbiviral hemorrhagic fevers is likely to be highly complex, and in vivo studies clearly will ultimately be required to confirm the significance of these in vitro findings.
Acknowledgements
The authors would like to thank Elana Chu for technical assistance.
This publication was made possible by grant number T32 RR07038 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. This publication was also supported by funds provided by the Center for Equine Health at the University of California-Davis and the Bernice Barbour Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atienza JM, Yu N, Kirstein SL, Xi B, Wang X, Xu X, Abassi YA. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev Technol. 2006;4:597–607. doi: 10.1089/adt.2006.4.597. [DOI] [PubMed] [Google Scholar]
- Backx A, Heutink CG, van Rooij EM, van Rijn PA. Clinical signs of bluetongue virus serotype 8 infection in sheep and goats. Vet Rec. 2007;161:591–592. doi: 10.1136/vr.161.17.591. [DOI] [PubMed] [Google Scholar]
- Barratt-Boyes SM, Rossitto PV, Stott JL, MacLachlan NJ. Flow cytometric analysis of in vitro bluetongue virus infection of bovine blood mononuclear cells. J Gen Virol. 1992;73:1953–1960. doi: 10.1099/0022-1317-73-8-1953. [DOI] [PubMed] [Google Scholar]
- Brewer AW, MacLachlan NJ. The pathogenesis of bluetongue virus infection of bovine blood cells in vitro: ultrastructural characterization. Arch Virol. 1994;136:287–298. doi: 10.1007/BF01321058. [DOI] [PubMed] [Google Scholar]
- Burrage TG, Laegreid WW. African horsesickness: pathogenesis and immunity. Comp Immunol Microbiol Infect Dis. 1994;17:275–285. doi: 10.1016/0147-9571(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Chiang ET, Persaud-Sawin DA, Kulkarni S, Garcia JG, Imani F. Bluetongue virus and double-stranded RNA increase human vascular permeability: role of p38 MAPK. J Clin Immunol. 2006;26:406–416. doi: 10.1007/s10875-006-9024-4. [DOI] [PubMed] [Google Scholar]
- Coetzer JAW, Guthrie AJ. Vol. 2nd Edition Vol. 2. Oxford University Press; Cape Town: 2004. pp. 1231–1246. [Google Scholar]
- Darpel KE, Batten CA, Veronesi E, Shaw AE, Anthony S, Bachanek-Bankowska K, Kgosana L, bin-Tarif A, Carpenter S, Muller-Doblies UU, Takamatsu HH, Mellor PS, Mertens PP, Oura CA. Clinical signs and pathology shown by British sheep and cattle infected with bluetongue virus serotype 8 derived from the 2006 outbreak in northern Europe. Vet Rec. 2007;161:253–261. doi: 10.1136/vr.161.8.253. [DOI] [PubMed] [Google Scholar]
- DeMaula CD, Jutila MA, Wilson DW, MacLachlan NJ. Infection kinetics, prostacyclin release and cytokine-mediated modulation of the mechanism of cell death during bluetongue virus infection of cultured ovine and bovine pulmonary artery and lung microvascular endothelial cells. J Gen Virol. 2001;82:787–794. doi: 10.1099/0022-1317-82-4-787. [DOI] [PubMed] [Google Scholar]
- DeMaula CD, Leutenegger CM, Bonneau KR, MacLachlan NJ. The role of endothelial cell-derived inflammatory and vasoactive mediators in the pathogenesis of bluetongue. Virology. 2002a;296:330–337. doi: 10.1006/viro.2002.1476. [DOI] [PubMed] [Google Scholar]
- DeMaula CD, Leutenegger CM, Jutila MA, MacLachlan NJ. Bluetongue virus-induced activation of primary bovine lung microvascular endothelial cells. Vet Immunol Immunopathol. 2002b;86:147–157. doi: 10.1016/s0165-2427(02)00012-0. [DOI] [PubMed] [Google Scholar]
- Drew CP, Gardner IA, Mayo CE, Matsuo E, Roy P, MacLachlan NJ. Bluetongue virus infection activates bovine monocyte-derived macrophages and pulmonary artery endothelial cells. Vet Immunol Immunopathol. 2010 doi: 10.1016/j.vetimm.2010.03.006. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JA, Coen ML, MacLachlan NJ, Wilson WC, Williams ES, Leudke AJ. Prevalence of bluetongue virus expression in leukocytes from experimentally infected ruminants. Am J Vet Res. 1993;54:1452–1456. [PubMed] [Google Scholar]
- Erasmus BJ. Bluetongue in sheep and goats. Aust Vet J. 1975;51:165–170. doi: 10.1111/j.1751-0813.1975.tb00048.x. [DOI] [PubMed] [Google Scholar]
- French TJ, Roy P. Synthesis of bluetongue virus (BTV) corelike particles by a recombinant baculovirus expressing the two major structural core proteins of BTV. J Virol. 1990;64:1530–1536. doi: 10.1128/jvi.64.4.1530-1536.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs EP, Greiner EC. The epidemiology of bluetongue. Comp Immunol Microbiol Infect Dis. 1994;17:207–220. doi: 10.1016/0147-9571(94)90044-2. [DOI] [PubMed] [Google Scholar]
- Gomez-Villamandos JC, Sanchez C, Carrasco L, Laviada MM, Bautista MJ, Martinez-Torrecuadrada J, Sanchez-Vizcaino JM, Sierra MA. Pathogenesis of African horse sickness: ultrastructural study of the capillaries in experimental infection. J Comp Pathol. 1999;121:101–116. doi: 10.1053/jcpa.1999.0305. [DOI] [PubMed] [Google Scholar]
- Gowen BB, Holbrook MR. Animal models of highly pathogenic RNA viral infections: hemorrhagic fever viruses. Antiviral Res. 2008;78:79–90. doi: 10.1016/j.antiviral.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Hemati B, Contreras V, Urien C, Bonneau M, Takamatsu HH, Mertens PP, Breard E, Sailleau C, Zientara S, Schwartz-Cornil I. Bluetongue virus targets conventional dendritic cells in skin lymph. J Virol. 2009;83:8789–8799. doi: 10.1128/JVI.00626-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerth EW, Tyler DE. Experimentally induced bluetongue virus infection in white-tailed deer: ultrastructural findings. Am J Vet Res. 1988;49:1914–1922. [PubMed] [Google Scholar]
- Inumaru S, Roy P. Production and characterization of the neutralization antigen VP2 of bluetongue virus serotype 10 using a baculovirus expression vector. Virology. 1987;157:472–479. doi: 10.1016/0042-6822(87)90289-3. [DOI] [PubMed] [Google Scholar]
- Jauniaux TP, De Clercq KE, Cassart DE, Kennedy S, Vandenbussche FE, Vandemeulebroucke EL, Vanbinst TM, Verheyden BI, Goris NE, Coignoul FL. Bluetongue in Eurasian lynx. Emerg Infect Dis. 2008;14:1496–1498. doi: 10.3201/eid1409.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar AK, Roy P. Defining the structure-function relationships of bluetongue virus helicase protein VP6. J Virol. 2003;77:11347–11356. doi: 10.1128/JVI.77.21.11347-11356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein SL, Atienza JM, Xi B, Zhu J, Yu N, Wang X, Xu X, Abassi YA. Live cell quality control and utility of real-time cell electronic sensing for assay development. Assay Drug Dev Technol. 2006;4:545–553. doi: 10.1089/adt.2006.4.545. [DOI] [PubMed] [Google Scholar]
- Laegreid WW, Burrage TG, Stone-Marschat M, Skowronek A. Electron microscopic evidence for endothelial infection by African horsesickness virus. Vet Pathol. 1992;29:554–556. doi: 10.1177/030098589202900615. [DOI] [PubMed] [Google Scholar]
- Li HB, Ge YK, Zhang L, Zheng XX. Astragaloside IV improved barrier dysfunction induced by acute high glucose in human umbilical vein endothelial cells. Life Sci. 2006;79:1186–1193. doi: 10.1016/j.lfs.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Loudon PT, Roy P. Assembly of five bluetongue virus proteins expressed by recombinant baculoviruses: inclusion of the largest protein VP1 in the core and virus-like proteins. Virology. 1991;180:798–802. doi: 10.1016/0042-6822(91)90094-r. [DOI] [PubMed] [Google Scholar]
- MacLachlan NJ, Crafford JE, Vernau W, Gardner IA, Goddard A, Guthrie AJ, Venter EH. Experimental reproduction of severe bluetongue in sheep. Vet Pathol. 2008;45:310–315. doi: 10.1354/vp.45-3-310. [DOI] [PubMed] [Google Scholar]
- MacLachlan NJ, Drew CP, Darpel KE, Worwa G. The pathology and pathogenesis of bluetongue. J Comp Pathol. 2009;141:1–16. doi: 10.1016/j.jcpa.2009.04.003. [DOI] [PubMed] [Google Scholar]
- MacLachlan NJ, Guthrie AJ. Re-emergence of bluetongue, African horse sickness, and other Orbivirus diseases. Vet Res. 2010;41:11–12. doi: 10.1051/vetres/2010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan NJ, Schore CE, Osburn BI. Antiviral responses of bluetongue virus-inoculated bovine fetuses and their dams. American Journal of Veterinary Research. 1984;45:1469–1473. [PubMed] [Google Scholar]
- Mahrt CR, Osburn BI. Experimental bluetongue virus infection of sheep; effect of vaccination: pathologic, immunofluorescent, and ultrastructural studies. Am J Vet Res. 1986;47:1198–1203. [PubMed] [Google Scholar]
- Mellor PS, Hamblin C. African horse sickness. Vet Res. 2004;35:445–466. doi: 10.1051/vetres:2004021. [DOI] [PubMed] [Google Scholar]
- Meyer G, Lacroux C, Leger S, Top S, Goyeau K, Deplanche M, Lemaire M. Lethal bluetongue virus serotype 1 infection in llamas. Emerg Infect Dis. 2009;15:608–610. doi: 10.3201/eid1504.081514. [DOI] [PubMed] [Google Scholar]
- Moulton JE. Pathology of bluetongue of sheep in California. J Am Vet Med Assoc. 1961;138:493–498. [PubMed] [Google Scholar]
- Oldfield S, Adachi A, Urakawa T, Hirasawa T, Roy P. Purification and characterization of the major group-specific core antigen VP7 of bluetongue virus synthesized by a recombinant baculovirus. J Gen Virol. 1990;71(Pt 11):2649–2656. doi: 10.1099/0022-1317-71-11-2649. [DOI] [PubMed] [Google Scholar]
- Pritchard LI, Sendow I, Lunt R, Hassan SH, Kattenbelt J, Gould AR, Daniels PW, Eaton BT. Genetic diversity of bluetongue viruses in south east Asia. Virus Res. 2004;101:193–201. doi: 10.1016/j.virusres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Purse BV, Brown HE, Harrup L, Mertens PP, Rogers DJ. Invasion of bluetongue and other orbivirus infections into Europe: the role of biological and climatic processes. Rev Sci Tech. 2008;27:427–442. [PubMed] [Google Scholar]
- Purse BV, Mellor PS, Rogers DJ, Samuel AR, Mertens PP, Baylis M. Climate change and the recent emergence of bluetongue in Europe. Nat Rev Microbiol. 2005;3:171–181. doi: 10.1038/nrmicro1090. [DOI] [PubMed] [Google Scholar]
- Spreull J. Malarial catarrhal fever (bluetongue) of sheep in South Africa. Journal of Comparative Pathology and Therapeutics. 1905;18:321–337. [Google Scholar]
- Stauber N, Martinez-Costas J, Sutton G, Monastyrskaya K, Roy P. Bluetongue virus VP6 protein binds ATP and exhibits an RNA-dependent ATPase function and a helicase activity that catalyze the unwinding of double-stranded RNA substrates. J Virol. 1997;71:7220–7226. doi: 10.1128/jvi.71.10.7220-7226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabachnick WJ. Culicoides and the global epidemiology of bluetongue virus infection. Veterinaria Italiana. 2004;40:145–150. [PubMed] [Google Scholar]
- Toussaint JF, Sailleau C, Mast J, Houdart P, Czaplicki G, Demeestere L, VandenBussche F, van Dessel W, Goris N, Breard E, Bounaadja L, Etienne T, Zientara S, De Clercq K. Bluetongue in Belgium, 2006. Emerg Infect Dis. 2007;13:614–616. doi: 10.3201/eid1304.061136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verwoerd DW, Erasmus BJ. Bluetongue. 2nd Edition Vol 2. Oxford University Press; Cape Town: 2004. pp. 1201–1220. [Google Scholar]
- Whetter LE, MacLachlan NJ, Gebhard DH, Heidner HW, Moore PF. Bluetongue virus infection of bovine monocytes. J Gen Virol. 1989;70:1663–1676. doi: 10.1099/0022-1317-70-7-1663. [DOI] [PubMed] [Google Scholar]
- Wilson A, Mellor P. Bluetongue in Europe: vectors, epidemiology and climate change. Parasitol Res. 2008;103(Suppl 1):69–77. doi: 10.1007/s00436-008-1053-x. [DOI] [PubMed] [Google Scholar]