

Abstract
Further refinement of the model using maximum likelihood procedures and re-evaluation of the native electron density map has shown that crystals of pig pancreatic α-amylase, whose structure we reported more than fifteen years ago, in fact contain a substantial amount of carbohydrate. The carbohydrate fragments are the products of glycogen digestion carried out as an essential step of the protein's purification procedure. In particular, the substrate-binding cleft contains a limit dextrin of six glucose residues, one of which contains both α-(1,4) and α-(1,6) linkages to contiguous residues. The disaccharide in the original model, shared between two amylase molecules in the crystal lattice, but also occupying a portion of the substrate binding cleft, is now seen to be a tetrasaccharide. There are, in addition, several other probable monosaccharide binding sites. To these results we have further reviewed our X-ray diffraction analysis of α-amylase complexed with α-cyclodextrin. α-Amylase binds three cyclodextrin molecules. Glucose residues of two of the rings superimpose upon the limit dextrin and the tetrasaccharide. The limit dextrin superimposes in large part upon linear oligosaccharide inhibitors visualized by other investigators. By comprehensive integration of these complexes we have constructed a model for the binding of polysaccharides having the helical character known to be present in natural substrates such as starch and glycogen.
Keywords: α-amylase, hydrolase, α-cyclodextrin, X-ray crystallography, limit dextrin
Pancreatic α-amylase (PPA, α-1,4-glucan-4-glucanohydrolase, EC 3.2.1.1) was among the first enzymes to be extensively analyzed in terms of substrate binding and catalytic properties (1, 2) and one of the first enzymes to be crystallized (3), yet many of its most salient features remain a mystery. This is due largely to the range of substrates that it will degrade and the diverse structures that those substrates assume. α-Amylase hydrolyzes, without anomeric inversion, α-(1,4) linkages between glucose residues in (1) amylopectin and amylose, which comprise starch, (2) glycogen, (3) oligosaccharides of various lengths, and (4) some cyclodextrins. Both amylopectin and glycogen contain α-(1,6) linkages, which α-amylase cannot hydrolyze, as well as α-(1,4) linkages and, therefore, exhibit indigestible branch points. Upon hydrolysis, the branched polysaccharides give rise to oligosaccharides termed “limit dextrins” (4-6). Cyclodextrins, also susceptible, in some cases, to cleavage by the enzyme (7-10), are covalently closed rings, the smaller of which are six to eight glucose residues in length and termed α-, β-, and γ-cyclodextrin, respectively.
The binding of substrates by α-amylase is further complicated by the complex structures formed by both linear amylose, and branched amylopectin and glycogen. The polysaccharides are not simply extended chains, but single and double helices (11-15). Amylopectin, for example, forms parallel double helices having six sugar residues on each strand per turn (or a total of 12 residues per turn) with a pitch of 21 Å. There are, in addition, higher orders of organization of the polysaccharides. They create condensed storage forms of high caloric carbohydrate by packing together in a partially ordered manner in what can be described as paracrystalline solids, forming dense and compact granules in cells that even water finds difficult to penetrate (16-19). Thus, α-amylase must contend with substrates that assume intricate and complex shapes that may not readily present themselves for binding and cleavage. These features of the substrates, and the necessity of amylase to address them, likely explain some of the more perplexing observations regarding the enzyme and its structure.
Pig pancreatic α-amylase is a protein of Mr = 55,436 daltons comprised of 496 amino acids, with an unusually high proportion (12.5%) of aromatic residues. It has one tightly bound calcium ion (1, 2, 8, 20, 21), and it is unusual in that it is catalytically activated by a chloride ion (22). X-ray crystallography has revealed that the cation is an integral component of the three dimensional structure (23, 24), and it has suggested a possible mechanism for activation by the anion (24). The protein is organized into two domains (23, 24), a large (β/α)8-barrel which is responsible for at least the major part of its catalytic activity, and a small eight stranded anti-parallel β-barrel, termed a carbohydrate binding module (CBM), which is common to most polysaccharide hydrolases (25). The large domain contains an excursion, which is sometimes considered as a third, or sub domain. The role of the CBM in α-amylase remains, as in other hydrolases, uncertain.
Catalysis, and the structure of the catalytic site are better understood than are substrate interactions. There is a prominent trough that traverses the entire face of the (β/α)8-barrel, and the active site lies in a deep depression near its center. We refer to this trough as the binding cleft, though it is not yet clear that every part of it participates in the binding of substrates. The catalytic center is characterized by three carboxylic acid containing residues, conserved among all α–amylases, Asp197, Asp300 and Glu233. These conspire to hydrolyze the α-(1,4) linkage and release a maltose unit from the reducing end of the polysaccharide fragment, though, depending on the length of the substrate, the product may also be a tri- or tetrasaccharide (26). X-ray crystallography suggests that a change in the conformation of a peptide loop that approaches the catalytic center, composed of residues 303 through 308, accompanies each cycle of binding and cleavage (27-30). The polysaccharide is associated with the enzyme in the binding cleft through hydrophobic interactions with aromatic residues and both direct and water-mediated hydrogen bonds.
One of the earliest, and most intriguing observations regarding α-amylase was that it is a processive enzyme (26, 31, 32). That is, the enzyme does not disengage from its polymeric substrate after release of product and subsequently bind at some arbitrary point elsewhere along the chain. Instead, it moves along the polysaccharide sequentially cleaving and releasing successive maltose or maltotriose units. Processivity of this sort is usually associated with toroidal enzyme complexes such as DNA or RNA polymerase, which trap their helical substrates in a center channel and move along it as they function (33). Unless α-amylase functions as a multimer and not a monomer, a feature that has otherwise escaped the attention of investigators, its processivity cannot arise from formation of a toroidal complex, but must result from some other properties of the enzyme.
The original structure of pancreatic α-amylase was determined in our laboratory (24) and independently by Qian et al. (23). In our paper we also described the presence in the native crystals of a disaccharide lying at one end of the substrate-binding cleft that linked neighboring protein molecules in the crystal lattice. We also described, based on crystal soaking and difference Fourier experiments, the binding of three α-cyclodextrin molecules to the enzyme. Two of the cyclodextrins appeared in the active site cleft of the amylase and could reasonably be expected to reflect the modes of binding of natural substrates. The third α-cyclodextrin was bound quite far from the active site, not in the binding cleft, and its presence remained unexplained. In addition to our results, a number of other X-ray diffraction investigations have been carried out in other laboratories (27-30, 34) that extend our knowledge of the interactions between the enzyme and its substrates.
To properly appreciate our results, it is essential to understand how the α-amylase that we crystallized and used in our X-ray studies was prepared. The procedure, whose significance has been largely overlooked, was, in fact, one of the first examples of affinity separation; it predated what we now refer to as affinity chromatography. Levitzki, Heller and Schramm (5) and Loyter and Schramm (6, 35) showed that if acetone powder of porcine pancreas were mixed with oyster glycogen suspended in 35% ethanol, then α-amylase would bind to the polysaccharide, non-covalently crosslink it, and subsequently form an insoluble mass. The enzyme, however, was inactive in 35% ethanol and would not hydrolyze the substrate. If the amylase/glycogen precipitate were collected by centrifugation and re-suspended in neutral buffer, then the enzyme became active again and proceeded to degrade the polysaccharide to limit dextrins, maltose, and other glycogen fragments. It was enzyme prepared by this procedure that we crystallized, and then re-crystallized for X-ray diffraction analysis. It is, therefore, not at all surprising that our crystals contained products of the degradation of glycogen.
At the time we reported the structure solution of PPA (24), our attention was principally focused on the structure of the protein and identifying the catalytic site. In addition, that model was refined using the mathematical procedures in the program X-PLOR (36, 37), which, although adequate at the time, have been superseded by the maximum likelihood approaches of today. Finally, much to our chagrin, we never deposited the coordinates for the model in the Protein Data Bank (38). To atone for our misdemeanor, and with the intention of now depositing the coordinates, we re-refined the model using the original X-ray diffraction data, but with a maximum likelihood-based program (39). The statistics for the structure determination and refinement are now notably superior to the original statistics, the coordinates for the putative native structure and the α-cyclodextrin complex have been deposited in the PDB, and we describe below the additional results that have emerged from this re-evaluation.
Materials and Methods
General Methods
The methods used for the preparation of α-amylase, its purification and crystallization are given in our previous papers (24, 40) and are essentially those of Loyter and Schramm (6, 35). The previous paper also includes the methods and procedures for the structure determination by isomorphous replacement, and the original refinement of the protein model using the program X-PLOR (36, 37). The same X-ray diffraction data described there was used for the refinement presented here, statistics for which are found in Table 1.
Table 1.
Data-processing, refinement and model statistics. Values in parentheses are for the highest resolution shell.
| Native | α-cyclodextrin complex | |
|---|---|---|
| Crystal data | ||
| Space group | P212121 | P212121 |
| Z | 4 | 4 |
| Unit-cell parameters (Å) | a=70.59, b=114.82, c=118.78 | a=70.65, b=114.88, c=118.85 |
| Data-processing statistics | ||
| Resolution (Å) | 14.01-2.11 (2.22-2.11) | 29.30-1.97 (2.07-1.97) |
| No. of unique reflections | 47149(2315) | 62592(5276) |
| Redundancy | NA | 4.84 (1.61) |
| <I/σ(I)> | 10.93(3.91) | 19.83 (3.07) |
| Completeness (%) | 82.6 (28.8) | 84.6 (36.6) |
| Rmerge † | 0.052 (NA) | 0.068 (0.178) |
| Structure-refinement statistics | ||
| Resolution (Å) | 14.01-2.11 (2.16-2.11) | 29.30-1.97 (2.02-1.97) |
| No. of reflections | 47147 (438) | 62084 (1218) |
| R/R free‡ (all data) | 0.115/0.157 (0.223/0.314) | 0.127/0.160 (0.240/0.279) |
| Reflections in test set (%) | 10.07 (11.16) | 10.11(8.46) |
| No. of refined parameters | 20378 | 20617 |
| No. of reflections | 42398 | 55805 |
| No. of restraints | 33319 | 33259 |
| Data-to-parameter ratio | 2.08 | 2.71 |
| Data/restraints-to-parameter ratio | 3.72 | 4.32 |
| Model statistics | ||
| Non-hydrogen atoms total | 5021 | 5044 |
| Protein (full/partial) | 3536/752 | 3601/628 |
| Ions (full) | 2 | 2 |
| Ligands (full/partial) | --/124 | 198/-- |
| Water (full/partial) | 508/99 | 549/66 |
| Geometry: r.m.s. deviations from ideal values | ||
| Bonds (Å) | 0.013 | 0.012 |
| Angles (°) | 1.49 | 1.39 |
| Planes (Å) | 0.007 | 0.006 |
| Chiral centers (Å3) | 0.11 | 0.09 |
| Average isotropic B factors (Å2) | ||
| B (Wilson plot) | 23.3 | 22.0 |
| Overall | 20.4 | 19.4 |
| Protein (full/partial) | 17.7/19.6 | 14.6/15.9 |
| Ions (full) | 15.6 | 15.6 |
| Ligands (full/partial) | --/34.8 | 80.3/-- |
| Water (full/partial) | 37.4/15.4 | 33.2/15.6 |
| Ramachandran plot | ||
| Most favored region | 374 (89.0%) | 374 (89.0%) |
| Allowed region | 45 (10.7%) | 45 (10.7%) |
| Generously allowed region | 1 (0.2%) | 1 (0.2%) |
| Disallowed regions | 0 (0.0%) | 0 (0.0%) |
Rmerge = Σh Σi | Ihi - <Ih> | / Σh Σi <Ih>, where Ihi is the ith used observation for unique hkl h and <Ih> is the mean intensity for unique hkl h.
R = Σh | Fo − Fc | / Σh Fo, where Fo and Fc are the observed and calculated structure factor amplitudes.
NA means data not available.
An important point is that the preparation of the enzyme for crystallization included a step in which the amylase was bound to glycogen under nonproductive conditions, and then transferred to accommodating media where the glycogen was subsequently hydrolyzed. Unlike the original procedure of Loyter and Schramm (6, 35), no attempt was made to remove hydrolytic products of the glycogen on an activated charcoal column. Recrystallization was used as the final step of purification. Thus, it is not unreasonable that substantial amounts of glycogen degradation products were carried forward into the crystals used for X-ray data collection. The experiments in which cyclodextrins were soaked into PPA crystals were preceded by washing those crystals 4-6 times with enzyme-free mother liquor.
Structure Refinement
For the purpose of discussion, although both structures have considerable bound carbohydrate, only the α-cyclodextrin complex was intentional and will be referred to as the complex or PPA complex while the structure described previously will be referred to as the native structure. The refinement of both the putative native structure and the α-cyclodextrin complex of PPA, upon which the results presented here were obtained, utilized the REFMAC5 maximum likelihood program (39). Model manipulation was performed with COOT (41). TLS parameters were refined for both structures; in each case, the enzyme was partitioned into three TLS groups based on the three domains A (aa 1-99 and 169-403), B (aa 100-168), and C (aa 404-496) and each glucose unit in each model was defined as an independent TLS group for a total of 14 groups for the native model and 21 groups for the cyclodextrin complex. Considerable disorder was found in both structures; 56 and 47 protein residues were modeled with multiple conformations in the native and complex structures, respectively, including the Cys70/Cys115 pair that forms a disulfide bridge. In addition, a number of glucose units had high B factors. In the native structure, a combination of high B factors and difference density suggestive of water overlaying the oligosaccharide structure lead us to a disordered water/saccharide model. However, even though two of the cyclodextrin rings had very high B factors, reduction of their occupancies did not improve the Rfree and produced positive ring-shaped difference density at the site of the cyclodextrin ring whose occupancy was reduced. Therefore, each ring in the final model of the cyclodextrin complex is at full occupancy. In each structure, occupancies of disordered protein residues were adjusted to equalize the B factors of corresponding atoms of the alternate conformers. For the glucose units in the native structure, the occupancies were adjusted to bring the B factors of each unit to about twice the average B factor of the enzyme. It is not unreasonable that the oligosaccharide fragments observed in the native structure have varying lengths.
The final statistics for the models of both the native structure and the α-cyclodextrin complex are found in Table 1. The final statistics compare very favorably with those previously reported; R and Rfree have been reduced from 0.168 and 0.213 to 0.115 and 0.157, respectively. Rms deviations in bonds and angles are 0.013 Å and 1.49°, respectively, versus the previously reported values of 0.010 Å and 2.10°. The percentage of residues in the most favored region of the Ramachandran plot has increased from 86% to 89%. The RCSB validation server (38) and PROCHECK (42) were used to assess the quality of the model and generate the Ramachandran statistics. Geometrical target values were those contained in the CCP4 dictionary (43) and consist of the values from Engh and Huber (44) for the protein structure and Saenger (45) for the carbohydrate structure. Figures were prepared using PyMol (46). Comparison of inhibitors and substrate analogs are based on Cα superpositioning of deposited structures onto our refined model. Relative displacements in the positions of corresponding saccharide residues are measured as the distance between the centers of their respective pyranose (or equivalent) rings. No effort was made to analyze differences in the orientation of the sugar rings, although, except for subsite −1, such differences appeared to be minimal.
Results
Upon re-refinement of the model against X-ray data using maximum likelihood procedures, much disordered density, previously seen but not modeled in the substrate binding cleft, as well as some ambiguous density present elsewhere in difference electron density maps, coalesced into identifiable carbohydrate molecules. Aside from several patches of density on the surface of the enzyme, which will be described below, there were notable appearances of oligosaccharide at two locations.
Tetrasaccharide
In the model originally presented (24), a disaccharide was identified at the interface of two α-amylase molecules related by a 21 screw axis along the crystallographic x direction. The disaccharide was of additional interest because it bridged an end of the substrate binding cleft on one molecule with the putative distal end of the binding cleft on another. In our current maps we can clearly see an additional glucose residue on each end of that disaccharide, so that it is now a tetrasaccharide.
The tetrasaccharide has a pronounced curvature, shown in Figure 1, that, as we discuss below, corresponds to the curvature of four glucose residues in a molecule of γ-cyclodextrin. The final model consists of an overlay of water structure and the tetrasaccharide; average B factors and occupancies for each of the glucose units is shown in Figure 1 and reflect the degree to which each glucose unit interacts with PPA. We assume that the tetrasaccharide model is really a composite of maltose, maltotriose and maltotetraose molecules and even interfaces devoid of saccharide molecules. According to Machius et al. (28), a bridging saccharide molecule is not necessary for the formation of this interface. As seen in Figure 1, primary PPA-saccharide interactions, which are both hydrogen bonding and hydrophobic in nature, occur between the central pair of glucose units; only water-mediated interactions are observed for the end glucose units. Supplementary Table 1 lists the interactions between the tetrasaccharide and PPA molecules.
Figure 1.
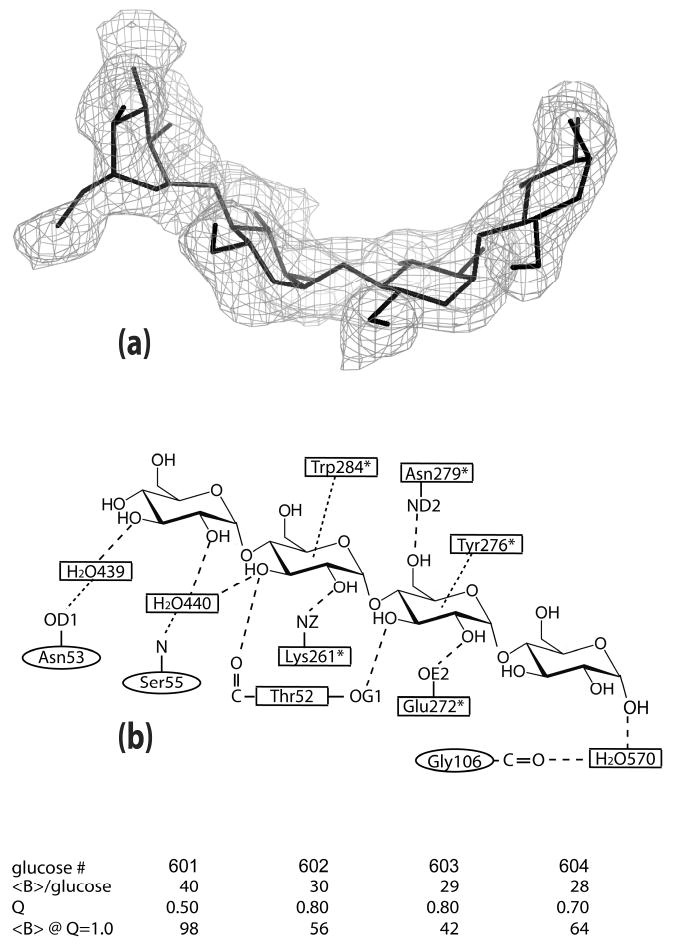
(a) The tetrasaccharide model is superimposed on the difference density calculated with the tetrasaccharide omitted from the structure factor calculations. The density around residues 602-604 is contoured at 2.5σ; around residue 601, the density is contoured at 1.0σ. (b) The interactions involving the tetrasaccharide are represented schematically. Residues with “*” are from the 21 screw-related PPA molecule. The residues in both the model and the schematic correspond to the glucose numbering in the table in which the occupancies and average B factors are given for each glucose unit.
Limit Dextrin
The second appearance of extensive oligosaccharide lay within the extended substrate binding cleft and consisted of six glucose units. The reducing end of this oligosaccharide occupied what is referred to as the -1 site (see Figure 2 for numbering system) according to the nomenclature of Davies et al. (47). This site immediately precedes the active center where cleavage of the scissile α-(1,4) glucosidic bond produces a maltose product. As shown in Figure 3, subsites -2 through -5 are also filled with glucose residues of the oligosaccharide, but the residue at subsite -3 also has an α-(1,6) linkage to a third glucose unit. Hence, this oligosaccharide is what is commonly termed a limit dextrin, and it serves to terminate processive degradation of a polysaccharide chain. The limit dextrin more or less fills the substrate-binding cleft between the active center and the location of the bridging tetrasaccharide. Again, we have assumed that the limit dextrin is a composite of various oligosaccharides by varying the occupancies of the constituent glucose residues. In this manner we can account, to some extent, for the presence of an overlying water structure and the large variation in temperature factors between the glucose units of the limit dextrin. Certainly, there are many oligosaccharide products that can result from the digestion of glycogen.
Figure 2.

Active site cleft subsite numbering and schematic representations of acarbose-derived inhibitors previously described and limit dextrin identified in this study.
Figure 3.
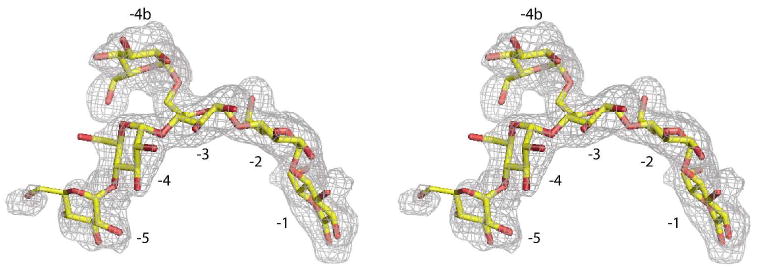
Stereoview of the limit dextrin superimposed on difference density generated from structure factors and phases calculated from the final model with the limit dextrin omitted. Subsites are numbered; the density is contoured at 2.5σ for subsites −1 through −4 and contoured at 1.5σ for subsites −5 and −4b.
The binding interactions between the limit dextrin and the protein are presented in Figure 4 (also see Supplementary Table 2). They consist of numerous hydrogen bonds and hydrophobic interactions, including contacts with a number of aromatic side chains. As does the bridging tetrasaccharide, the limit dextrin too exhibits a pronounced curvature that, as we describe further below, also corresponds well with the course of glucose residues in a γ-cyclodextrin molecule.
Figure 4.
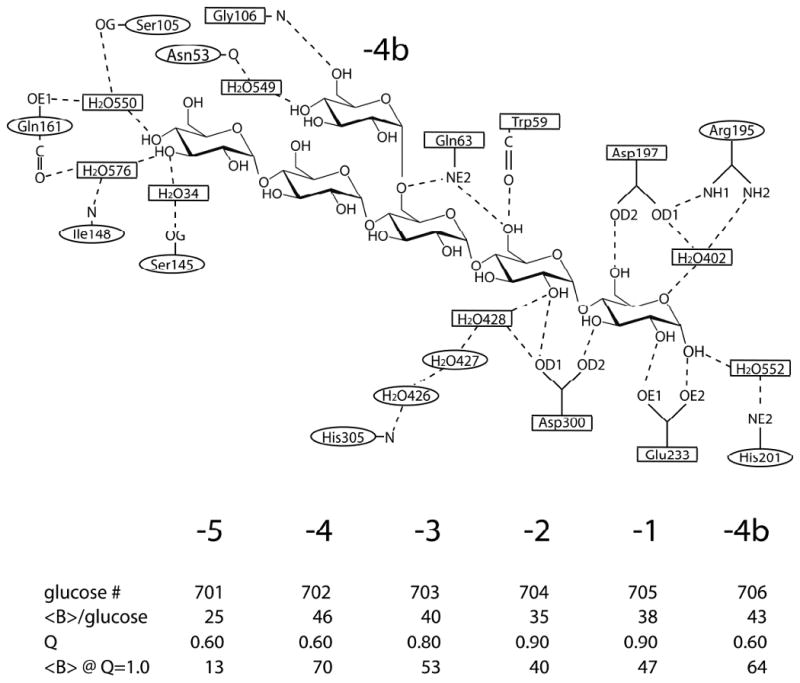
Schematic diagram of the hydrogen bonded interactions between the limit dextrin and PPA.
Saccharide structure
Generally, the electron density maps provided a good image of the saccharide ligands but not the detail seen for the enzyme, especially for weakly interacting glucose residues. While the path and extent of the oligosaccharides are clear, the atomic structure is less so. This is evident by the average B factors <B> for the glucose residues which, even at partial occupancy, are as high as 40 Å2 for Glc601 at 50% occupancy; residues in cyclodextrins II and III at full occupancy have <B> exceeding 100 Å2 (<B> for the oligosaccharides are listed in Figures 1 and 4; for the cyclodextrin rings, see Supplementary Table 3). Although all glucose rings in the initial models had 4C1 conformations, some rings refined to a different conformation. Glc601 in the tetrasaccharide has a twisted chair conformation. Glc703 of the limit dextrin, from which the α-(1,6) branch emanates, has an E3 conformation while the branched Glc706 has a 4E conformation. Even α-cyclodextrin III has one glucose ring that refined to the boat conformation 2,5B. We do not believe that these deviations, none of which are in the active site, necessarily represents reality but rather are a function of disorder or partial occupancy. Puckering parameters have been calculated and deposited as Supplementary Table 3.
The oligosaccharides have a left-handed helical bias. Using the O4 (or O1) atoms to determine the helical parameters, we obtained a left-handed helix with a radius of 4.2 Å, a rise per residue of 2.1 Å and 10.4 residues per turn for the tetrasaccharide. The limit dextrin (neglecting O1 on Glc705) has a curvature corresponding to a left-handed helix with a radius of 3.1 Å, a rise of 3.3 Å per residue and 7.2 residues per turn. We omitted O1 because it refined to a position midway between the two anomeric positions resulting in O4′...O1 distance of 5.46 Å which is much greater than any other equivalent O...O distance. The average O4′...O4 distances were 4.72 Å for the tetrasaccharide and 4.48 Å for the limit dextrin. The comparable distances in the cyclodextrins, which approximate the curvature of single stranded helical V amylose, were 4.20, 4.19, and 4.15 Å, demonstrating that the oligosaccharides are more open than α-cyclodextrin and V amylose.
The glycosidic linkage torsion angles ϕ(O5′-C1′-O4-C4) and ψ(C5-C4-O4-C1′) have the following ranges: tetrasaccharide, 69.2 to 127.6° for ϕ and -109.6 to -113.9° for ψ; limit dextrin, 77.9 to 122.2° for ϕ and -100.4 to -136.6° for ψ; α-cyclodextrin I, 101.8 to 117.9 for ϕ and -102.1 to -120.5° for ψ; α-cyclodextrin II, 104.4 to 145.2 for ϕ and -107.1 to -132.8° for ψ; α-cyclodextrin III, 109.8 to 157.2 for ϕ and -80.6 to -151.6° for ψ. For the oligosaccharides, the links between the first and second residues are abnormal, most likely resulting from disorder due to minimal interactions involving the first residue of the chain. However, in the case of the limit dextrin there was overlying solvent that could also affect the refinement and produce distortions. As for the cyclodextrins, ring I conforms best to the expected values; this is anticipated since it is a fairly well behaved molecule with reasonable B factors. However, the poorly defined cyclodextrins II and III, with B factors exceeding 100 Å2, have wide ranges of torsion angles, most probably indicating severe disorder. Many of the hydroxyls of cyclodextrins II and III are poorly defined in electron density maps.
Finally, most successive glucose units have O2′...O3 distances less than 3.25 Å, suggesting intrachain hydrogen bonding. Only 601-602 in the tetrasaccharide, 701-702 in the limit dextrin, and 805-806 in cyclodextrin III have O2′...O3 distances greater than this value and they all exceed 3.5 Å. Supplementary Table 4 provides greater detail on O...O distances, linkage torsion angles and glucopyranosyl ring twisting torsion angles.
Loop 303-308
Other investigators observed that the polypeptide segment comprised of amino acid residues 303 through 308 exhibited some movement as a consequence of substrate binding (27-30, 48). The two distinct conformations appeared when the enzyme devoid of any bound ligand was compared with the structure of the enzyme complexed with the non–hydrolyzable acarbose-derived inhibitors (28, 29, 34, 48). These inhibitors fill not only subsites −1 up to −4, but also subsites +1 up to +3. The same loop movement was observed in the complex of PPA with the substrate analog methyl-4,4′-dithio-α-maltotrioside which contains two substrate molecules in the active site cleft, one in subsites −2 and −3 and the other in subsites +1 and +2 (49). It has been suggested that this loop movement is driven by the formation of a strong hydrogen bond between His305 and the sugar residue at subsite −2 (28). In our limit dextrin complex, we see electron density for both conformations of the loop. The predominant conformation is the “open” form corresponding to the unliganded enzyme, but, as seen in Figure 5, there is reasonably well-defined density for the closed conformation as well. The fact that we observe density corresponding to the closed conformation of the loop suggests that there may be residual sugars in the product sites of some of the PPA molecules in our crystals. Indeed, we do observe some density near subsite +1, but it appears to be disordered and not clearly recognizable as a carbohydrate moiety. In all likelihood, there is some residual sugar in some subsites +1 in our crystals, sufficient to induce the loop movement, but not well enough ordered to be recognizable. In any case, we have modeled no carbohydrate residues in subsites +1 and +2. Payan and Qian (30) made a similar observation and drew a similar conclusion in their structure of PPA with a maltotriose bound in subsites −1 to −3. Clearly, the closed conformation occurs only in those structures that have subsites +1 and +2 occupied. This suggests that, contrary to the initial suggestions that the His305 interaction with the saccharide moiety at subsite −2 produced the shift, the true driving force behind the loop shift is the presence of moieties in the +1 and/or +2 subsites. This suggests that only after the product is released does the loop relax to its unliganded position simultaneously releasing its hold on the sugar residue at subsite −2 to allow repositioning of the upstream oligosaccharide. His305, it may be noted in passing, could possibly be the histidine identified by Ishikawa and Hirata (50) as participating in catalysis. Furthermore, the His305 interaction may assist in substrate repositioning by pulling on the substrate as the loop relaxes to its conformation in the unliganded state.
Figure 5.
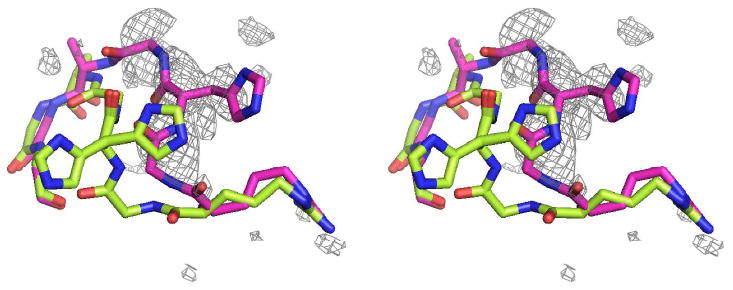
Loop 303-309 from the present study is in green and from 1HX0 (29) is in magenta. Electron difference density, which covers the 1HX0 loop fairly well, was calculated with water molecules in the vicinity of the 1HX0 loop removed from the final model. The density suggests an unmodeled alternate conformation for this loop that is consistent with the loop conformation observed in all the acarbose-derived inhibitor complexes. The loop sequence is Arg303-Gly-His-Gly-Ala-Gly-Gly309. The difference density is contoured at 2.0σ.
Cyclodextrin
In our earlier paper (24) we described an experiment in which we diffused α-cyclodextrin into α-amylase crystals and carried out difference Fourier analyses. From these, we identified and described the binding of three α-cyclodextrin molecules to the protein, shown in Figure 6. Two of these were found in the substrate-binding cleft and superimpose reasonably well on the bridging tetrasaccharide in one case (I), and upon portions of the limit dextrin in the second case (II).
Figure 6.
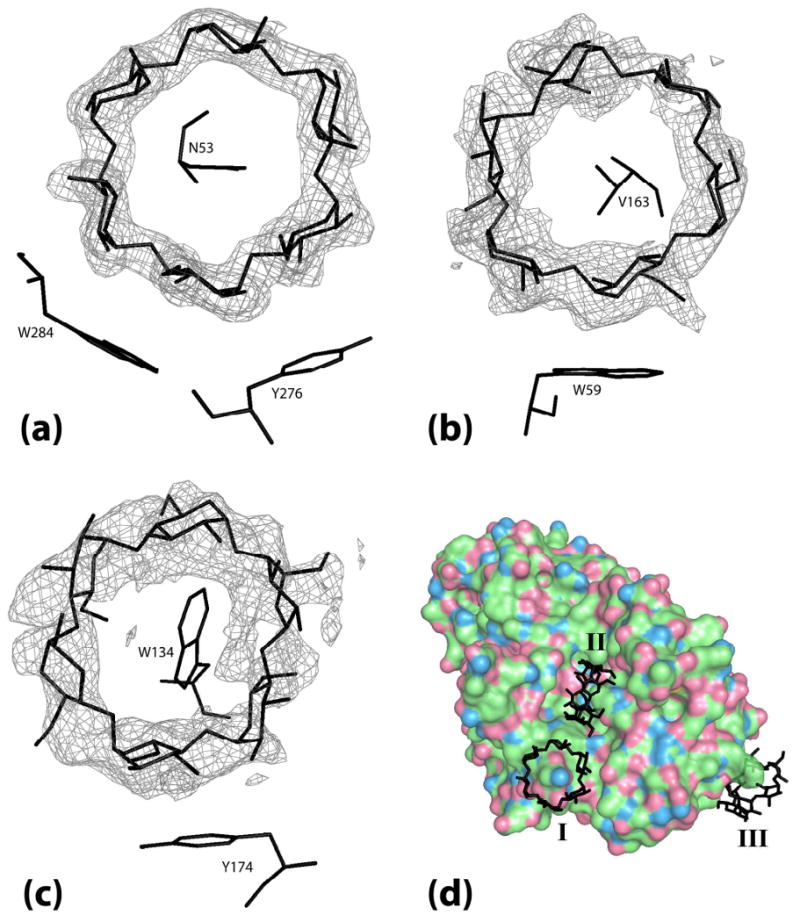
The electron density for each α-cyclodextrin ring is from a Fo-Fc map phased by the final model with the α-cyclodextrin ring that the density covers removed. (a) α-cyclodextrin I bridging between two PPA molecules superimposed on the map density contoured at 4.0 σ. (b) α-cyclodextrin II bound in the binding site cleft superimposed on map density contoured at 2.0 σ. (c) α-cyclodextrin III bound at Trp134 superimposed on map density contoured at 1.8 σ. (d) Full view of the 3 molecules of α-cyclodextrin bound on the surface of PPA with α-cyclodextrins labeled I, II, and III.
The third α-cyclodextrin (III) was not associated with the substrate-binding cleft but was at the opposite end of the major, catalytic domain from the CBM. The purpose and nature of this “accessory site” was not clear, and remains so now. The association, however, is clearly with a single amylase molecule in the crystal lattice and its binding is not assisted by contributions from symmetry related protein molecules. Its binding is unlikely to be simply fortuitous as its influence on the catalyzed reaction is detectable by solution studies (9).
We subsequently carried out identical diffusion and difference Fourier experiments using β-cyclodextrin and γ-cyclodextrin. In those two experiments, no cyclodextrin rings were observed in difference Fourier maps. There was density present in subsites -1 and −2 of the γ-cyclodextrin maps that could well be sugar residues, but none otherwise. This, however, might have been anticipated, as α-amylase hydrolyzes both of these two cyclodextrins. Amylase present in the mother liquor of the crystals alone might have been sufficient to cleave these to maltose even if enzyme molecules in the crystal itself were incapable.
The binding of the cyclodextrin is instructive. Only four consecutive glucose residues of a ring (less in the case of the “accessory” cyclodextrin) make close interactions with the protein. This alleviates, to some extent, the problem of binding cyclodextrins of significantly different sizes. The issue does remain, however, that the curvature of the four interacting glucose residues in the α-, β-, and γ-cyclodextrins are substantially different. The outside diameters of the three cyclodextrins are 13.7 Å, 15.3 Å, and 16.9 Å, respectively (51), and the angles subtended by 4 residues are 240°, 206°, and 180°, respectively. That is, as shown in Figure 7, relative to the four residues in α-cyclodextrin, those in γ-cyclodextrin have substantially less curvature. The implication is that the binding sites for the cyclodextrins are tolerant and non-specific in terms of variations in substrate structure. Further, because two of the cyclodextrin binding sites overlap closely with crystallographically observed substrate binding sites, an additional implication is that the substrate binding cleft must share these same characteristics vis-à-vis natural substrates.
Figure 7.
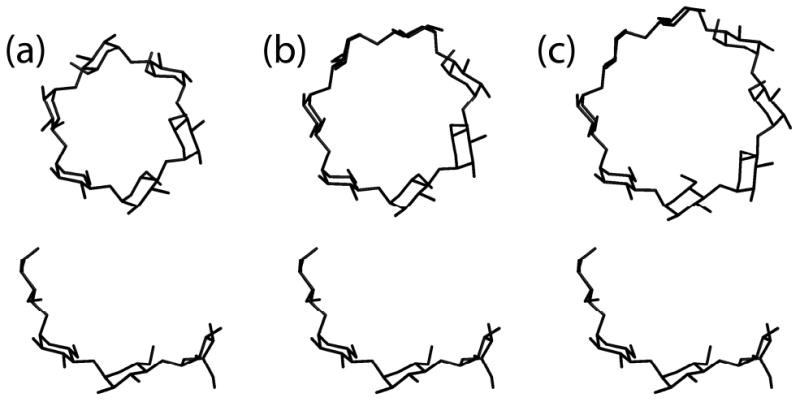
(a-c) α-, β-, and γ-Cyclodextrin structures, respectively, with the tetrasaccharide below each for comparison of curvature. The exterior diameters are 13.7, 15.3, and 16.9 Å for α-, β-, and γ-cyclodextrin, respectively.
The “accessory” cyclodextrin is primarily fixed to the protein by an intimate inclusion interaction in which the side chain of Trp134 is inserted into the cyclodextrin cavity and through a stacking interaction with the phenol ring of Tyr174. Trp134 is particularly interesting. The tryptophan side chain is the most hydrophobic of all of the amino acids and tryptophan is generally sequestered in the interior of a protein, a part of its core. Trp134, however, is on the protein surface and its side chain is extended into the solvent where it waves like a flag. A variety of conformations for this side chain have been observed. Qian et al. (48), Gilles et al. (34) and Machius et al. (28) modeled the side chain in a similar conformation, Qian et al. (49) modeled it entirely differently and in their liganded structure, Machius et al. (28) found no density for the side chain at all. We have modeled the side chain with two conformations, both of which are similar to the two observed by Qian et al. (29). It is clearly mobile. In the α-cyclodextrin complex, however, the side chain has a single, ordered position by virtue of its interaction with the cyclodextrin. It imposes itself into the cavity of the cyclodextrin.
The interaction of Trp134 with the sugar ring, as seen in Figure 8, is reasonable from the perspective of cyclodextrin properties. Cyclodextrins are known, and widely used in many chemical applications (8, 52), to sequester a broad variety of molecules in the cavity of the toroids. The small molecules are frequently, but not always, hydrophobic. The insertion of the indole side chain of Trp134 into the cyclodextrin cavity is a good example. Inspection of the other two cyclodextrins bound by PPA shows that, in the case of cyclodextrin II, the side chain of Val163 is inserted into the cavity, and, in the case of cyclodextrin I, it is the side chain of Asn53.
Figure 8.
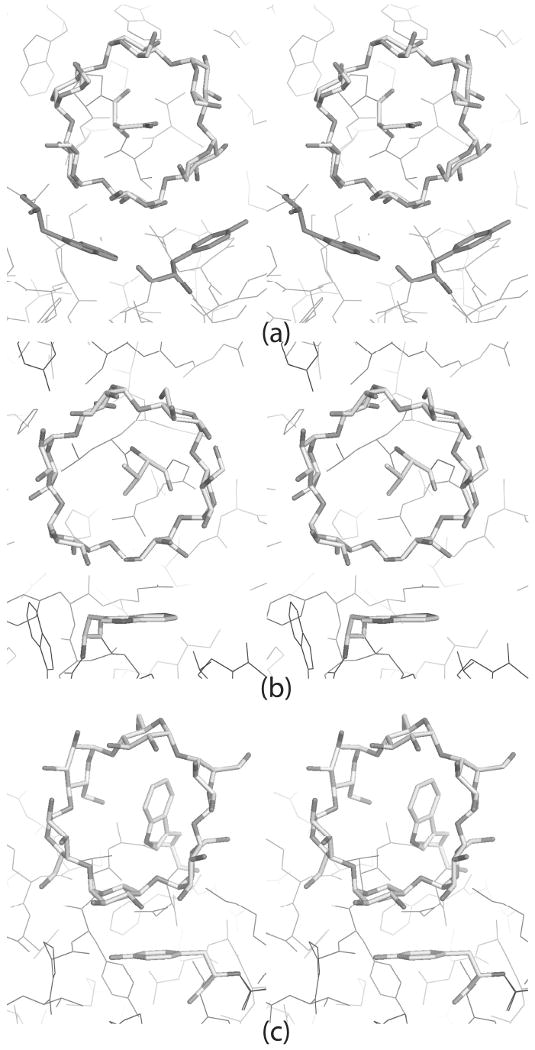
α-Cyclodextrins are shown acting as hosts to various side chains within their cavities. Key platform-forming aromatic residues are also highlighted. (a) α-Cyclodextrin ring I bridges between 21 screw related molecules. Asn53 is the guest side chain; Tyr276 and Trp284 of the symmetry related molecule (in magenta) are the platform residues. (b) α-Cyclodextrin ring II is in the active site cleft. Val163 is the guest side chain for this ring. Trp59 at the bottom of the active site cleft is the platform residue. (c) Trp134 is the guest side chain for α-cyclodextrin ring III. Tyr174 is the platform residue.
Previous inhibitor studies
The most detailed description of the binding subsites for the glucose residues in an extended substrate chain, and the most extensive in terms of the amount of substrate binding cleft it describes, is that reported by Machius et al. (28) based on the binding of an octameric inhibitor V-1532. This has a non-hydrolyzable N linkage joining the residue at the reducing end of a linear oligosaccharide in subsite -1 to the 4-amino–4,6–dideoxyglucose in subsite +1. Three additional residues precede the scissile bond as shown in the diagram of Figure 2. In the PPA/V-1532 complex structure, six inhibitor residues were clearly seen in electron density maps and modeled, occupying subsites −3 to +3, while at subsite -4, there was density that suggested the presence of a disordered residue.
In other inhibitor studies, two different inhibitors were used, acarbose (34) and acarviosine-glucose (29), yielding in each case the same inhibitor occupying subsites -4 through +2 (see Figure 2). There was good agreement between the atomic positions of the overlapped glucose residues of this inhibitor and V-1532 at subsites −3 through +2. Taken together, the three analyses show that the binding cleft can accommodate at least seven glucose residues of α-(1,4) linkage in a linear chain and fill subsites −4 through +3. It may be relevant that the work of Robyt and French (26, 32) using linear oligosaccharides as substrates showed that trisaccharides and tetrasaccharides are also products of the α-amylase catalyzed reaction, as well as maltose. Their data further indicated that as progressively longer oligosaccharides were used as substrates, the proportion of longer products increased.
Limit Dextrin Disposition
The glucose residue of the limit dextrin in subsite -2 of the complex superimposes well (0.42 Å average displacement) on the glucose residues in that subsite for the acarbose-derived inhibitors that have been studied. There is a small, marginally significant displacement (average of 0.81 Å) of the limit dextrin glucose in subsite -3 from those in the inhibitors. At subsite -4, however, there is a clear displacement of the limit dextrin glucose of more than 2 Å from the corresponding glucose residues in the inhibitors that have this subsite occupied. No glucose residue has previously been seen in subsite -5, though one is clearly evident in the limit dextrin. Because of the displacements at subsites −3 and −4, which are likely due to the presence of the α-(1,6) linked glucose on the glucose residue at subsite −3, the position of the subsite −5 residue may be different than what would be found for the non-branched amylose substrate. The α-(1,6) linked glucose is tucked well into the binding cleft and is clearly associated with the protein.
Most interesting, is the limit dextrin glucose in subsite -1. When compared with the corresponding glucose residues from the inhibitors that also had residues in subsites +1 and +2, there is a pronounced movement. The -1 subsite glucose of the limit dextrin appears to have pulled away from the binding cleft, and away from the positions of the corresponding residues of the inhibitors by 1.7 Å. It appears almost to be initiating disassociation from the enzyme. In complexes with acarbose-derived inhibitors, the conduritol ring in subsite -1 is closely juxtaposed with the underlying Tyr62. In the limit dextrin complex there are, in fact, two water molecules interposed between the glucose and tyrosine rings. However, in two complexes in which maltotriose occupies subsites −1 through −3 (30, 53), the glucose in subsite −1 has the same disposition with respect to Tyr62 as the conduritol rings in the inhibitors.
The disposition of the limit dextrin glucose in subsite -1 suggests to us that the presence of glucose residues (or analogues) in subsites +1 and +2 may be necessary to compel a glucose residue of the remaining oligosaccharide into the -1 subsite. Forcing that glucose into subsite -1 by the binding of residues in subsites +1 and +2 could have the effect of straining the scissile α-(1,4) bond linking the glucose residues in subsites -1 and +1, and thereby contribute to catalysis.
As noted previously, the bridging tetrasaccharide and the limit dextrin (as well as extended inhibitors studied by others) have distinct curvatures. When we examined the binding of α-cyclodextrin, and by extrapolation, β- and γ-cyclodextrin, by making optimal correspondence of protein interacting glucose residues, and compared those with the complex containing the bridging tetrasaccharide and the limit dextrin, we found that both the limit dextrin and the tetrasaccharide superimposed very well upon four residues of a γ-cyclodextrin. The radii of curvatures were virtually identical. The coincidence of substrates and inhibitors with γ-cyclodextrin probably explains why α-amylase hydrolyzes γ-cyclodextrin rapidly, β-cyclodextrin with difficulty, but fails to hydrolyze α-cyclodextrin to any appreciable extent.
Active Site Interactions Summary
If all of the data from this and previous analyses are integrated in a comprehensive manner, then an extended polysaccharide chain of eight residues can be placed within the active site cleft of the enzyme. This then allows definition of the interactions responsible for association of enzyme and substrate. These interactions are presented in Table 2 (also see Supplementary Figure 1).
Table 2.
Possible interactions of natural substrates based on limit dextrin and inhibitor structures. A “Y” indicates that the interaction is present in the structure.
| subsite | atom from glucose or equivalent | interacting atom | residue | number of mediating waters | Present study | 1PPI | 1OSE | 1PIG | 1HX0 |
|---|---|---|---|---|---|---|---|---|---|
| -1 | O1 (+1 N4) | OEx | E233 | Y | Y | Y | Y | Y | |
| O1 (+1 N4) | ODx | D300 | Y | Y | Y | Y | |||
| O1 | NE2 | H201 | 1 | Y | |||||
| O2 | ODx | D300 | Y | Y | Y | Y | |||
| O2 | NH2 | R195 | Y | ||||||
| O3 | ODx | D300 | Y | Y | Y | Y | Y | ||
| O3 | NE2 | H299 | Y | Y | Y | Y | |||
| O5 | NHx | R195 | 1 | Y | |||||
| O5 | ODx | D197 | 1 | Y | |||||
| O6 | NE2 | H101 | Y | Y | Y | Y | Y | ||
| O6 | ODx | D197 | Y | Y | Y | Y | Y | ||
| Ring | Stack | Y62 | Y | Y | Y | Y | |||
| -2 | O2 | ODx | D300 | Y | |||||
| O2 | ODx | D300 | 1 | Y | |||||
| O2 | ND1 | H305 | Y | Y | Y | Y | |||
| O3 | N | G306 | 2 | Y | Y | ||||
| O3 | O | G304 | 3 | Y | |||||
| O3 | N | H305 | 3 | Y | |||||
| O6 | O | W59 | Y | Y | Y | Y | Y | ||
| O6 | NE2 | Q63 | Y | Y | Y | Y | Y | ||
| Ring | Stack | W59 | Y | Y | Y | Y | Y | ||
| -3 | O2 | NE1 | W59 | 2 | Y | Y | Y | Y | |
| O2 | ODx | D356 | 2 | Y | Y | Y | Y | ||
| O3 | O | N53 | 2 | Y | |||||
| O4 (N4) | O | V163 | Y | Y | Y | Y | |||
| O6 | NE2 | Q63 | Y | ||||||
| Ring | Stack | W59 | Y | Y | Y | Y | Y | ||
| -4 | O3 | O | Q161 | 2 | Y | ||||
| O3 | O | I148 | 2 | Y | |||||
| O3 | N | G164 | 2 | Y | |||||
| O6 | N | G106 | Y | Y | |||||
| O6 | O | G164 | Y | ||||||
| O6 | N | S105 | Y | ||||||
| -4b | O3 | O | N53 | 1 | Y | ||||
| O6 | N | G106 | Y | ||||||
| -5 | O3 | OG | S145 | 1 | Y | ||||
| O3 | N | I148 | 1 | Y | |||||
| O3 | O | Q161 | 1 | Y | |||||
| O4 | OG | S105 | 1 | Y | |||||
| O4 | OE1 | Q161 | 1 | Y | |||||
| +1 | O2 | NE2 | H201 | Y | Y | Y | Y | ||
| O2 | N | I235 | 1 | Y | Y | Y | Y | ||
| O2 | O | E233 | 1 | Y | Y | Y | Y | ||
| O3 | OEx | E233 | Y | Y | Y | Y | |||
| O3 | O | E233 | 1 | Y | |||||
| O3 | N | K200 | 1 | Y | |||||
| N4 (-1 O1) | ODx | D300 | Y | Y | Y | Y | |||
| N4 (-1 O1) | OEx | E233 | Y | Y | Y | Y | |||
| O5 | N | G306 | 3 | Y | |||||
| +2 | O1 | O | G306 | 1 or 2 | Y | Y | |||
| O1 | N | L237 | 2 | Y | |||||
| O2 | NZ | K200 | Y | Y | Y | Y | |||
| O2 | OEx | E240 | Y | Y | Y | Y | |||
| O2 | O | G238 | 2 | Y | |||||
| O2 | N | L237 | 2 | Y | Y | ||||
| O3 | NZ | K200 | Y | Y | Y | Y | |||
| O5 | O | G306 | 1 | Y | |||||
| O6 | O | G306 | Y | ||||||
| O6 | N | G306 | 1 | Y | |||||
| O6 | N | V163 | 2 | Y | |||||
| O6 | OH | Y151 | 1 | Y | |||||
| O6 | O | I148 | 3 | Y | |||||
| Ring | Stack | Y151 | Y | Y | Y | Y | |||
| +3 | O1 | symmetry molecule | Y | ||||||
| O2 | O | G238 | 1 | Y | |||||
| O6 | symmetry molecule | Y |
We have noted that there is a significant difference in the position of the moiety at subsite -1 in the five structures we are comparing. In the acarbose-derived inhibitor structures, the 6-hydroxymethylconduritol ring stacks on Tyr62, deep in the active site, whereas in the limit dextrin, the glucose ring is shifted approximately 1.7 Å away from Tyr62. This changes a few interactions. As seen in Table 2, the conserved interactions involve O6 of the moiety with NE2 of His101 and one carboxyl O of Asp197, O3 with a carboxyl O atom of Asp300 and O1 (N4 of +1 subsite moiety) with carboxyl O of Glu233. However, the interactions of O3 with His299, O2 with Asp300, and N4 of subsite +1 with Asp300 are lost in the limit dextrin while water-mediated interactions between O1 and His201 and O5 with Arg195 and Asp197 are gained. A direct hydrogen-bonded interaction between O2 and Arg195 is only seen in 1HX0 (29), the structure determined at the highest resolution of the set.
There is very little difference between the saccharide unit at subsite −2 in the limit dextrin and those seen in the inhibitors. The only conserved interactions are the partial stacking of the ring on Trp59 and the involvement of O6 with carbonyl O of Trp59 and the amide N of Gln63. The differences in the interactions at this site relate to the shift in loop 303-308 that occurs in the inhibitor structures that is not present in the limit dextrin structure. Hence, there is an interaction of the side chain of His305 with O2 in the inhibitor structures resulting from this shift, which is not seen in the limit dextrin structure. On the other hand, the hydrogen bond and the water-mediated interactions involving Asp300 and O2 of this saccharide moiety of the limit dextrin results because the 303-308 loop did not shift. Likewise, other water-mediated interactions between O3 of subsite −2 and loop 303-308 are changed, although there is inconsistency in these interactions within the set of inhibitor structures. However, it is clear that the interactions of the protein with the moiety at subsite −2 varies with the position of loop 303-308, the position of which seems directly related to the state of subsites +1 and/or +2 as previously discussed.
There is only one conserved interaction between protein and subsite −3 for the five structures being compared here. This is the partial stacking of the saccharide ring on the indole ring of Trp59. The inhibitors have a hydrogen-bonding interaction between N4 and the carbonyl O of Val163 that is not present in the limit dextrin structure, since there is no N atom at this position. On the other hand, O6 interacts with the amide nitrogen of Gln63. The inhibitor structures all have water-mediated interactions between O2 and the PPA residues Trp59 and Asp356. The 1HX0 structure has one additional water-mediated interaction with Asn53, at the extreme end of the active site cleft with which the α-cyclodextrin and the tetrasaccharide interact. The differences between the limit dextrin and the inhibitors most likely result from the 0.8 Å shift of the −3 moiety in the limit dextrin due to the presence of the α-(1,6) branch on this moiety which lies between the subsite −3 ring and the floor of the active site cleft (see Supplementary Figure 1c).
In the limit dextrin structure, subsite −4 has no interaction, direct or water-mediated, with the enzyme, which partially accounts for the fact that this moiety has the highest average B factor of all the sugar residues of the limit dextrin when at full occupancy. It is shifted by more than 2 Å from the positions of the corresponding moieties in the two inhibitor structures that have this subsite occupied (1HX0, 29; 1OSE, 34)(see Supplementary Figure 1d). Because of the differences in the positioning of the O6 atom at subsite −4, the direct interactions of the two inhibitors are not exactly the same. They both have a hydrogen bond between the peptide N of Gly106, but 1OSE has two other potential hydrogen bonds between O6 and PPA residues Gly164 and Ser105. The higher resolution structure of 1HX0 has an additional three water-mediated interactions between the sugar O3 atom of subsite −4 and PPA residues Ile148 (O), Gln161 (O) and Gly164 (N). If we consider the glucose that is α-(1,6) branched from the glucose at subsite −3 to be subsite −4b, since it is parallel to the subsite −4 moiety along a continuous chain from the reducing end, we can compare its interaction with those of subsite −4 found in the inhibitor structures. There is a difference of about 5 Å between the ring centers of the moieties in the inhibitors and the subsite −4b sugar ring center. The subsite −4b moiety has a direct hydrogen bond to the carbonyl O of Gly106, a PPA residue that is involved in subsite −4 interactions in the inhibitors albeit through the peptide N. There is also a water-mediated interaction with the carbonyl O of Asn53, an interaction that was seen with subsite −3 in the 1HX0 structure.
Subsite −5, if indeed it is a valid substrate subsite, has no peer in any reported structure. All the interactions between this subsite and PPA are water-mediated, several of which involve residues involved in the subsite −4 interactions of 1HX0 (Ile148 (N) and Gln161 (O and OE1)) and of 1OSE (Ser105 (OG)). In addition, there is an interaction with Ser145 (OG). The −4 and −4b subsites are directed toward the tetrasaccharide position and, indeed, as shown below, can be extended to utilize the entire nonreducing end of the substrate binding cleft. The degree of interaction and the trend of increasing B factors for the individual subsites suggest a diminishing binding strength from the −1 subsite to the −4 subsite. Subsite −5, although apparently more securely bound than subsite −4, still is only weakly bound since all interactions are indirect through water. The direction of the saccharide chain at this point is toward cyclodextrin III. However, the chain would have to continue up the binding cleft wall and follow the enzyme surface toward Trp134, about 20 Å away.
Although the limit dextrin structure has no identified sugar residues on the reducing end of the binding cleft, the +1 and +2 subsite interactions of the inhibitors are quite consistent, especially for the direct hydrogen bonded interactions. For subsite +1, these include possible hydrogen bonds between O2 and PPA residues His201, O3 and Glu233, N4 and Asp300 and Glu233. There are also water-mediated interactions between O2 and Ile235 and Glu233 that appear in all structures. Other water-mediated interactions are found in 1HX0 for Glu233 and Lys200, with a third involving Gly306 that is found in two structures, 1HX0 and 1PPI.
The key direct interactions at subsite +2, found in all four inhibitor structures, involve O2 with Lys200 and Glu240, O3 with Lys200 and the stacking of the sugar ring against Tyr151. Less conserved among these structures are the water mediated interactions involving Gly306, Leu237, Gly238, Val163, Tyr151 and Ile148. Only the V-1532 complex has an additional direct interaction between O6 and Gly306 (O). The number of interactions at these subsites (+1 and +2) rival those of subsites −1 and −2, but only one structure (49, 53) has been determined for PPA in which these subsites are occupied but not covalently linked to subsite −1. This seems to be a curious observation.
Finally, subsite +3, occupied only in the V-1532 complex, has only a single water-mediated interaction with residues lining the active site cleft. This is between O2 and Gly238 (O), which was involved in a water-mediated interaction at subsite +2 in 1PPI, the first acarbose-derived inhibitor structure to be reported. However, although not meaningful from a substrate binding point of view, there are several interactions between this subsite (O1 and O6) with a symmetry-related PPA molecule, which may account for the sugar ring being observed in this subsite.
Additional Carbohydrate Binding Sites
In addition to the oligosaccharides in the substrate-binding cleft, there is direct evidence in difference Fourier maps for a number of other sites where carbohydrate is bound. These are illustrated in Figure 9. These sites presumably represent points of association with other polysaccharide chains in hydrated starch or glycogen granules, or where the enzyme binds to chains joined to the substrate oligosaccharide by α-(1,6) linkages. It is argued, for example, that this is precisely the function of the ubiquitous CBM (25). The binding of cyclodextrin well away from the active site is likely another example.
Figure 9.
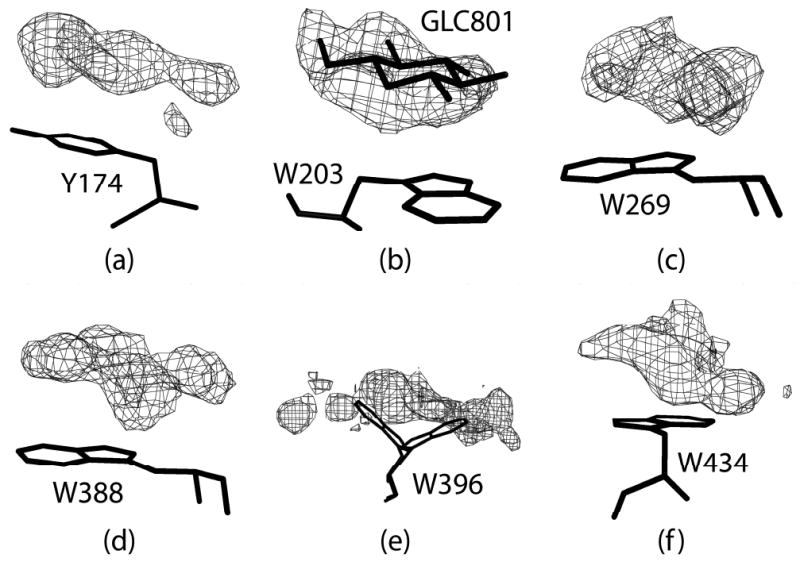
Additional or potentially additional carbohydrate surface binding sites. Electron difference density was calculated from the phases and structure factors using the final model for parts (b), (c), (e), and (f). For parts (a) and (d) water molecules in the vicinity of the density were remove from the final model before the map calculation. The density in parts (a), (b), (c), (e) and (f) are contoured at 2.0σ; in part (d) it is contoured at 2.5σ.
In addition to Trp134 in the α-cyclodextrin complex, discussed above, we observe convincing density for a glucose stacked upon Trp203 in the native structure (also seen by Payan and Qian (30)). Several other exposed aromatic side chains (see Supplementary Table 5) are also associated, as seen in Figure 9, with significant density, but density that is not sufficiently distinct and well defined that we can be sure that it represents carbohydrate. These aromatic groups include Trp434 on the CBM, Trp269 at the interface between the CBM and the catalytic domain, and possibly Trp396 and Trp388. It should be noted that several PPA structures have been reported with maltose bound to Trp388 (28, 30, 49, 53). There is also suspicious density that may be glucose closely associated with Tyr118 and Phe348.
Aromatic Residues
There is an interesting, and somewhat unique feature of pig pancreatic α-amylase that deserves some attention. The extinction coefficient in the ultraviolet for α-amylase is remarkably high; 1 mg/ml of amylase yields an OD280 of about 2.6 OD units. This is due to an unusually high percentage of aromatic amino acids, about 13%. Among these are 19 tryptophans, 19 tyrosines, and 24 phenylalanines. Most of the aromatic residues are formed in discrete dimer and trimer clusters, or in larger networks of interacting side chains. The predominant constellations of residues were parallel stacks of ring on ring, or edges perpendicular to ring faces. Supplementary Table 6 presents those found in α-amylase. Antigenic binding sites on antibodies that bind carbohydrates have frequently been observed to also exhibit a high proportion of aromatic amino acids (54-57). The same has been observed to be true of many enzymes that bind oligo- and polysaccharides.
Discussion
Structure of the Natural Substrates
It is questionable whether solution and crystallographic studies focused on linear oligosaccharides as the substrates, or as inhibitors, accurately or completely reflect the mechanisms by which α-amylase binds and degrades its natural substrates. These are, in general, considerably more complex in structure. The natural substrates of α-amylase are starch and glycogen, which are polymers of glucose linked by α-(1,4) glucosidic linkages, with occasional branch points where another chain is initiated through an α-(1,6) linkage. Starch has two polymeric components. Depending on the source, 5-40% is amylose, which is completely linear in a covalent sense and generally contains no α-(1,6) branches. The remainder is amylopectin, which exhibits an α-(1,6) branch point about every 20 glucose residues.
Glycogen, produced by animals, is more highly branched with about one in twelve residues containing both α-(1,4) and α-(1,6) links. Both starch and glycogen condense into compact, dehydrated granules with about 1 water molecule per glucose residue (58). The granules exhibit a paracrystalline structure of closely packed chains. Before hydrolysis can commence, the granule must be hydrated and transformed into a gel. In the course of this process α-amylase has been shown to participate in the disruption of the polymeric chains and the unwinding of its helices (18). While α-amylase can hydrolyze α-(1,4) bonds in a processive manner, it cannot hydrolyze α-(1,6) bonds at all, and limit dextrins, as we see in this study, are a common product of amylase digestion of starch and glycogen (4, 5).
The polymeric backbones of both starch and glycogen, and even short, linear glucose polymers, assume distinctive helical conformations. The turns of these helices are rather stiff and present contiguous hydrophobic surfaces. The helices are left-handed and generally double helical (with a 21 Å pitch, and 12 total glucose residues per turn)(11, 12), though the polymers also exist as single stranded helices with 6 residues per turn and 8 Å pitch (14, 15). When double helices are unwound, as they must be for α-amylase to catalyze hydrolysis, then the polymeric substrates are single stranded helices. The turns of these single stranded helices have been likened in their properties to cyclodextrins, which are closed rings of, generally, six to eight residues joined by α-(1,4) glucosidic linkages (14). Cyclodextrins of various sorts and sizes can, in fact, be made from amylase degradation products of starch (8, 52, 59, 60). The binding of three cyclodextrin molecules to α-amylase observed in our crystalline complex is consistent with enzymatic data based on solution studies. Kinetic analyses indicate the binding of three β-cyclodextrin molecules to α-amylase, and, furthermore, show that all three bind with very close to the same affinity, as all have virtually the same dissociation constant (9). Cyclodextrins are not natural substrates of α-amylase. They would rarely be encountered under physiological circumstances, yet they are bound in multiple copies and, in the cases of β- and γ-cyclodextrins, hydrolyzed like other oligosaccharides containing α-(1,4) glucosidic linkages. The association of cyclodextrins and their hydrolysis is not so difficult to understand, however, in light of the gross structures of the natural polymeric substrates, starch (amylose and amylopectin) and glycogen. It is quite plausible that the association with cyclodextrins reflects the binding by the enzyme of helical turns of natural substrates. There are several observations that lend support to this hypothesis.
Cyclodextrins, in two of three cases, bind in the substrate binding cleft, and they do so using the same interactions as are used to bind other oligosaccharides, e.g. the limit dextrin, the “bridging” tetrasaccharide, acarbose-derived inhibitors. As shown crystallographically, here and elsewhere, natural substrates, or their inhibitor analogues, are not flat, but have a distinctive curvature that is consistently the same. That curvature corresponds very well to that of the ring of glucose residues in γ-cyclodextrin, a readily hydrolyzable substrate (7). This curvature likely corresponds to the preferred curvature of natural, helical polysaccharides as they bind to the enzyme.
The fact that the larger cyclodextrins can be hydrolyzed by α-amylase further confirms that they not only bind, but that they bind productively. The extended binding site for α-amylase is clearly very plastic and tolerant of diverse substrate structure and local structural variation as shown by its ability to associate productively with helical polysaccharide, polysaccharides containing α-(1,6) glucose linkages that produce branch points, and circular cyclodextrin molecules having a range of sizes and properties. It is important to note, in this regard, that the diameter range of the cyclodextrins is about the same diameter as the helical turns found in starch and glycogen.
Proposed Model for Total Binding
If the linear oligosaccharide inhibitors and cyclodextrin binding observed in crystals are reflective of polysaccharide binding to the enzyme, then we can attempt to reconcile them with the binding of the natural, helical substrates. It should be noted that the limit dextrin, inhibitors and cyclodextrin II have the same direction as the tetrasaccharide and cyclodextrin I, so it is feasible that these two fragments could be linked together to give an extended polysaccharide substrate. Because γ-cyclodextrin most closely superimposes with the glucose chains of crystallographic inhibitor complexes, we initiated a modeling effort based on two γ-cyclodextrins in the binding cleft where they are observed. We assume that the cyclodextrins are surrogates for helical turns within polysaccharides.
Based on this two-turn concept, we derived a model based on the cycloamylose 26 structure (PDB entry 1C58 (61)) as shown in Figure 10a. This molecule consists of 26 glucose residues that form two anti-parallel left-handed helices of approximately two turns each with between 6 and 7 glucose residues per turn. Each helix is connected to the other at both ends by a bridging glucose to form a cyclic structure. The helical turns in cycloamylose 26 closely approximate the helical turns in V6 or V7 amylose chains; the radius is slightly greater that 4 Å and the rise per residue is about 1.35 Å.
Figure 10.
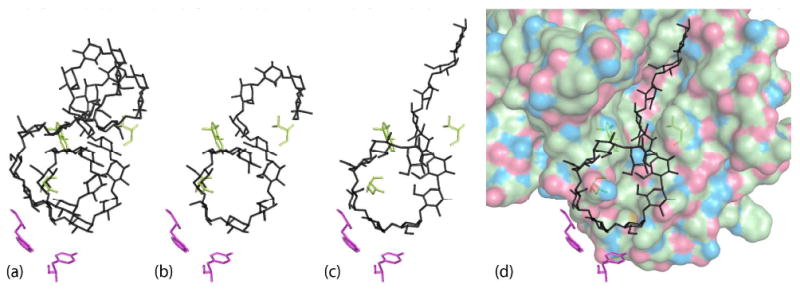
Cycloamylose-derived model for extended substrate binding. (a) Cycloamylose 26 (PDB entry: 1C58) roughly placed on cyclodextrins I and II. (b) 13 glucose units extracted from the cycloamylose model. (c) 13 glucose units after manual adjustments to relieve bad interatomic contacts and refinement against native data to get a better fit of the most tightly bound glucose residues in subsites −2 and −3 and the two central glucose units of the tetrasaccharide. (d) The final extended substrate model in the active site binding cleft.
We extracted a fragment of 13 glucose units (residues 9-21) and superimposed it onto cyclodextrins I and II (Figure 10b). Glucose 14 of the extracted fragment is the connecting residue between the helices and initiates the change of glucose orientation. This residue in our model serves the same purpose in transitioning between cyclodextrin I and cyclodextrin II. Minor adjustments were made to the oligosaccharide to alleviate penetration of atoms of the fragment into the enzyme model. The reducing end was straightened to align more closely with the inhibitors in subsites −1, +1 and +2. This fragment was refined using CNS (62) against our native data while holding constant the enzyme, the glucose ring at Trp203, and the water model that was not in conflict with the fragment being refined. This refinement gave a better fit of glucose units at subsites −1, -2 and −3 and at the tetrasaccharide binding site while reducing undesirable van der Waals contacts. The final refined model is shown in Figure 10c and d.
The model in Figure 10 is composed of 13 contiguous α-(1,4) glucosidically linked residues. It has a number of attractive features. First, it uses virtually all of the protein–carbohydrate interactions that have been observed in the amylase–oligosaccharide, inhibitor, and amylase–cyclodextrin complexes studied by X-ray crystallography. Second, it possesses a left-handed helical turn of approximately the correct diameter. Third, it is consistent with what we know of the stereochemistry of oligosaccharides and polysaccharides. Finally, the model oligosaccharide superimposes on nearly all of the corresponding glucose residues of the crystallographic complexes. We propose that the model in Figure 10c may serve as a fair representation of the complex between α-amylase and a realistic, helical, polymeric substrate.
Unwinding Double Helices
Proteins that passively (without expenditure of ATP) unwind extended, double helical molecules such as DNA and dsRNA, do so by cooperatively binding in a contiguous manner along the two complementary single chains once a “bubble” in the helix occurs. They bind contiguously because of energetically favorable protein–protein interactions between adjacent protein molecules. By altering the equilibrium between double and single stranded forms they unwind the double helices and make them available for other enzymes to operate on the nucleic acid (63). The Gene 5 protein from bacteriophage fd, the bacteriophage T4 gene 32 protein, and ssB from E. coli are examples.
α-Amylase has a similar task in that it must disrupt and unwind double helical polysaccharide. Because it expends no ATP, it likely does so by a similar mechanism. In the case of α-amylase, as with the proteins above, cooperativity would depend on favorable intermolecular interactions between neighbors along a polysaccharide chain, and these could be the interactions between 21-screw related molecules that we see in the crystal.
Bridging binding site—implications for processivity
It is noteworthy that the binding site for the tetrasaccharide and the “bridging” cyclodextrin I is a composite binding site at the juncture of two protein molecules related by a 21 screw axis. The binding of the cyclodextrin, or what we would contend would be a helical turn of a polysaccharide, benefits from significant interactions from both protein molecules. This suggests that if the model in Figure 10c is correct, or nearly so, then α-amylase may function by multiple protein molecules binding contiguously along a polysaccharide chain such that the sequential molecules are related by the same 21 operation that exists in the crystals.
Inspection of the contacts between protein molecules along the x direction in the crystal shows that several potentially favorable interactions are in fact likely. These include both hydrogen bonds and hydrophobic interactions (see Supplementary Table 7). In addition, about 634 Å2 of surface area on the two protein molecules is buried. With the bridging tetrasaccharide in place, the buried surface area increases to 1030 Å2, which suggests an attractive interface for cooperative binding (64). This is in addition to all of the favorable protein–carbohydrate interactions that would accrue by the coordinated and cooperative binding of a helical turn of the polysaccharide substrate, as represented in crystals by the “bridging” tetrasaccharide or the cyclodextrin (see Supplementary Table 1).
We appreciate the danger of interpreting lattice interactions between protein molecules in crystals physiologically. These can often be misleading and one must approach their significance with caution. Often, however, crystals do incorporate interactions having functional relevance and, thereby, provide valuable information regarding the mechanisms operative in vivo.
Further support for cooperative binding, based on the model of Figure 10c, is founded on the observation that α-amylase is a processive enzyme. With a processive mechanism of catalysis, the enzyme essentially never releases the chain that it is degrading from its grasp. Most processive enzymes are large complexes of different proteins that often have toroidal structures. The extended, linear or helical substrate is captured and brought into the center of the toroid where it cannot escape. This insures that the substrate will be processed continuously and without release. In most of these complexes, ATP is expended to provide directionality and drive function. α-Amylase is not a toroid and does not use ATP, though some energy may be provided to the process by the hydrolysis of the α-(1,4) sugar linkages.
Breyer and Matthews (33) have argued that in the absence of toroidal trapping, a processive enzyme must have an alternative property. This would be an extended binding region with numerous subsites, none of which have high specificity or high affinity for the units of the extended substrate, but which collectively bind the polymer tightly to the enzyme. This would allow the enzyme and substrate to locally loosen their grip on one another and permit the enzyme to slide along the substrate. The binding cleft of α-amylase, including the binding site for the bridging tetrasaccharide, has exactly those characteristics.
Cyclodextrin at Trp134
Cyclodextrin III in our complex is not in the substrate binding cleft, but an appreciable distance away. It is generally agreed, however, that α-amylase, like other polysaccharide hydrolases, must function in the dense environment of hydrated starch and glycogen granules, some of which are paracrystalline in nature (16-19). Hence, an amylase molecule likely interacts not only with the chain it is currently hydrolyzing, but must assist as well in the progressive disruption of the polysaccharide network. Hence, they almost certainly associate with multiple polysaccharide chains. It has been postulated, for example, that this is the explicit function of the amylase CBM (25).
The “accessory” cyclodextrin (III), which has about the same dissociation constant with the enzyme as the other two cyclodextrins (9), binds to an unusual feature of the protein, a fully solvated and disordered surface tryptophan. We find not only the cyclodextrin at this site, but nearby we have identified a glucose residue associated with Trp203 as was observed by Payan and Qian (30). The observations support the contention that the “accessory” cyclodextrin is, indeed, physiologically relevant. It possibly indicates the mode of interaction of the enzyme with other polysaccharide chains during the process of disruption of condensed substrate.
Carbohydrate binding module
Most polysaccharide hydrolases, such as chitinases and xylanases, but also including α-amylase, possess a small domain or module which are currently classified into seven structural folds predominated by the antiparallel β-barrel or β-sandwich fold (25). In PPA this is composed of the carboxy terminal residues 404 through 496. The function of this domain is not certain. Generally, of those hydrolases possessing such a domain, that domain is a considerable distance from the substrate-binding cleft. Nevertheless, it is commonly believed to be involved in the binding of polysaccharide away from the substrate cleft, and probably assists in the disruption of densely packed polysaccharide strands. As a consequence, it has been given the name of “carbohydrate binding module”, or CBM. For some other hydrolases, complexes with oligosaccharides have been observed in crystals of their CBMs (25).
In α-amylase, in spite of the ready availability of oligosaccharides of considerable variety, we did not find, with one minor exception, carbohydrate bound by the amylase CBM. Certainly, it forms no extensive interactions with oligosaccharides in our crystals, and this is not due to occlusion by other molecules in the lattice (the crystals contain about 70% solvent). The only exception is the possible presence of a glucose residue bound to Trp434 (see Figure 9f). While surface-exposed aromatic residues are signatures of oligosaccharide binding proteins, the CBM of PPA, which contains eleven aromatic residues (∼12%), has only two that are accessible for binding carbohydrate, Trp434 and Tyr468. Perhaps the amylase CBM does not bind carbohydrate of arbitrary structure, but is specific for local structural features of the polysaccharide substrate that are eliminated by the hydrolysis of glycogen that occurs prior to crystallization.
Tetrasaccharide
An interesting question is raised by the shared tetrasaccharide regarding the crystallization of PPA. The tetrasaccharide cannot be shared in solution, as this would imply the existence of extended strings of weakly linked molecules having screw symmetry, which subsequently crystallized. This is impossible. The only explanation is that the tetrasaccharide is bound to, and is carried through the purification process by a single PPA molecule in solution, and that sharing occurs as PPA associates along the x direction as the crystal is formed.
This being the case, then the question becomes: Is the tetrasaccharide bound to the half site involving amino acids Tyr276 and Trp284 at the reducing end of the binding cleft, or by the half site containing amino acids Thr52 and Asn53 at the nonreducing end of the binding cleft? Given the extensive participation of aromatic, and particularly tryptophan residues in the binding of carbohydrate elsewhere on PPA in addition to three hydrogen bonds involving residues Lys261, Glu272, and Asn279, it seems most likely that the tetrasaccharide is carried in solution by the half site at the reducing end of the cleft.
Supplementary Material
Abbreviations
- PPA
pig pancreatic α-amylase
- CBM
carbohydrate binding module
- aa
amino acids
- PDB
Protein Data Bank
Footnotes
This work was supported by NIH grant GM 080412 to AM.
Atomic coordinates and structure factors for the structures presented here have been deposited in the Protein Data Bank at the Research Collaboratory for Structural Bioinformatics with PDB codes 3L2L and 3L2M.
Supporting Material Available: Supplementary Tables 1-7 and Supplementary Figure 1 provide additional results in support of topics included in the text. These include lists of: specific interactions at the tetrasaccharide binding site (Tables 1 and 7); specific interactions between the limit dextrin and PPA (Table 2); glucopyranosyl ring puckering parameters (Table 3); glycosidic linkage torsion angles and O...O distances (Table 4); calculations of solvent accessible aromatic residues (Table 5); and, contacts between aromatic residues as a description of the constellations of the aromatic residues in PPA (Table 6). Figure 1 illustrates the specific PPA-limit dextrin interactions at each subsite along the limit dextrin. This material can be accessed free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fischer E, Stein E. Alpha amylases. In: Boyer P, Lardy H, Myrback K, editors. The Enzymes. Vol. 4. Academic Press; New York: 1960. pp. 333–343. [Google Scholar]
- 2.Thoma JA, Spradlin JE, Dygert S. Plant and animal amylases. In: Boyer PD, editor. The Enzymes. Vol. 5. Academic Press; New York: 1971. pp. 115–199. [Google Scholar]
- 3.Caldwell ML, Booher LE, Sherman HC. Crystalline amylase. Science. 1931;74:37–37. doi: 10.1126/science.74.1906.37. [DOI] [PubMed] [Google Scholar]
- 4.Heller J, Schramm M. α-Amylase limit dextrins of high molecular weight obtained from glycogen. Biochim Biophys Acta. 1964;81:96–100. [Google Scholar]
- 5.Levitzki A, Heller J, Schramm M. Specific precipitation of enzyme by its substrate: the α-amylase–macrodextrin complex. Biochim Biophys Acta. 1964;81:101–107. [Google Scholar]
- 6.Loyter A, Schramm M. Multimolecular complexes of α-amylase with glycogen limit dextrin. J Biol Chem. 1966;241:2611–2617. [PubMed] [Google Scholar]
- 7.Braun PJ, French D, Robyt JF. The effect of substrate modification on porcine α-amylase subsite binding: hydrolysis of substrates containing 2–deoxy-D-glucose and 2-amino-2-deoxy-D-glucose. Arch Biochem Biophys. 1985;242:231–239. doi: 10.1016/0003-9861(85)90497-7. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto T, Kitahata S, Hiromi K, Ohnishi M, Minamiura N, Shinke R, Okada S, Komaki T, editors. Handbook of Amylases and Related Enzymes; Their Sources, Isolation Methods, Properties and Applications. Pergamon Press; New York: 1988. pp. 1–274. [Google Scholar]
- 9.Mora S, Simon I, Elodi P. Studies on the active center of pancreatic amylase I. binding of β–cyclodextrin. Mol Cell Biochem. 1974;4:205–209. doi: 10.1007/BF01731482. [DOI] [PubMed] [Google Scholar]
- 10.Koukiekolo R, Desseaux V, Moreau Y, Marchis-Mouren G, Santimone M. Mechanism of porcine pancreatic alpha-amylase. Inhibition of amylose and maltopentaose hydrolysis by alpha-, beta- and gamma-cyclodextrins. Eur J Biochem. 2001;268:841–848. doi: 10.1046/j.1432-1327.2001.01950.x. [DOI] [PubMed] [Google Scholar]
- 11.Imberty A, Perez S. A revisit to the 3-dimensional structure of B-type starch. Biopolymers. 1988;27:1205–1221. [Google Scholar]
- 12.Imberty A, Chanzy H, Perez S, Buléon A, Tran V. Double-helical nature of the crystalline part of A-starch. J Mol Biol. 1988;201:365–378. doi: 10.1016/0022-2836(88)90144-1. [DOI] [PubMed] [Google Scholar]
- 13.Ring SG, Miles MJ, Morris VJ, Turner R, Colonna P. Spherulitic crystallization of short chain amylose. Intl J Biol Macromol. 1987;9:158–160. [Google Scholar]
- 14.Buléon A, Delage MM, Brisson J, Chanzy H. Single crystals of V amylose complexed with isopropanol and acetone. Int J Biol Macromol. 1990;12:25–33. doi: 10.1016/0141-8130(90)90078-o. [DOI] [PubMed] [Google Scholar]
- 15.Brisson J, Chanzy H, Winter WT. The crystal and molecular structure of VH amylose by electron diffraction analysis. Int J Biol Macromol. 1991;13:31–39. doi: 10.1016/0141-8130(91)90007-h. [DOI] [PubMed] [Google Scholar]
- 16.Holtzer RL, van Lancker JL, Swift H. Release of amylase from zymogen granules and microsomes. Arch Biochem Biophys. 1963;101:439–444. doi: 10.1016/0003-9861(63)90500-9. [DOI] [PubMed] [Google Scholar]
- 17.Levitt MD, Duane WC, Cooperba SR. Study of macroamylase complexes. J Laboratory and Clinical Medicine. 1972;80:414–419. [PubMed] [Google Scholar]
- 18.Leloup V, Colonna P, Marchis-Mouren G. Mechanism of the adsorption of pancreatic alpha-amylase onto starch crystallites. Carbohydr Res. 1992;232:367–374. doi: 10.1016/0008-6215(92)80069-d. [DOI] [PubMed] [Google Scholar]
- 19.Walker GJ, Hope PM. The action of some α-amylases on starch granules. Biochem, J. 1963;86:452–462. doi: 10.1042/bj0860452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muus J, Brockett FP, Connelly CC. The effect of various ions on stability of crystalline salivary amylase in solution. Arch Biochem Biophys. 1956;65:268–277. doi: 10.1016/0003-9861(56)90193-x. [DOI] [PubMed] [Google Scholar]
- 21.Vallee BL, Stein EA, Sumerwell WN, Fischer EH. Metal content of α-amylases of various origins. J Biol Chem. 1959;234:2901–2905. [PubMed] [Google Scholar]
- 22.Levitzki A, Steer ML. Allosteric activation of mammalian α-amylase by chloride. Eur J Biochem. 1974;41:171–180. doi: 10.1111/j.1432-1033.1974.tb03257.x. [DOI] [PubMed] [Google Scholar]
- 23.Qian M, Haser R, Payan F. Structure and molecular model refinement of pig pancreatic α-amylase at 2.1 Å resolution. J Mol Biol. 1993;231:785–799. doi: 10.1006/jmbi.1993.1326. [DOI] [PubMed] [Google Scholar]
- 24.Larson SB, Greenwood A, Cascio D, Day J, McPherson A. Refined molecular structure of pig pancreatic α–amylase at 2.1 Å resolution. J Mol Biol. 1994;235:1560–1584. doi: 10.1006/jmbi.1994.1107. [DOI] [PubMed] [Google Scholar]
- 25.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robyt JF, French D. Multiple attack and polarity of action of porcine pancreatic α-amylase. Arch Biochem Biophys. 1970;138:662–670. doi: 10.1016/0003-9861(70)90394-2. [DOI] [PubMed] [Google Scholar]
- 27.Nahoum V, Roux G, Anton V, Rougé P, Puigserver A, Bischoff H, Henrissat B, Payan F. Crystal structures of human pancreatic α-amylase in complex with carbohydrate and proteinaceous inhibitors. Biochem J. 2000;346:201–208. [PMC free article] [PubMed] [Google Scholar]
- 28.Machius M, Vértesy L, Huber R, Wiegand G. Carbohydrate and protein-based inhibitors of porcine pancreatic α-amylase: structure analysis and comparison of their binding characteristics. J Mol Biol. 1996;260:409–421. doi: 10.1006/jmbi.1996.0410. [DOI] [PubMed] [Google Scholar]
- 29.Qian M, Nahoum V, Bonicel J, Bischoff H, Henrissat B, Payan F. Enzyme–catalyzed condensation reaction in a mammalian α-amylase. High resolution structural analysis of an enzyme–inhibitor complex. Biochemistry. 2001;40:7700–7709. doi: 10.1021/bi0102050. [DOI] [PubMed] [Google Scholar]
- 30.Payan F, Qian M. Crystal structure of the pig pancreatic α-amylase complexed with malto-oligosaccharides. J Prot Chem. 2003;22:275–284. doi: 10.1023/a:1025072520607. [DOI] [PubMed] [Google Scholar]
- 31.Robyt JF, French D. Multiple attack hypothesis of α-amylase action: action of porcine pancreatic, human salivary, and aspergillus oryzae α-amylases. Arch Biochem Biophys. 1967;122:8–16. doi: 10.1016/0003-9861(67)90118-x. [DOI] [PubMed] [Google Scholar]
- 32.Robyt JF, French D. The action pattern of porcine pancreatic α-amylase in relationship to the substrate binding site of the enzyme. J Biol Chem. 1970;245:3917–3927. [PubMed] [Google Scholar]
- 33.Breyer WA, Matthews BW. A structural basis for processivity. Protein Sci. 2001;10:1699–1711. doi: 10.1110/ps.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilles C, Astier JP, Marchis-Mouren G, Cambiiau C, Payan F. Crystal structure of pig pancreatic α-amylase isoenzyme II, in complex with the carbohydrate inhibitor acarbose. Eur J Biochem. 1996;238:561–569. doi: 10.1111/j.1432-1033.1996.0561z.x. [DOI] [PubMed] [Google Scholar]
- 35.Loyter A, Schramm M. The glycogen–amylase complex as a means of obtaining highly purified α-amylases. Biochim Biophys Acta. 1962;65:200–206. doi: 10.1016/0006-3002(62)91039-9. [DOI] [PubMed] [Google Scholar]
- 36.Brünger AT. Crystallographic refinement by simulated annealing; application to a 2.8 Å resolution structure of aspartate aminotransferase. J Mol Biol. 1988;203:803–816. doi: 10.1016/0022-2836(88)90211-2. [DOI] [PubMed] [Google Scholar]
- 37.Brünger AT. Simulated annealing in crystallography. Annu Rev Phys Chem. 1991;42:197–223. [Google Scholar]
- 38.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.McPherson A, Rich A. X-ray crystallographic analysis of swine pancreas α–amylase. Biochim Biophys Acta. 1972;285:493–497. doi: 10.1016/0005-2795(72)90340-6. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. Procheck: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 43.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallographica. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallographica. 1991;A47:392–400. [Google Scholar]
- 45.Saenger W. Principles of Nucleic Acid Structure. Verlag-Springer; New York: 1983. p. 70.p. 86. [Google Scholar]
- 46.DeLano WL. The PyMOL Molecular Visualization System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- 47.Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321:557–559. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian M, Haser R, Buisson G, Duée E, Payan F. The active center of a mammalian α-amylase. Structure of the complex of a pancreatic α-amylase with a carbohydrate inhibitor refined to 2.2-Å resolution. Biochemistry. 1994;33:6284–6294. doi: 10.1021/bi00186a031. [DOI] [PubMed] [Google Scholar]
- 49.Qian M, Spinelli S, Driguez H, Payan F. Structure of a pancreatic α-amylase bound to a substrate analogue at 2.03 Å resolution. Protein Sci. 1997;6:2285–2296. doi: 10.1002/pro.5560061102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishikawa K, Hirata H. New substrate specificity of modified porcine pancreatic α-amylase. Arch Biochem Biophys. 1989;272:356–363. doi: 10.1016/0003-9861(89)90229-4. [DOI] [PubMed] [Google Scholar]
- 51.Szejtli J. Introduction and General Overview of Cyclodextrin Chemistry. Chem Rev. 1998;98:1743–1753. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 52.Szejtli J. Cyclodextrin Technology. Vol. 1. Springer; New York: 1988. pp. 1–484. [Google Scholar]
- 53.Qian M, Ajandouz EH, Payan F, Nahoum V. Molecular basis of the effects of chloride ion on the acid-base catalyst in the mechanism of pancreatic α-amylase. Biochemistry. 2005;44:3194–3201. doi: 10.1021/bi048201t. [DOI] [PubMed] [Google Scholar]
- 54.Cygler M, Rose DR, Bundle DR. Recognition of a cell-surface oligosaccharide of pathogenic salmonella by an antibody Fab fragment. Science. 1991;253:442–445. doi: 10.1126/science.1713710. [DOI] [PubMed] [Google Scholar]
- 55.Cygler M. Recognition of carbohydrates by antibodies. Res Immunol. 1994;145:36–40. doi: 10.1016/s0923-2494(94)80040-5. [DOI] [PubMed] [Google Scholar]
- 56.Larson SB, Day JS, Glaser S, Braslawsky G, McPherson A. The structure of an antitumor CH2–domain-deleted humanized antibody. J Mol Biol. 2005;348:1177–1190. doi: 10.1016/j.jmb.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 57.McCallum RM, Martin ACR, Thornton JM. Antibody–antigen interactions: contact analysis and binding site topography. J Mol Biol. 1996;262:732–745. doi: 10.1006/jmbi.1996.0548. [DOI] [PubMed] [Google Scholar]
- 58.Copeland L, Blazek J, Salman H, Tang MC. Form and functionality of starch. Food Hydrocolloids. 2009;23:1527–1534. [Google Scholar]
- 59.Biwer A, Antranikian G, Heinzle E. Enzymatic production of cyclodextrins. Appl Microbiol Biotechnol. 2002;59:609–617. doi: 10.1007/s00253-002-1057-x. [DOI] [PubMed] [Google Scholar]
- 60.Wind RD, Liebl W, Buitelaar RM, Penninga D, Spreinat A, Dykhuizen L, Bahl H. Cyclodextrin formation by the thermostable alpha amylase of T. thermosulfurigens EM1 and reclassification of the enzyme as a cyclodextrin glycosyltransferase. Appl Environ Microbiol. 1995;61:1257–1265. doi: 10.1128/aem.61.4.1257-1265.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gessler K, Uson I, Takaha T, Krauss N, Smith SM, Okada S, Sheldrick GM, Saenger W. V-amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucose residues (cyclomaltohexaicosaose) Proc Natl Acad Sci USA. 1999;96:4246–4251. doi: 10.1073/pnas.96.8.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 63.von Hippel PH, Jensen DE, Kelly RC, McGhee JD. Molecular approaches to the interaction of nucleic acids with “melting” proteins. In: Vogel HJ, editor. Nucleic Acid–Protein Recognition. Academic Press; New York: 1977. pp. 66–89. [Google Scholar]
- 64.Dasgupta S, Lyer GH, Bryant SH, Lawrence CE, Bell JA. Extent and nature of contacts between protein molecules in crystal lattices and between subunits of protein oligomers. Proteins. 1997;28:494–514. doi: 10.1002/(sici)1097-0134(199708)28:4<494::aid-prot4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.