

Abstract
Assembly of retrovirus particles is promoted by interaction of the Gag polyprotein with RNA. Nonspecific RNA association with the nucleocapsid domain (NC) of Gag induces the dimerization of Gag through protein-protein contacts in the capsid domain (CA), followed by higher-order assembly to form the immature virus particle. NMR relaxation studies were conducted to investigate the initial steps of Rous sarcoma virus (RSV) assembly by examining the association with nucleic acid of a fragment of Gag comprising the C-terminal domain of CA (CTD) postulated to mediate Gag dimerization, the spacer region between CA and NC (SP), and NC. This fragment, CTD-SP-NC (residues 394 to 577), spans the critical SP region and allows assessment of this key Gag-nucleic acid interaction in the context of the Gag polyprotein rather than the isolated domains. Mainchain amide relaxation of CTD-SP-NC was measured in the absence and presence of (GT)4, an 8-mer DNA oligonucleotide that binds tightly to the polyprotein but is too short to promote Gag dimerization. The results show that the CTD and NC domains tumble independently. In contrast, the two zinc finger domains within NC are rotationally coupled in both the unbound and bound states, even though only the first zinc finger appears to make direct contact with (GT)4. In addition, the NMR data indicate that SP and flanking residues undergo a conformational exchange process that is slowed in the presence of (GT)4. This region around SP where relaxation is strongly affected by (GT)4 binding is nearly identical to the assembly domain defined previously by mutagenesis studies. Other changes in relaxation induced by (GT)4 implicate conformational perturbations of helices 1 and 4 in CTD. Based on the combined data, we propose a model for the promotion of Gag dimerization by RNA association in which NC-RNA binding disrupts an assembly-inhibitory, intramolecular interaction involving SP and CTD. Disruption of this intramolecular interaction is proposed to enhance the accessibility of the Gag dimer contact surface and release the assembly domain to promote intermolecular oligomerization.
Keywords: retrovirus, virus assembly, gag polyprotein, multidomain protein dynamics
The Gag polyprotein, comprising matrix (MA), capsid (CA), nucleocapsid (NC) and other smaller proteins depending on the viral genus, is the necessary and sufficient protein component for retroviral assembly. In immature virus particles, the Gag polyprotein is arranged radially with the N-terminus of Gag associated with the membrane (1, 2). Assembly of the immature particle involves interaction of the Gag NC domain with viral RNA, accumulation of Gag at the plasma membrane through the association of the N-terminal MA domain, and oligomerization through Gag-Gag interactions in the CA domain and closely adjoining regions to form the protein core of an immature virion. NC-RNA association promotes by an unknown mechanism the oligomerization mediated by CA-CA domain contacts and higher-order assembly to form the immature viral core. Late in the budding process Gag is cleaved by the viral protease into the three structural proteins, MA, CA, and NC. This proteolytic processing leads to a change in virus particle morphology, and maturation of the virus into its infectious form. During maturation the liberated CA forms a shell that surrounds the NC-RNA complex and the viral enzymes reverse transcriptase and integrase.
The amino-acid sequence similarity between the different genera of retroviruses is less than 25-30%; however, the tertiary structures of the three major structural proteins of Gag are highly conserved (3, 4, 5, 6, 7). The isolated domain structures of individual Gag proteins have been determined in both solid and solution states for diverse retroviruses. MA is predominantly α-helical (8, 9, 10, 11). CA is a two domain α-helical protein consisting of an N-terminal (NTD) and C-terminal (CTD) domain that are connected by a flexible linker (3, 4, 12, 13, 14). Isolated NC is a highly flexible protein possessing one or two 14-residue CCHC zinc finger domains, which are the only structured regions of NC (15, 16, 17, 18, 19).
Besides HIV-1 (genus lentiviruses), one of the most important model systems to study retrovirus assembly is Rous sarcoma virus (RSV, genus alpharetroviruses or avian sarcoma and leukemia viruses). In vitro assembly studies have established that association of Gag with nucleic acid creates an assembly “building block” to initiate RSV assembly. Gag interaction with either RNA or DNA for in vitro assembly promotes dimerization of two Gag molecules through protein-protein contacts postulated to occur in the CTD (20). Artificially induced dimerization of Gag molecules lacking an NC domain also leads to assembly and budding in cells (21). Oligomerization and assembly to regular higher-order structure require as well the presence of the NTD, and it is suggested that NTD-NTD contacts lead to Gag assembly into virion-like particles (VLP) while CTD-CTD contacts stabilize the transient dimer building block (22, 23, 24). Due to structural similarities between retroviral CA and SCAN domains, it has been suggested that CA dimerization may involve domain swapping, similar to that occurring between two SCAN domains (25). Clearly, dimerization of CA is not a simple process. While RSV CTD and CA are monomeric in solution even at millimolar concentrations, HIV-1 CA dimerizes at modest concentrations. Further, RSV CTD dimers observed from crystals obtained at different conditions of pH form five unique interfaces (26), one of which is similar to that observed for HIV-1 CTD crystals (6), and two closely resemble the interface of HIV-1 CTD in crystals of the complex with a peptide that inhibits assembly (27).
In many retroviruses, including HIV-1 and RSV, a short “spacer” sequence separates the CA and NC domains of Gag, and is proteolytically removed during virus maturation. This sequence, called SP in RSV (12 residues) and SP-1 or p2 in HIV-1 (14 residues) has been shown by mutagenesis to be absolutely required for proper immature assembly. RSV SP also contains an internal protease cleavage site (28). In HIV-1 (29, 30) the short stretch of 11 or 12 residues between the last helix of the CTD and SP-1 is also critical for the correct formation of immature virus particles, while in the case of RSV, an ‘assembly domain’ consisting of the last residues of CA, all of SP, and the first 4 residues of NC is critical (31). In structures determined for CA or CTD from either HIV-1 (6) or RSV (3, 4), these residues are disordered and without stable secondary or tertiary structure. The mechanistic role of spacer in assembly is unknown, although part or all of this sequence in HIV-1 is predicted to be helical, which implicates a possible helix-coil transition (32, 33, 34).
In electron cryotomography studies of immature HIV-1 particles, density was observed below the hole made by the CA NTD hexamer corresponding to SP and appears to be holding the Gag hexamer together (35, 36). The density was fit with a 6-helix bundle, consistent with previous data suggesting that SP-1 forms an α-helix (29-31). Briggs et al. point out that this is a working model and at the current resolution other arrangements of SP1 can be fit into the density (35).
We report the results from NMR studies to investigate the initial steps of assembly by examining the association of RSV Gag with nucleic acid. 15N relaxation studies were conducted to characterize the dynamics of an RSV Gag fragment in the absence and presence of (GT)4, an 8-mer DNA oligonucleotide of nonspecific sequence that binds tightly to the polyprotein but is too short to promote Gag dimerization (22, 37, 38). The aim was to examine properties in the context of the Gag polyprotein, rather than the isolated domains, and thus a Gag fragment that spans the critical SP region and comprises CTD (394 - 476), SP (477 - 488), and NC (489 - 577) (CTD-SP-NC) (Figure 1) was used. An analogous Gag fragment from HIV-1 has been characterized in the free state by NMR relaxation (39). The overall trends observed for unbound RSV CTD-SP-NC relaxation are generally consistent with the conclusions for HIV-1, although certain differences exist in the details of the motional properties, particularly those of SP. New to the work reported here are the results that characterize the effect of binding (GT)4 on CTD-SP-NC dynamics. These data, along with the identification of an assembly domain by mutagenesis (21, 31), and the discovery of inhibition of HIV-1 assembly by a peptide that binds to the CTD (27, 40), suggest a model for how nucleic acid binding to Gag initiates Gag dimerization, and in turn leads to assembly.
Figure 1.

Schematic representation of the RSV Gag polyprotein and the CTD-SP-NC polyprotein construct used in this study. CTD is from residue 394 to 474, SP from 475 to 488, and NC from 489 to 577. NC has two zinc fingers, ZF1 (509 – 522) and ZF2 (535 – 548) that together makeup the NC core (509 – 548). The horizontal lines in SP represent the PR cleavage sites between CTD and NC at 476 and 488. A third cleavage site also exists within SP at 479. The amino acid sequence comprising the “assembly domain” from residue 469 at the C-terminus of CTD to residue 492 at the N-terminus of NC is shown.
Materials and Methods
Expression and purification of Recombinant RSV CTD-SP-NC polyprotein
Protein expression and purification of RSV CTD-SP-NC were carried out as described by Ma and Vogt (22) with some modifications. For protein expression in Escherichia coli, the RSV pET-CTD-NC plasmid was transformed into BL21(DE3) pLysS cells. Uniformly 15N- and 15N/13C-labeled proteins were prepared by growing the cells in M9 minimal media with 15NH4Cl (1 g/L) and 12C6-D-glucose (2 g/L) or 15NH4Cl (1 g/L) and 13C6-D-glucose (2 g/L), respectively, supplemented with 100 μg/L ampicillin and 34 μg/L cholramphenicol. A starter culture of 25 mL of M9 inoculated from a single colony was grown at 37°C overnight. The starter culture was added to 1 L of M9 supplemented with 0.1 mM CaCl2 and grown at 37°C to an OD600 of 0.6 before induction with 1mM IPTG (isopropyl-b-D-thiogalactopyransoide). Induction was carried out for 4 hours before the cells were harvested by centrifugation (6,000 × g) at 4°C and stored at −20°C. The cells were thawed on ice, suspended in lysis buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 10% Glycerol, 0.1% N-P40, 50 mM DTT, 20 μM ZnCl2, and protease inhibitor cocktail for His tag proteins (Sigma-Aldrich)), and sonicated on ice by three bursts of 2 min. Cell debris was pelleted by centrifugation at 45K for 3 hours in a Type 60 Ti rotor (Beckman). The remaining nucleic acid was removed by polyethylenimine (PEI) precipitation using final concentration of 0.3%(w/v) PEI. The protein was then precipitated with a final concentration of 40%(w/v) ammonium sulfate and dialyzed against low salt buffer (20mM Tris pH 7.5, 100mM NaCl, 50mM DTT, 20μM ZnCl2, and protease inhibitor cocktail) overnight. After dialysis, the protein was applied to a pre-equilibrated Phosphocellulose column and eluted from the column with high salt buffer (20mM Tris, pH 7.5, 500mM NaCl, 50mM DTT, 20μM ZnCl2). Purified protein was concentrated to approximately 1mM and dialyzed against NMR buffer (10mM Sodium Phosphate, 150mM NaCl, 20μM ZnCl2, 1mM DTT). Prior to NMR data collection, 10% (v/v) 2H20 was added to the samples.
NMR Spectroscopy
All NMR spectra were acquired at 25°C on a Varian Inova 600 spectrometer equipped with 5-mm [1H, 15N, 13C] triple-resonance z axis pulsed-field gradient probes. Backbone assignments were made by using standard triple-resonance experiments 3D HNCACB, CBCA(CO)HN, C(CO)NH, and HC(CO)HN recorded on uniformly 15N/13C-labeled RSV CTD-SP-NC. NMR data were processed with NMRPIPE (41) and analyzed with SPARKY (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Longitudinal relaxation rates (R1), transverse relaxation rates (R2), and {1H}-15N steady-state heteronuclear NOE (XNOE) for backbone 15N nuclei were measured with 2D inverse-detected water flip-back pulse sequences using conventional procedures (42, 43) on uniformly 15N-labeled RSV CTD-SPNC. The R1 data were collected with 10, 20, 40, 80, 160, 320, 640, 1200, and 2000 ms relaxation recovery delays, and the R2 data were collected with 10, 30, 50, 70, 90, 110, 130, and 190 ms relaxation recovery delays. A recovery delay of 1.5 s was applied between scans. The {1H}-15N NOEs were measured from 15N-HSQC spectra recorded with and without 1H presaturation. A window function with Gaussion line broadening of −15 Hz was applied in the 1H dimension to resolve resonances in the crowded core region of the 2D 1H-15N spectra.
Heteronuclear Relaxation Rates
15N relaxation data were measured at 600 MHz field strength and in duplicate. The intensities of the signals of the cross-peaks in the 2D 1H-15N spectra were measured with SPARKY (T. D. Goddard and D. G. Kneller, University of California, San Francisco). The R1 and R2 relaxation rates of the 15N nuclei were estimated by a least-squares fit of a two-parameter exponential function. Only well resolved peaks with well-fit decay curves were selected for further analysis. Uncertainties of the R1 and R2 rates were estimated from the root-mean-square noise level in the spectra. The {1H}-15N steady-state heteronuclear NOE interactions are reported using the relationship NOE-1 = I/I0, where I and I0 are signal intensities corresponding to spectra with and without 1H saturation respectively. Uncertainties in the {1H}-15N steady-state heteronuclear NOE values were estimated from individual data sets collected in duplicate.
Heteronuclear relaxation analysis
R1 and R2 relaxation rates and the steady-state nuclear Overhauser enhancement of the 15N nucleus from dipolar interaction with the bonded proton and from 15N chemical shift anisotropy are given by (44)
| (1) |
where the dipolar constant, d, and chemical shift anisotropy (CSA) interaction constant, c, are:
| (2) |
μ0 is the permeability of free space, γH and γN are the gyromagnetic ratio of 1H and 15N, respectively, ħ is Planck's constant divided by 2π, rNH is the average N-H bond length, 1.02Å, and σP−σ⊥ is the difference between parallel and perpendicular components of the axially symmetric chemical shift tensor for amide 15N and equal to −160 ppm. In the event that a conformational exchange process occurs on a timescale longer than the overall rotational correlation time, an additional term, Rex, (not shown in eqn. 1) contributes to the measured R2.
Motional Models
General relaxation behavior and the trends associated with (GT)4 were considered using four models for molecular motion: a single correlation time where overall rotation is either (1) isotropic or (2) anisotropic with axial symmetry; (3) Lipari-Szabo model-free analysis of two independent motions, one for overall rotation and one for internal motion on a much faster timescale (>50x) than overall rotation; and (4) was the same as 3 except that the internal motion is only somewhat faster than overall rotation (10-50x).
Motional model 1
In the simplest analysis, relaxation is accounted for by one rotational correlation function with correlation time τm. For a globular domain, this model corresponds to spherical, rigid body that tumbles in solution with isotropic rotation and no significant internal motion. For non-globular portions of the polyprotein, the model applies to flexible regions with dynamics that can be fit by a single time dependence on a relatively fast timescale, less than 1 to 2 ns. The spectral density for isotropic rotation:
| (3) |
The overall rotational correlation time, τm, is related to the isotropic rotational diffusion rate, Diso, by τm = 1/ 6Diso.
Motional model 2
A rigid-body, axially symmetric, folded domain tumbles anisotropically, as described by two diffusion constants. For an axially symmetric diffusion tensor, Dzz = D□ and Dxx = Dyy = D⊥ , where Dxx, Dyy and Dzz are the principal components of the molecular rotational diffusion tensor. The spectral density comprises three components (45, 46):
| (4) |
where
| (5) |
and the coefficients
| (6) |
with zd being the z-component of the NH unit vector in the principal axis frame of the diffusion tensor. The relative correlation time is maximal or minimal for an N-H vector aligned along xy or z, respectively.
Motional model 3
Internal mobility is taken into account using the model-free approach (47). Internal motion alters the spectral density depending on the amplitude and the timescale of the motion. For very fast (relative to τm) internal motion, the spectral density is scaled by the generalized order parameter, S2, with no time dependence. We examine the effect of internal motion for the case of isotropic overall rotation. (Fast internal motion with anisotropic overall rotation is also a reasonable model, but, the combined effects of internal motion and rotational anisotropy are not required for the analysis here.) For fast internal motion and isotropic rotation, J (ω) becomes
| (7) |
Motional model 4
For the case of longer timescale internal motion, an additional time dependent term is required (47):
| (8) |
where and τint is the correlation time of the internal motion. The Lipari-Szabo formalism requires that the internal and overall tumbling motions are uncoupled. The condition that τint is much shorter than τm satisfies this requirement and is needed to accurately determine τint from experiment (48). Given τm ~ 1 to 10 ns, eqn. (7) applies for τint < 10 ps, while eqn. (8) holds for τint ~ 10 to 100 ps.
Application
Model 1 or 2 are applicable to rigid core regions of globular domains with spherical or axially symmetric shape, respectively, and correlation times typically longer than 4-5 ns. Models 3 and 4 correspond to regions within a globular domain that exhibit internal mobility on timescales faster than the overall rotation of the domain. Flexible linkers that behave as random-flight chains, without folded structure, are treated using model 1 with correlation times less than 1-2 ns. More elaborate analyses of domain motions are the “extended” model-free approach (49) and wobble-in-a-cone model (50, 51), which in principal could be applied to CTD-SP-NC. Nevertheless, the differences in predicted relaxation rates compared to the extended model-free approach are small considering the descriptive analysis sought in this study. Analysis using the wobble-in-a-cone model demands additional relaxation data and is an area for future studies. An overall rotational correlation time (τm) was estimated from the 15N relaxation data for structured domains in CTD-SP-NC by the numerical procedure in TENSOR (52). Motional parameters for linker regions were estimated graphically based on agreement of the 15N relaxation data with theoretical plots.
Molecular weight dependence of τm
The expected value of τm for a folded domain of a given molecular weight polypeptide is taken from the data reported by Garcia de la Torre (53). Correlation times were determined from experimental measurements of rotational diffusion and were predicted by hydrodynamic calculations for proteins ranging in molecular weight from 3 kDa to 27 kDa, and the values are plotted in Figure 2. The data were fit by a least-squares linear regression to give the dependence of τm on molecular weight: τm= 0.68 ns/kDa.
Figure 2.

Dependence of molecular correlation time on molecular weight determined empirically. Values taken from reference (53) for estimates from analysis of experimental data (solid squares) and calculated from structure (open diamonds). Linear regression: τ (ns) = 0.68 MW (kDa).
Results and Discussion
Heteronuclear Relaxation of Unbound CTD-SP-NC
15N longitudinal (R1) and transverse (R2) relaxation rates and steady-state nuclear Overhauser effects (NOE+1=I/I0) were measured in duplicate at a magnetic field strength of 600 MHz (11.74 T) for resolved peaks of CTD-SP-NC in the absence (Figure 3) and presence of (GT)4 (see below). In both the free and (GT)4 complex of the polyprotein, data points are absent for residues lacking backbone assignments, which include residues in the inter zinc finger linker (527, 528, 531, 532) and residues at the carboxy terminus of NC (552 – 559, and 564 – 574). Values are also missing because of peak overlap (residues 414, 462, 430, 490, 482, 509) or peak broadening upon addition of (GT)4 (residues 478 – 496).
Figure 3.
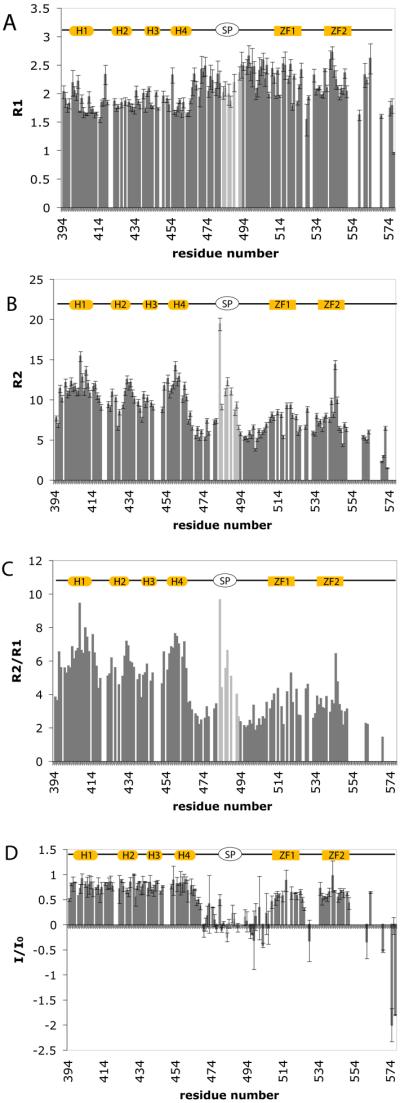
15N relaxation data at 600 MHz for RSV CTD-SP-NC in the unbound state. A. Longitudinal relaxation rates, R1 (1/T1). B. Transverse relaxation rates R2 (1/T2). C. R2/R1 ratio. D. I/I0 values. α1=402 to 416; α2=420 to 436; α3=437 to 447; α4=453 to 465. Helices in CTD are indicated at the top with cylinders, SP by an oval, and ZF1 and ZF2 by rectangles. Residues 483-493 (light gray) at the boundary of SP and N-terminus of NC have a peak-like R2 profile with a maximum at I487.
As expected for a polyprotein, CTD-SP-NC exhibits complex relaxation behavior (Figure 3) consistent with multiple domains of differing degrees of flexibility, rather than a single globular unit. The rates plotted by residue for unbound CTD-SP-NC in Figure 3 show large variations in R1, R2 and I/I0 values. In particular, SP and the flanking regions of the C-terminus of CTD and N-terminus of NC (residues 468 – 507) have I/I0 values near 0.0 while CTD and the NC core, comprising ZF1 and ZF2, have near limiting values of 0.8 for I/I0. The overall trends in dynamics define three regions in RSV CTD-SPNC: the CTD core region (residues 394 – 467), the NC core region (residues 509 – 548), and finally SP and its flanking regions (residues 468 – 507). Average values for these regions, grouped based on their dynamics, are shown in Table 1.
Table 1.
Average 15N backbone relaxation rates and I/Io (=NOE-1) for regions of RSV CTD-SP-NC in the unbound state. The standard deviation of the average value is listed.
| residues | R̄1 (s−1) | R̄2 (s−1) | τm (ns)* | |||
|---|---|---|---|---|---|---|
| CTD | 397 – 467 | 1.8 ± 0.2 | 10.6 ± 2.1 | 5.9 ± 1.4 | 0.8 ± 0.1 | 6.7 |
| SP+flanking | 468 – 507 | |||||
| C-term CTD | 468 - 477a | 2.2 ± 0.3 | 6.4 ± 0.6 | 2.8 ± 1.5 | 0.2 ± 0.2 | N.D. |
| SP* | 479 - 493 a ,b | 2.2 ± 0.1 | 10.2 ± 4.2 | 4.9 ± 2.2 | 0.06 ± 0.2 | N.D. |
| N-term NC | 495 - 507a | 2.4 ± 0.2 | 5.5 ± 1.6 | 2.3 ± 0.8 | −0.02 ± 0.2 | N.D. |
| NC core | 509 - 548 | 2.2 ± 0.3 | 7.4 ± 1.7 | 3.4 ± 0.9 | 0.6 ± 0.01 | 5.1 |
| ZF 1 | 509 - 522 | 2.3 ± 0.3 | 8.0 ± 1.1 | 3.7 ± 0.9 | 0.6 ± 0.1 | 4.9 |
| ZF 2 | 535 - 548 | 2.2 ± 0.2 | 7.7 ± 2.2 | 3.5 ± 0.9 | 0.6 ± 0.1 | 5.1 |
Calculated using TENSOR by an overall fit of relaxation data from residues listed in column 2.
Regions defined by common relaxation behavior.
Region colored in gray in Figure 3. SP* residue range is based on relaxation behavior and differs somewhat from SP residues 477-488.
Relaxation Rates Predicted for Alternative Motional Models
The objective of the study reported here is to characterize the interaction of CTD-SP-NC with nucleic acid by examining hydrodynamic behavior of various segments of RSV Gag polyprotein. We therefore focus on dynamic properties averaged over these three polypeptide regions, rather than a detailed interpretation of residue-level mobility. Correlation times for a domain are estimated for the clearly globular CTD and NC core regions by numerically fitting a subset of R1, R2 and I/I0 values from residues within these regions to dynamic models with anisotropic rotation and internal motion described in Methods. For SP and flanking regions, it cannot be assumed a priori that the dynamics of this linker region are similar to that of a flexible loop in a globular protein with two characteristic time constants for overall slow tumbling and a faster internal motion. Alternatively, SP motion may be appropriately modeled as an independent linker by using a single, fast timescale correlation function. Dynamics of this apparently unstructured, highly flexible Gag region is therefore interpreted with theoretical curves for R1, R2 and I/I0 values calculated for the four motional models to obtain a region-level characterization. Overall, this assessment of CTD-SP-NC yields conclusions in terms of an effective hydrodynamic molecular weight and general mobility of regions composing CTD-SP-NC. Of particular interest are the changes in dynamics upon (GT)4 binding assessed in this manner for giving insights into the assembly mechanism. Spectral density mapping (54), a structure-independent approach, was also conducted and those results are provided in supplementary material.
We recall the theoretical relaxation behavior for four motional models (see Methods for details) to guide the interpretation of the relaxation exhibited by each of the three regions of CTD-SP-NC. Theoretical curves to illustrate the dependence of R2/R1 and I/I0 on anisotropic rotation for nonspherical shapes and ps-to-ns internal motion are compared in Figure 4 to curves for the reference rigid-body spherical rotation. The curves were calculated from spectral densities corresponding to the four motional models described in Methods.
Figure 4.

Theoretical R2/R1 and I/I0 at 600 MHz 1H frequency as a function of overall correlation time,τm, for four motional models. Model 1, isotropic rotation with a single overall rotational correlation time (solid, red curve) is shown in all panels for reference. A) and B) Axially symmetric rotation with Dzz = D□=1.3 and Dxx = Dyy = D⊥ =0.85, and τm = 1/[2(Dxx + Dyy + Dzz)]. Two curves delimit the relaxation for N-H bond vectors aligned parallel (dashed, green) or perpendicular (dotted, blue) to the z-axial component of the diffusion tensor. C) and D) Overall isotropic rotation with a fast internal motion. Model 3, very fast internal motion squares, S2=0.5 (filled squares, red). Model 4, internal motion with τint=0.1 ns and S2=0.85 (dashed, green), 0.6 (dash-dot, cyan) and 0.5 (dotted, blue). The vertical lines denote τm values estimated for CTD and NC core regions in unbound CTD-SP-NC.
Relaxation rates for isotropic (model 1) and anisotropic, axially symmetric (model 2) rigid rotation are compared in Figure 4A and 4B where R2/R1 and I/I0, respectively, are plotted as a function of overall correlation time τm = 1/ 6Diso for isotropic (solid), and τm = 1/[2(Dxx + Dyy + Dzz)] for axially symmetric (dashed and dotted) diffusion. The two curves for axially symmetric rotation are for an NH bond vector aligned parallel (dashed) or perpendicular (dotted) to the z-axial component of the diffusion tensor, and delimit the range of possible R2/R1 values. The anisotropy for model 2 is DP/D⊥ = 1.75, which is an upper limit based on previously reported values for retroviral CA and NC proteins. Studies report DP/D⊥ values of 1.4 for CTD in the context of CA (4) and 1.2-1.6 for individual zinc finger domains in the context of NC (16, 55). With anisotropic (axially symmetric) rotation, R2/R1 is observed in Figure 4A to depend strongly on the NH vector orientation relative to the diffusion axes when τm>3 ns, while the effect on I/I0 due to anisotropic rotation (Figure 4B) is insignificant in this slow tumbling regime. For small values of τm where the slope is steep, I/I0 does depend on the NH vector orientation so that somewhat larger I/I0 values result for vectors oriented parallel to the z diffusion-tensor component while perpendicular vectors exhibit smaller I/I0 values.
Internal motion within a globular domain (models 3 and 4) is evaluated using the model-free formalism of Lipari and Szabo (47). Theoretical curves calculated for very fast internal motion, with no dependence on the timescale of the internal motion (model 3, eqn (7), squares), and for a time-dependent fast internal motion (model 4, eqn (8), dotted or dashed), are compared to those for rigid-body rotation (model 1, solid curves) in Figures 4C and 4D. The parameters for model 4 are τint=0.1 ns and S2=0.85 (dashed), 0.6 (dot-dash), or 0.5 (dotted). The value τint=0.1 ns is the slowest value reasonably consistent with the assumption in Lipari-Szabo theory of fast internal motion with respect to overall rotation on a timescale of approximately 1 to 10 ns, and S2=0.5 indicates considerable reorientation of the NH bond vector within the molecular frame work, and thus together, these parameters reasonably illustrate limiting effects that arise from reorientation of the amide NH bond due to internal motion.
As previously recognized (56), R2/R1 (Figure 4C) is insensitive to fast internal motion (model 3, squares), but when the internal motion timescale approaches overall tumbling, R2/R1 is somewhat reduced at longer τm (model 4, dashed, dot-dash or dotted). (The reader is reminded that the theoretical plots for R2/R1 in Figure 4 do not include Rex, which accounts for slower exchange processes and that μs-ms motions would contribute by addition of an Rex component to an observed R2.) As with R2/R1, I/I0 (Figure 4D) is quite insensitive to very fast internal motion (τint<0.1τm) of model 3: no change is observed even though S2 << 1. On the other hand, I/I0 is diminished significantly if internal motion occurs on a timescale such that τint approaches the overall correlation time. This time-dependent behavior depends on the amplitude S2 and internal correlation time τint (Figure 4D, dashed, dash-dot or dotted); the diminution in I/I0 increases with slower, larger amplitude internal motion, i.e. as τint increases and S2 decreases. It is noted that the curves in Figure 4D show that the internal motion must result in extensive reorientation (S2<0.6, τint~0.1 ns) in order to reduce I/I0 to values below 0.3 for τm between 5 and 10 ns.
Dynamics of the CTD core in unbound CTD-SP-NC
The heteronuclear relaxation data of CTD core residues in CTD-SP-NC were analyzed using the previously determined structure of RSV CTD (PDB code 1EOQ) (3) and axially symmetric diffusion, model 2. The chemical shifts of CTD in the Gag construct are similar to those in the isolated CTD domain, excluding the C-terminal residues, indicating that CTD remains close to the 3D structure determined for the isolated domain. Axially symmetric diffusion was also found previously to be the best fit to relaxation rates of CTD in HIV-1 (4). Values of R2 and R1 for residues 394 – 467 of CTD are well fit by the program TENSOR with τm = 6.7 ns (Table 1). (Residues in the C-terminus of CTD exhibit different dynamics and are discussed below.) The large range of R2/R1 ~4 to 7 observed for CTD core residues 394 to 464 (Figure 3C) is consistent with the theoretical curves for axially symmetric rotation of a well-ordered domain given τm ≈ 7 ns (Figure 4A). The average I/I0 equals 0.75 with small variation among these residues. This value is only slightly less than the limiting value of 0.83, indicating that CTD core residues have only limited internal mobility. Given the empirical plot for the dependence of τm on molecular weight in Figure 2 (53), the value τm ≈ 7 ns corresponds to a 90-residue protein, only slightly larger in size than CTD alone. A value for τm previously determined from RSV CTD relaxation was in the context of full length CA and is a larger value, ~ 12 ns (4), which implies that rotational diffusion of the two domains NTD and CTD is coupled. By contrast, in the context of CTD-SP-NC, CTD rotates faster, more like an isolated domain and is marginally affected by the presence of NC. Thus CTD is a well-ordered globular domain with axially symmetric rotational diffusion (model 2) nearly independent of SP and NC.
Dynamics of the NC core in unbound CTD-SP-NC
The NC core, comprising ZF1 and ZF2, of RSV is folded and likely non-spherical, but because the structure is unknown, the relaxation rates are analyzed based only on an isotropic rotation (model 1) to estimate an overall correlation time for each zinc finger separately and together with the connecting segment. The purpose of this analysis is to determine the degree of rotational coupling of the two zinc fingers by comparing the diffusional behavior of the two ZF domains analyzed independently and combined.
The τm value fitted using TENSOR from R1 and R2 for residues in ZF1 (509 – 522), ZF2 (535 – 548), or the longer NC core region (509 – 548) is 4.9, 4.9, and 5.1 ns, respectively. The globally fit value for τm of 5.1 ns is confirmed with the theoretical plot (Figure 4A); the average R2/R1 values for ZF1, ZF2, and NC core equal to 3.7, 3.5, and 3.4, respectively, correspond to τm ≈ 5 ns. The similarity in the correlation time for both ZF1 and ZF2, as well as the combined NC core indicates that this central region of NC rotates as a unit and that the two ZFs are coupled. Further, the large τm of 4.9 ns for NC core corresponds to a 65-residue protein based on the hydrodynamic data of de la Torre (53) (Figure 2). NC is 89 residues and the NC core region spans approximately 40 amino acids, with each ZF being 14 residues. Thus the magnitude of the correlation time also supports the notion that ZF1 and ZF2 rotate more as a single unit than two independent units, and are hydrodynamically coupled. In the case of HIV-1 NC, τm ~ 6 and 5.7 ns are reported for ZF1 and ZF2, respectively, and it was concluded that the two ZF's are rotationally independent based on consistently lower values of R1 for ZF2 with respect to ZF1 (55). We do not find this type of systematic variation in the data for RSV Gag.
The average for I/I0 over the NC core residues is approximately 0.6, somewhat smaller than the limiting high molecular weight value of 0.83 expected for τm = 5 ns. Neither anisotropic rotation nor very fast internal motion (τint <<τm, model 3) reduce I/I0, therefore we conclude the NC core experiences slower-timescale internal mobility with more extensive motional averaging than CTD.
Dynamics of SP and flanking regions in unbound CTD-SP-NC
The relaxation rates of the SP linker and flanking regions from the C-terminus of CTD and the N-terminus of NC (residues 468 – 507) indicate local conformational flexibility for this region of CTD-SP-NC. High mobility can result from a fully flexible linker described by a single, fast correlation time, or by a more complex motion with a longer overall domain-like rotation time plus an internal motion on a faster timescale. We assess the local dynamics of this region based on general relaxation behavior and consistent agreement for I/I0, R1 and R2 between predicted and experimental values.
A notable feature of the SP and flanking region (Figure 3D) is the near zero I/I0 values, whereas I/I0 values exhibited by residues in CTD and the NC core are close to the maximal limit expected for globular proteins. Near zero values for I/I0 are often observed for terminal residues of a folded protein but are less common for internal residues. This small value can be accounted for by either model 1, with a single rotational correlation time near τm ~ 1 ns (Figure 4B, solid) representative of random and flexible chains, or model 4, with a longer overall τm and a time-dependent internal motion. To consider viable estimates of the motional parameters of model 4, we use for τm the range 5 to 7 ns for the NC core and CTD (Table 1), and find that S2=0.5-0.6 and τint ~ 0.1 ns (Figure 4D, dashed and dot-dash) are consistent with I/I0~0. Other combinations of S2 and τint are of course also possible. Anisotropic rotation and very fast internal motion have negligible effect on I/I0 (Figure 4B and 4D, squares), and thus model 2 and 3 cannot account for the near zero value for I/I0.
A second notable feature in the relaxation behavior of unbound CTD-SP-NC is the peak-like profile in R2 with a maximum at I487 (Figure 3B, gray bars) for residues 483 to 493 at the boundary of SP and NC. This region also has larger R2/R1 than the immediate upstream or downstream residues, which suggests that exchange (Rex) contributes to R2. A maximum in the residue profile for R2 has been interpreted to correspond to the site at which a conformational transition takes place (57). Here, the maximum in the R2 profile occurs at residue I487. RSV has a conserved proline residue at position 485 (Figure 1) near the maximum in the profile, so that exchange could involve a cis – trans isomerization of P485 in SP. This conformational exchange process is affected by (GT)4 binding, as discussed below.
To decipher the motional properties in general terms of the SP and flanking regions of Gag polyprotein, we consider the R1, R2 profiles (Figure 3) and the fact that near zero I/I0 values are consistent with either a single, shorter correlation time (model 1, τm ~ 1 ns) for a fully flexible linker, or a longer overall τm plus a time-dependent internal motion (model 4). Of these, model 4 is most consistent based on the R2/R1 values observed for SP and the flanking regions; the R2/R1 values are too large to be consistent with fast tumbling τm ~ 1 ns (Figure 4C, Table 1). The individual R2 and R1 values also rule out model 1 as detailed in supplementary material. Further, the presence of a conformational exchange process cannot be readily reconciled with the behavior of a fully flexible linker associated with model 1.
We conclude relaxation of SP and the flanking regions (residues 468 to 507) is most consistent with a motional model described by two correlation times (model 4). For a limiting case of the overall rotational diffusion similar to either CTD or NC, the slow internal motion is approximated by S2=0.5-0.6 and τint ~ 0.1. Importantly, a conformational exchange process is needed to account for the residue variation and peak maximum in R2.
CTD-SP-NC association with (GT)4
Perturbations in chemical shift (CSP) of CTD-SP-NC in the presence of (GT)4 were used to identify the likely site of interaction with (GT)4. Chemical shifts were measured from 15N-HSQC (Figure 5A) at a ratio of (GT)4 to protein of 0.5, 1.0, and 2.0. At a ratio of (GT)4 to protein of 0.5, two sets of frequencies are observed for residues involved in binding while a single set of frequencies is observed at ratios of 1.0 and 2.0 indicating that binding is in slow exchange. The observation of slow exchange and that no differences in resonance frequencies were observed for equimolar and 2-fold excess molar equivalents of (GT)4, is consistent with the previous estimate for the (GT)4 binding association constant Ka = 1.5 to 1.7 × 105 M−1 (20), and that (GT)4 binds to a single site.
Figure 5.

Chemical shift perturbation of RSV CTD-SP-NC upon (GT)4 binding. A) Representative region of 15N-HSQC of CTD-SP-NC bound (red) and unbound (blue). B) Plot of chemical shift perturbation of RSV CTD-SP-NC upon binding to (GT)4 oligonucleotide. . Structural elements drawn at the top are as in Figure 3.
Upon (GT)4 binding, the resonances for SP and the N-terminus of NC are broadened so that no intensity is observed for most of the residues 478 - 496. (GT)4 binding also affects chemical shifts for NC resonances; however only the resonances from ZF1 residues 509 – 524 have large perturbations in chemical shift upon binding (GT)4 (Figure 5B). Small perturbations are observed for resonances from ZF2 while those of CTD are relatively unaffected by the presence of (GT)4. These data suggest that the (GT)4 binding site is localized to ZF1 and are consistent with previous biochemical data showing that ZF1 of HIV-1 plays a more significant role in RNA encapsidation (58).
Effects of (GT)4 binding on heteronuclear relaxation
Nucleic acid association is thought to induce conformational changes in Gag that are necessary to promote assembly (20, 22). Alterations in protein dynamics can reflect such conformational changes.
The 15N relaxation rate constants R1, R2 and steady-state enhancement I/I0 were measured for CTD-SP-NC plus (GT)4 at a 1:1 molar ratio, and a 600 MHz Larmor frequency. The residue profiles for these values, along with the R2/R1 ratio, are shown in Figure 6, and the averages over regions of the protein are listed in Table 2. The differences in relaxation rates between free and bound states are indicated with black bars in Figure 6 to facilitate the comparison of these two states of CTD-SP-NC.
Figure 6.
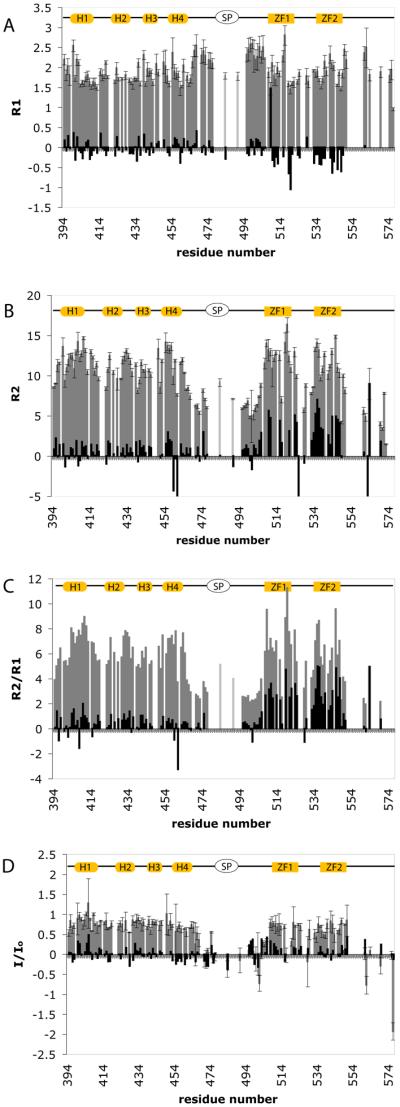
15N relaxation data at 600 MHz for RSV CTD-SP-NC in the bound form (gray bars) and the difference equal to bound minus unbound values (black bars). A. Longitudinal relaxation rates, R1. B. Transverse relaxation rates R2. C. R2/R1 ratio. D. 1H-15N I/I0 values. Structural elements drawn at the top are as in Figure 3. Except for Q484 and V491, residues 479-493 in SP and N-terminus of NC, colored light gray in Figure 3, are exchange broadened and not detected.
Table 2.
Average 15N backbone relaxation rates and I/Io (=NOE-1) for regions of RSV CTD-SP-NC in the (GT)4 bound state. The standard deviation of the average value is listed.
| residues | R̄1 (s−1) | R̄2 (s−1) | τm (ns)* | |||
|---|---|---|---|---|---|---|
| CTD | 397 - 467 | 1.9 ± 0.3 | 11.2 ± 2.1 | 6.0 ± 1.5 | 0.7 ± 0.1 | 7.2 |
| SP+flanking | 468 - 507 | |||||
| C-term CTD | 468 - 477a | |||||
| SP* | 479 - 493 a ,b | |||||
| N-term NC | 495 - 507a | 2.4 ± 0.2 | 6.2 ± 0.8 | 2.6 ± 0.5 | 0.02 ± 0.2 | N.D. |
| NC core | 509 - 548 | 1.8± 0.3 | 11.3 ± 2.4 | 6.3 ± 1.9 | 0.6 ± 0.2 | 7.1 |
| ZF 1 | 509 - 522 | 1.9 ± 0.4 | 12.1 ± 2.2 | 6.4 ± 2.2 | 0.6 ± 0.2 | 7.9 |
| ZF 2 | 535 - 548 | 1.8 ± 0.3 | 11.2 ± 1.7 | 6.1 ± 1.6 | 0.6 ± 0.1 | 7.8 |
Calculated using TENSOR by an overall fit of relaxation data from residues listed in column 2.
Regions defined by common relaxation behavior.
Region colored in gray in Figure 3. SP* residue range is based on relaxation behavior and differs somewhat from SP residues 477-488.
Association affects apparent mass of both ZF1 and ZF2, but not CTD
Nucleic acid binding alters R2/R1 relaxation of not only ZF1, the region that exhibits large chemical shift changes, but also ZF2 (Figure 6 and Table 2). The value of τm estimated from R2/R1 for ZF1 and ZF2 is 7.9 and 7.8 ns, an increase of 3.0 and 2.7 ns, respectively, for the (GT)4 complex relative to unbound CTD-SP-NC. For the NC core in the presence of (GT)4, R2/R1 becomes 7.1 ns, an increase of 2.0 ns. This increase in the R2/R1 ratios is roughly consistent with the predicted increase in the overall correlation time of approximately 1.8 ns for the larger molecular weight of 2.7 kDa corresponding to (GT)4 (Figure 2). Further, the concomitant increase in R2 and decrease in R1 values expected for this change in τm are observed (Table 2 and supplementary material). That the presence of (GT)4 similarly alters both ZF1 and ZF2 relaxation indicates a comparable increase in apparent mass for both zinc fingers. On the other hand, only ZF1 appears to make direct contact based on chemical shift perturbations. Together, these observations strongly support the postulate that the NC core is a rotationally linked domain.
In the case of the CTD domain, the changes in overall rotational time observed upon (GT)4 binding are minimal. There is a small increase in the average R2/R1 from 5.9 to 6.0, which corresponds to an increase in τm from 6.7 to 7.2 ns (Table 2). We view this small change in the estimated value of τm to be attributed to an increase in the mass of CTD-SP-NC by association of (GT)4 at a remote site on the polyprotein, but CTD and NC remain largely rotationally uncoupled in the complex.
Slowed exchange for SP
A result of (GT)4 association on the relaxation properties of CTD-SP-NC is that the peaks for residues in SP and the N-terminus of NC broaden and are no longer detected. This intermediate exchange behavior is observed for the amide resonances of residues 479 to 493, excluding residues Q484 and V491. The broadening of peaks for SP and the N-terminus of NC supports the proposal that the relatively large R2 values measured in the free state for this region are due to exchange contributions to R2. That is, these resonances in the free state are in a fast exchange regime and the broadened linewidths in the bound state indicates that (GT)4 binding alters the underlying conformational exchange and slows the timescale to the intermediate regime. The reason resonances Q484 and V491 are not broadened is unknown at this time.
Effect of (GT)4 binding on two CTD helices
Significant features due to (GT)4 binding are the decreases in I/I0 and R2/R1 observed for two CTD helices (Figure 6C, D). For I/I0, helix 4, as well as residues in the C-terminus of CTD, exhibit a small but consistent trend to smaller I/I0 values (Figure 6D). Given τm ~ 7 ns, I/I0 can be diminished only by a time-dependent internal motion indicating that the mobility of helix 4 and the C-terminus is enhanced upon binding. In the case of R2/R1, association of (GT)4 leads to reduced R2/R1 values for two residues in helix 1 and two residues in helix 4 (Figure 6C), while all other residues in CTD and the NC core show an increase. As discussed above, a small increase in R2/R1 values is expected from the added mass of (GT)4 at a distant site. For axially symmetric rotation, assuming the domain structure of CTD and principal axis system of the diffusion tensor are unchanged by (GT)4 binding, so that the orientation of an NH bond vector is constant within this framework, then R2/R1 should increase uniformly according to τm (Figure 4A). Two explanations are proposed for the observed decrease in R2/R1 of certain residues. First, if an NH vector reorients within the principal axis framework, then R2/R1 is not predicted by a change in τm, and can either increase or decrease. As such, a decrease in R2/R1 upon ligand binding can occur by reorientation of the NH group, so that the smaller values of R2/R1 for residues in helix 1 and 4 infers that (GT)4 binding induces a conformational rearrangement of these helices within the frame of CTD. Second, an exchange process could exist in the free state of CTD-SP-NC, leading to increased R2 values due to Rex. Binding of (GT)4 could therefore diminish an exchange contribution and reduce R2. Together these data suggest that (GT)4 binding has a subtle effect on the conformational equilibrium for an area that encompasses helix 1, helix 4 and the C-terminus of CTD, and thereby implicates this area to have a role in assembly.
Model for Autoinhibition of Assembly
We propose a molecular model for autoinhibition of assembly and how nucleic acid binding releases this inhibition to initiate assembly. The model is based on the NMR results reported here and earlier mutagenesis studies (20, 22), and is also motivated by a discovered inhibition of HIV-1 assembly conferred by a peptide that binds CTD (27, 40).
Nearly an identical region near SP is distinguished by both NMR relaxation behavior and earlier mutagenesis studies of Gag polyprotein. As described here, the NMR relaxation behavior of SP and flanking regions (residues 468 to 507) differs distinctly from that of CTD and NC; collectively, the relaxation data are best fit by motion modeled with two correlation times corresponding to a slower overall rotation similar to the CTD and NC domains, plus an internal motion (model 4). In addition, the NMR data show that (GT)4 binding slows a conformational exchange process involving the residues 479 to 493 (SP*). Previously reported mutagenesis of Gag polyprotein identified the ‘assembly domain’ (residues 469 - 492) ; the insertion or substitution of the flexible peptide sequences GSGSG or GG into SP, the eight C-terminal residues of CTD, or the first four residues of NC alters budding VLPs from an immature spherical to a mature tubular morphology, and the same mutations totally disrupt assembly in vitro (21, 31). Thus the assembly domain identified by mutagenesis to be critical for assembly, is nearly identical to the region of SP and the flanking residues where an exchange process is observed. The exchange process reported on by NMR relaxation is therefore likely a functional change in conformational equilibrium associated with assembly.
The residues found to have relaxation rates altered by (GT)4 binding in a manner that suggests a conformation change by reorientation with respect to the overall diffusion tensor are located in a region that is key for protein interactions of assembly. These residues are in helix 1 and 4, which lie on the same surface of CA and form two edges of a hydrophobic patch (Figure 7, blue surface) conserved among retroviral CA proteins (3). CTD residues with smaller R2/R1 values in the presence of (GT)4 are colored in green in Figure 7. Recent electron cryocrystallography studies of two-dimensional crystals of HIV-1 CA demonstrate that this conserved hydrophobic patch forms a heterodimerization interface between intermolecular NTD-CTD domains in the mature hexameric lattice (24). The conserved hydrophobic patch was also found by crystallography and NMR to bind a peptide (CAI) (27) that inhibits both immature and mature HIV-1 assembly in vitro (40). The inhibitory CAI peptide alters the homodimerization interface of CTD observed in the crystal (27, 40). This CTD-CTD homodimeric interface is involved in connecting neighboring hexamers in the mature CA lattice of HIV (24) and may be involved in formation of the immature hexameric lattice (23). The crystallographic structure of RSV CTD at low pH (26) finds a dimeric interface that closely resembles the HIV-1 interface, and thus similar interfaces likely stabilize the RSV lattice. Mutagenesis of conserved residues within the CAI binding site of HIV-1 support the idea that this hydrophobic patch is important in regulating the conformation necessary for virus assembly and maturation (59). It was concluded that the inhibitory activity from CAI binding may result from direct steric hindrance of NTD-CTD contacts or indirectly by stabilizing a CTD conformation that is not compatible with the CTD-CTD dimer formed in the mature particle.
Figure 7.

Model for initiation of assembly. In the free state of Gag, C-terminal residues of CTD associate with the hydrophobic area of CTD between helix 1 and helix 4 (colored blue, residues L410, L411, L419, P421, A423, A425, I428, I429, F432, G455, and I458) and localize SP at the top of the groove near helix 1 and 2. The intramolecular association disfavors a functional dimer contact (as does the inhibitory CAI peptide). Binding of NC (ZF1) to RNA ‘drags on’ SP and its flanking region to dislodge the C-term and SP from CTD. This action allows a proper dimerization unit to form. Residues with reduced R2/R1 values in helix 1 and 4 are colored in green (residues Q397, S402, N408, E415, K459, V461).
Together, these results suggest a model for autoinhibition of assembly and release triggered by RNA association with Gag. We postulate that, in the absence of nucleic acid, formation of assembly-competent CTD dimers is autoinhibited by an interaction of residues at the C-terminus of CTD with the vicinity of the conserved hydrophobic patch between helices 1 and 4 (Figure 7, left hand side), and localizes SP at the top of the groove near the loop between helix 1 and 2. This groove contact is speculated to disfavor Gag dimerization in a manner similar to that achieved by the inhibitory CAI peptide (40). NMR relaxation of SP and flanking regions is consistent with this region of Gag being associated with CTD in that the overall relaxation is best fit by model 4 with a longer (~5 ns) rotational correlation time similar to CTD, plus a faster timescale motion corresponding to a conformational exchange process. Although the nature of this conformational change cannot yet be defined, it is clear that nucleic acid binding alters the exchange given the loss of these peaks by exchange broadening in the complex of CTD-SP-NC with (GT) 4.
Intramolecular association of the C-terminus of CTD and the assembly domain with the hydrophobic patch between helix 1 and 4 of CTD would destabilize the Gag dimeric interface and sequester the assembly domain from intermolecular associations compatible with higher-order oligomerization for assembly. The ZF1-mediated association of RNA with NC is postulated to dislodge the C-terminus of CTD and SP from the hydrophobic groove (Figure 7, right hand side), thereby affecting helices 1 and 4 consistent with the observed changes in NMR relaxation, and would promote the proper dimer contact surface of Gag necessary for immature assembly. It is noted that the system studied here corresponds only to the initial steps in assembly in that oligonucleotides as short as (GT)4 do not actually lead to RSV Gag dimerization shown in Figure 7 given that the formation of the dimer building block requires (GT)8 (20) or oligomers long enough to bind two Gag molecules. During assembly, the close proximity of Gag upon binding one (GT)8, or longer oligonucleotide, increases the effective local Gag concentration and is thought to stabilize a Gag-Gag association that is too low affinity to form otherwise. As such, the function of Gag association with nucleic acid may be two-fold: first, to remove the inhibitory intramolecular interaction between the assembly domain and the hydrophobic groove of CTD, and second, to drive a low-affinity intermolecular association.
Conclusions
15N heteronuclear relaxation and chemical shifts indicate that the CTD and NC regions of the Gag polyprotein are rotationally independent domains. The correlation time of CTD in CTD-SP-NC is only slightly longer than that estimated for isolated CTD, and varies independent of that for NC. Chemical shifts of CTD in CTD-SP-NC and from the isolated CTD domain indicate the CTD structure is largely preserved in the context of the polyprotein. Small disparities in chemical shifts, however, do suggest that the conformation of the C-terminal helix 4 of CTD is altered by the covalent attachment of SP and NC. Based on the characterization of rotational tumbling in both the free and (GT)4-bound state, the NC core appears to be a single domain so that (GT)4 binding slows the tumbling and increases the apparent mass of both ZF1 and ZF2 even though direct contact is suggested by chemical shift perturbation to occur only with ZF1.
Residues in SP and the flanking regions exhibit greater variation in relaxation rates, and thus more complex dynamics, than either CTD or the NC core. Moreover, the dynamics are sensitive to (GT)4 binding, even though no direct interaction with (GT)4 is suggested from chemical shift perturbations; the only region of CTD-SP-NC with large perturbations (>0.1 ppm) is ZF1. In particular, SP and the flanking regions, which corresponds to the assembly domain defined by mutagenesis (31), undergo a relatively fast conformational exchange process in the free state and a slower one in the presence of (GT)4. Comparison of the RSV relaxation profiles reported here with those of HIV-1 (39) finds that SP and the analogous SP-1 region of HIV are both conformationally flexible, but the R2 relaxation profile for RSV SP shows a strong maximum at residue I487 while the SP-1 profile is notably uniform. Cis – trans isomerization of Pro485, a conserved residue in RSV located at the C-terminus of SP, may be the origin of the observed R2 maximum. There are no conserved Pro residues in SP-1 of HIV-1. RSV SP also differs from HIV-1 SP-1 in that a trend in chemical shifts of residues in the N-terminal region of SP-1suggests transient formation of a helix (39). Such a trend is not observed in the chemical shift profile of RSV SP. Whether the observed differences in NMR relaxation behavior reflect significant distinctions between RSV and HIV-1 or are due to slight differences in equilibrium or time dependences of transitions, or dependences on other factors such as pH, is unknown at this time. It is important to recall that virus assembly is driven by the combined forces of many weak interactions, so that a structural feature important for stabilizing the assembled particle may not be realized under the conditions in which the isolated protein or protein complex is studied.
A central outcome of this work is the proposal for a model on the autoinhibition of assembly and how nucleic acid binding to Gag polyprotein can trigger dimerization to promote assembly. The fundamental features of the model (Figure 7), suggested by the NMR relaxation data and mutagenesis effects on assembly, is the intramolecular association of the SP assembly domain with CTD, and the disruption of this association by intermolecular binding of RNA to NC. SP binding to the hydrophobic surface patch of CTD is postulated to inhibit dimerization, analogous to the inhibition of assembly by the CAI that binds in a similar location on CTD (27, 40), and to sequester the assembly domain to prevent premature intermolecular association. The model provides a new framework to test with future experiments and should aid progress in defining the molecular mechanism of retroviral assembly.
Supplementary Material

Acknowledgments
This work was supported by the National Institutes of Health (AI45976 to C.B.P. and CA20081 to V.M.V.), the Markey Center for Structural Biology, and the Purdue University Center for Cancer Research (CA 23568).
1Abbreviations
- CA
capsid
- CSA
chemical shift anisotropy
- CTD
C-terminal domain of capsid
- CTD-SP-NC
C-terminal domain of capsid-spacer-nucleocapsid
- DTT
DL dithiothreitol
- EM
electron microscopy
- HIV-1
Human immunodeficiency virus – 1
- I/Io
NOE – 1
- IPTG
Isopropyl-β-D-thiogalactoside
- kDa
kilodalton
- MA
matrix
- MHz
megahertz
- NC
nucleocapsid
- NH4Cl
ammonium chloride
- NMR
nuclear magnetic resonance
- NTD
N-terminal domain of capsid
- NaCl
sodium chloride
- PEI
polyehtyleneimine
- RSV
rous sarcoma virus
- R1
longitudinal relaxation rates
- R2
transverse relaxation rates
- SP
RSV spacer
- SP-1
HIV-1 spacer
- S2
generalized order parameter
- τm
correlation time
- VLP
virion-like particle
- XNOE
{1H} – 15N steady-state heteronuclear NOE
- ZF1
N-terminal zinc finger
- ZF2
C-terminal zinc finger
- ZnCl2
zinc chloride
Footnotes
Supplemental Information
Values from spectral density mapping and generalized order paramters averaged over domains for CTDSP-NC unligated and bound to (GT)4 are provided in supplementary information. Theoretical curves of R1 and R2 relaxation rates are also shown as a function of correlation time. This supplemental material may be accessed free of charge online at http://pubs.acs.org.
References
- 1.Fuller SD, Wilk T, Gowen BE, Kräusslich H-G, Vogt VM. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Current Biology. 1997;7:729–738. doi: 10.1016/s0960-9822(06)00331-9. [DOI] [PubMed] [Google Scholar]
- 2.Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: Implications for retroviral assembly mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7299–7304. doi: 10.1073/pnas.95.13.7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kingston RL, Fitzon-Ostendorp T, Eisenmesser EZ, Schatz GW, Vogt VM, Post CB, Rossmann MG. Structure and self-association of the Rous sarcoma virus capsid protein. Structure. 2000;8:617–628. doi: 10.1016/s0969-2126(00)00148-9. [DOI] [PubMed] [Google Scholar]
- 4.Campos-Olivas R, Newman JL, Summers MF. Solution structure and dynamics of the Rous sarcoma virus capsid protein and comparison with capsid proteins of other retroviruses. Journal of Molecular Biology. 2000;296:633–649. doi: 10.1006/jmbi.1999.3475. [DOI] [PubMed] [Google Scholar]
- 5.Murray PS, Li Z, Wang J, Tang CL, Honig B, Murray D. Retroviral Matrix Domains Share Electrostatic Homology: Models for Membrane Binding Function throughout the Viral Life Cycle. Structure. 2005;13:1521–1531. doi: 10.1016/j.str.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the Carboxyl-Terminal Dimerization Domain of the HIV-1 Capsid Protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 7.Darlix J-L, Lapadat-Tapolsky M, de Rocquigny H, Roques BP. First Glimpses at Structure-function Relationships of the Nucleocapsid Protein of Retroviruses. Journal of Molecular Biology. 1995;254:523–537. doi: 10.1006/jmbi.1995.0635. [DOI] [PubMed] [Google Scholar]
- 8.McDonnell JM, Fushman D, Cahill SM, Zhou W, Wolven A, Wilson CB, Nelle TD, Resh MD, Wills J, Cowburn D. Solution structure and dynamics of the bioactive retroviral M domain from rous sarcoma virus. Journal of Molecular Biology. 1998;279:921–928. doi: 10.1006/jmbi.1998.1788. [DOI] [PubMed] [Google Scholar]
- 9.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional Structure of the Human Immunodeficiency Virus Type 1 Matrix Protein. Journal of Molecular Biology. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 10.Christensen AM, Massiah MA, Turner BG, Sundquist WI, Summers MF. Three-Dimensional Structure of the HTLV-II Matrix Protein and Comparative Analysis of Matrix Proteins from the Different Classes of Pathogenic Human Retroviruses. Journal of Molecular Biology. 1996;264:1117–1131. doi: 10.1006/jmbi.1996.0700. [DOI] [PubMed] [Google Scholar]
- 11.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature. 1995;378:743–747. doi: 10.1038/378743a0. [DOI] [PubMed] [Google Scholar]
- 12.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal Structure of Human Cyclophilin A Bound to the Amino-Terminal Domain of HIV-1 Capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 13.Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. Structure of the Amino-Terminal Core Domain of the HIV-1 Capsid Protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 14.Nandhagopal N, Simpson AA, Johnson MC, Francisco AB, Schatz GW, Rossmann MG, Vogt VM. Dimeric Rous Sarcoma Virus Capsid Protein Structure Relevant to Immature Gag Assembly. Journal of Molecular Biology. 2004;335:275–282. doi: 10.1016/j.jmb.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Kaluarachchi K, Giedroc DP. Solution structure and backbone dynamics of Mason-Pfizer monkey virus (MPMV) nucleocapsid protein. Protein Science. 1998;7:2265–2280. doi: 10.1002/pro.5560071104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein DJ, Johnson PE, Zollars ES, De Guzman RN, Summers MF. The NMR Structure of the Nucleocapsid Protein from the Mouse Mammary Tumor Virus Reveals Unusual Folding of the C-Terminal Zinc Knuckle. Biochemistry. 2000;39:1604–1612. doi: 10.1021/bi9922493. [DOI] [PubMed] [Google Scholar]
- 17.Summers MF, Henderson LE, Chance MR, Bess JW, Jr., South TL, Blake PR, Sagi I, Perez-Alvarado G, Sowder RC., III Nucleocapsid zinc fingers detected in retroviruses: EXAFS studies of intact viruses and the solution-state structure of the nucleocapsid protein from HIV-1. Protein Science. 1992;1:563–574. doi: 10.1002/pro.5560010502. al., e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Summers MF, South TL, Kim B, Hare DR. High-resolution structure of an HIV zinc fingerlike domain via a new NMR-based distance geometry approach. Biochemistry. 1990;29:329–340. doi: 10.1021/bi00454a005. [DOI] [PubMed] [Google Scholar]
- 19.Morellet N, Jullian N, Derocquigny H, Maigret B, Darlix JL, Roques BP. Determination of the Structure of the Nucleocapsid Protein NCP7 from the Human-immunodeficiency-virus Type-1 by H-1-NMR. Embo Journal. 1992;11:3059–3065. doi: 10.1002/j.1460-2075.1992.tb05377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma YM, Vogt VM. Rous Sarcoma Virus Gag Protein-Oligonucleotide Interaction Suggests a Critical Role for Protein Dimer Formation in Assembly. J. Virol. 2002;76:5452–5462. doi: 10.1128/JVI.76.11.5452-5462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson MC, Scobie HM, Ma YM, Vogt VM. Nucleic Acid-Independent Retrovirus Assembly Can Be Driven by Dimerization. J. Virol. 2002;76:11177–11185. doi: 10.1128/JVI.76.22.11177-11185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma YM, Vogt VM. Nucleic Acid Binding-Induced Gag Dimerization in the Assembly of Rous Sarcoma Virus Particles In Vitro. J. Virol. 2004;78:52–60. doi: 10.1128/JVI.78.1.52-60.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO. 2007;26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131:70–79. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Ivanov D, Stone JR, Maki JL, Collins T, Wagner G. Mammalian SCAN domain dimer is a domain-swapped homolog of the HIV capsid C-terminal domain. Mol. Cell. 2005;17:137–143. doi: 10.1016/j.molcel.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Bailey GD, Hyun JK, Mitra AK, Kingston RL. Proton-Linked Dimerization of a Retroviral Capsid Protein Initiates Capsid Assembly. Structure. 2009;17:737–748. doi: 10.1016/j.str.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Ternois F, Sticht J, Duquerroy S, Krausslich H-G, Rey FA. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat Struct Mol Biol. 2005;12:678–682. doi: 10.1038/nsmb967. [DOI] [PubMed] [Google Scholar]
- 28.Pepinsky RB, Papayannopoulos IA, Chow EP, Krishna NK, Craven RC, Vogt VM. Differential proteolytic processing leads to multiple forms of the CA protein in avian sarcoma and leukemia viruses. J. Virol. 1995;69:6430–6438. doi: 10.1128/jvi.69.10.6430-6438.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C, Hu J, Whitney JB, Kleiman L, Wainberg MA. A Structurally Disordered Region at the C Terminus of Capsid Plays Essential Roles in Multimerization and Membrane Binding of the Gag Protein of Human Immunodeficiency Virus Type 1. J. Virol. 2003;77:1772–1783. doi: 10.1128/JVI.77.3.1772-1783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melamed D, Mark-Danieli M, Kenan-Eichler M, Kraus O, Castiel A, Laham N, Pupko T, Glaser F, Ben-Tal N, Bacharach E. The Conserved Carboxy Terminus of the Capsid Domain of Human Immunodeficiency Virus Type 1 Gag Protein Is Important for Virion Assembly and Release. J. Virol. 2004;78:9675–9688. doi: 10.1128/JVI.78.18.9675-9688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller PW, Johnson MC, Vogt VM. Mutations in the Spacer Peptide and Adjoining Sequences in Rous Sarcoma Virus Gag Lead to Tubular Budding. J. Virol. 2008;82:6788–6797. doi: 10.1128/JVI.00213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheslock SR, Poon DTK, Fu W, Rhodes TD, Henderson LE, Nagashima K, McGrath CF, Hu W-S. Charged Assembly Helix Motif in Murine Leukemia Virus Capsid: an Important Region for Virus Assembly and Particle Size Determination. J. Virol. 2003;77:7058–7066. doi: 10.1128/JVI.77.12.7058-7066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a Putative {alpha}-Helix across the Capsid-SP1 Boundary That Is Critical for the Multimerization of Human Immunodeficiency Virus Type 1 Gag. J. Virol. 2002;76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. Helical structure determined by NMR of the HIV-1 (345-392)Gag sequence, surrounding p2: Implications for particle assembly and RNA packaging. Protein Science. 2005;14:375–386. doi: 10.1110/ps.041087605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Briggs JAG, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. Structure and assembly of immature HIV. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11090–11095. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. Embo Journal. 2007;26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell S, Vogt VM. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 1995;69:6487–6497. doi: 10.1128/jvi.69.10.6487-6497.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher RJ, Rein A, Fivash M, Urbaneja MA, Casas-Finet JR, Medaglia M, Henderson LE. Sequence-Specific Binding of Human Immunodeficiency Virus Type 1 Nucleocapsid Protein to Short Oligonucleotides. J. Virol. 1998;72:1902–1909. doi: 10.1128/jvi.72.3.1902-1909.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newman JL, Butcher EW, Patel DT, Mikhaylenko Y, Summers MF. Flexibility in the P2 domain of the HIV-1 Gag polyprotein. Protein Science. 2004;13:2101–2107. doi: 10.1110/ps.04614804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich H-G. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 43.Ma L, Hass MAS, Vierick N, Kristensen SM, Ulstrup J, Led JJ. Backbone Dynamics of Reduced Plastocyanin from the Cyanobacterium Anabaena variabilis: Regions Involved in Electron Transfer Have Enhanced Mobility. Biochemistry. 2003;42:320–330. doi: 10.1021/bi020553h. [DOI] [PubMed] [Google Scholar]
- 44.Abragam A. The Principles of Nuclear Magnetism. Clarendon Press; Oxford: 1961. [Google Scholar]
- 45.Ghose R, Fushman D, Cowburn D. Determination of the rotational diffusion tensor of macromolecules in solution from NMR relaxation data with a combination of exact and approximate methods - Application to the determination of interdomain orientation in multidomain proteins. Journal of Magnetic Resonance. 2001;149:204–217. doi: 10.1006/jmre.2001.2295. [DOI] [PubMed] [Google Scholar]
- 46.Woessner DE. Nuclear Spin Relaxation in Ellipsoids Undergoing Rotational Brownian Motion. Journal of Chemical Physics. 1962;37:647–654. [Google Scholar]
- 47.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. Journal of the American Chemical Society. 1982;104:4546–4559. [Google Scholar]
- 48.Jin D, Figueirido F, Montelione GT, Levy RM. Impact of the Precision in NMR Relaxation Measurements on the Interpretation of Protein Dynamics. Journal of the American Chemical Society. 1997;119:6923–6924. [Google Scholar]
- 49.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. Journal of the American Chemical Society. 1990;112:4989–4991. [Google Scholar]
- 50.Baber JL, Szabo A, Tjandra N. Analysis of Slow Interdomain Motion of Macromolecules Using NMR Relaxation Data. Journal of the American Chemical Society. 2001;123:3953–3959. doi: 10.1021/ja0041876. [DOI] [PubMed] [Google Scholar]
- 51.Chang S-L, Tjandra N. Analysis of NMR Relaxation Data of Biomolecules with Slow Domain Motions Using Wobble-in-a-Cone Approximation. Journal of the American Chemical Society. 2001;123:11484–11485. doi: 10.1021/ja016862x. [DOI] [PubMed] [Google Scholar]
- 52.Dosset P, Hus J-C, Blackledge M, Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. Journal of Biomolecular NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 53.García de la Torre J, Huertas ML, Carrasco B. HYDRONMR: Prediction of NMR Relaxation of Globular Proteins from Atomic-Level Structures and Hydrodynamic Calculations. Journal of Magnetic Resonance. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 54.Peng JW, Wagner G. Mapping of the spectral densities of nitrogen-hydrogen bond motions in Eglin c using heteronuclear relaxation experiments. Biochemistry. 1992;31:8571–8586. doi: 10.1021/bi00151a027. [DOI] [PubMed] [Google Scholar]
- 55.Lee BM, De Guzman RN, Turner BG, Tjandra N, Summers MF. Dynamical behavior of the HIV-1 nucleocapsid protein. Journal of Molecular Biology. 1998;279:633–649. doi: 10.1006/jmbi.1998.1766. [DOI] [PubMed] [Google Scholar]
- 56.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 57.Schwalbe H, Fiebig KM, Buck M, Jones JA, Grimshaw SB, Spencer A, Glaser SJ, Smith LJ, Dobson CM. Structural and Dynamical Properties of a Denatured Protein. Heteronuclear 3D NMR Experiments and Theoretical Simulations of Lysozyme in 8 M Urea. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 58.Gorelick RJ, Chabot DJ, Rein A, Henderson LE, Arthur LO. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J. Virol. 1993;67:4027–4036. doi: 10.1128/jvi.67.7.4027-4036.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bartonova V, Igonet S. b., Sticht J, Glass B. r., Habermann A, Vaney M-C, Sehr P, Lewis J, Rey FA, Kraüsslich H-G. Residues in the HIV-1 Capsid Assembly Inhibitor Binding Site Are Essential for Maintaining the Assembly-competent Quaternary Structure of the Capsid Protein. Journal of Biological Chemistry. 2008;283:32024–32033. doi: 10.1074/jbc.M804230200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.