

Abstract
The murine chronic GVH (cGVH) model of SLE is induced by allo-recognition of foreign major histocompatibility complex (MHC) Class II determinants. Previous studies have shown that syngeneic CD4+ T cells are needed during B cell development in order to induce cGVH response in CD4KO mice. Our present studies show that B cells require “nurturing” by CD4 T cells through much of their ontogeny in order to respond to allo-signaling and become autoreactive. The nurturing process does not require antigen-specific cognate interactions between CD4 T cells and B cells. It is mediated by IL-4, but not IL-10, IL-6 and IFN-γ. The CD4 T-cell nurturing may be supplanted by large doses of IL-4 and or by agonistic anti-CD40 mAb. Understanding the mechanism of this “nurturing” process may yield clues to the role of CD4 T cells in lupus and in host defense in general.
Keywords: Graft versus host disease, B cells, T cells, Systemic Lupus Erythematosus, Autoantibodies
INTRODUCTION
Systemic lupus erythematosus (SLE) is a multi-organ autoimmune disease that is associated with a variety of auto-abs that target normal proteins and nucleic acids such as chromatin, Sm, DNA and other histones [1]. The chronic GVH (cGVH) system in mice has been developed as one of the best experimentally induced models of SLE. It is established by transferring I-A-incompatible spleen cells from non-autoimmune B6.C-H2bm12/KhEg (bm12) mice into co-isogenic C57BL/6 (B6) recipients [2; 3]. The bm12 strain has a mutant form of I-A that differs by three amino acids from the I-Ab of B6, so that CD4+ donor T cells reactive to the wild type I-A interact with host B cells to provoke the disease [4; 5]. The gamut of autoantibodies produced includes specificities that are characteristic of SLE. The immunopathological changes are also typical of lupus, including diffuse proliferative glomerulonephritis with deposition of immunoglobulin and complement in the glomerular capillary loops [6].
A considerable body of literature shows that T cells play a crucial role in promoting the development of human lupus by rendering help to autoreactive B cells and by facilitating tissue damage in end organs [7]. The inducible cGVH model, on the other hand, does not predicate any role of endogenous CD4 T cells, since it is mediated mainly by cognate recognition of recipient B cells by allo-reactive donor CD4 T cells [2; 8]. However, several studies have indicated that recipient T cells may also be involved in mediating the autoimmune disease. Merino et. al., showed in an in vitro system that the ability of B cells from F1 hybrid mice to respond to allogeneic help from parental T cells depended on the presence of CD4+ T cells in the F1 B cell recipients [9]. Studies from our laboratory also revealed that cGVH reactions could not be established in CD4KO mice, indicating a central role for endogenous CD4 T cells in development of autoreactivity [10]. Our more recent study further highlighted that absence of endogenous (host) CD4 T cells leads to certain underlying functional aberrations in B cells in CD4KO mice and renders them resistant to allo-stimulation. These intrinsic B-cell defects could be remedied only if syngeneic CD4 T cells were provided during B-cell development [11].
Our present studies were undertaken to explore further the mechanisms of this endogenous CD4 T cell requirement in promoting B cells responsive to allo-stimulation. Our results show that the presence of CD4 T cells is essential during B cell ontogeny for these B cells to respond to cGVH reactions. We have termed this process “nurturing”, and we show further that this mechanism does not require cognate interactions between CD4 T cells and B cells and is mediated by IL-4, but not by IL-10, IL-6 or IFN-γ. The CD4 T cell nurturing process can also be mimicked by high doses of IL-4 or by an anti-CD40 agonistic mAb.
MATERIALS AND METHODS
Mice
C57BL/6-Cd4tm1Mak (CD4KO), C57BL/6J (B6) and B6.C-H2bm12/KhEg (bm12) mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were subsequently bred and maintained in our mouse colony at the University of Pennsylvania Medical Center. B6/Thy1.1, IFN-γ KO, IL-10 KO, IL-4 KO and IL-6 KO, all on C57BL/6 background, were purchased from The Jackson Laboratory (Bar Harbor, ME). AND x RAG−/− (on C57BL/6 background) was kindly provided by Dr. Terri Laufer, University of Pennsylvania. Recipient and donor mice were sex and age-matched within each independent experiment. All the experimental procedures performed on these animals were conducted according to the guidelines of the Institutional Animal Care and Use Committee.
Adoptive cell transfer and experimental cGVH disease
CD4KO recipients were given sub-lethal dose of 5 Gy (using Cs-137), according to Allman et al [12; 13] to remove their peripheral B cells. Five to ten million purified syngeneic CD4 T cells were transferred intravenously on the following day. cGVH disease was induced, in most cases, on day 22 post-irradiation by injecting (i.p) 40–50 × 106 bm12 CD4 T cells as previously described [11]. Blood samples were obtained from experimental mice before the induction of cGVH disease, at one week afterward, and at 2- to 4-wk intervals thereafter. Sera were stored at −20°C for later analysis.
Cell separation using magnetic beads
Splenic B cells were purified by using a B-cell isolation kit, and CD4 T cells were purified by using anti-CD4 magnetic beads, purchased from Miltenyi Biotec (Auburn, CA), and the AutoMACS magnetic column. Briefly, splenic cell suspensions were prepared by pressing donor spleens through a wire mesh screen in HBSS and lysing red blood cells with ACK buffer. Cells were incubated with magnetic beads at 6–12° C for 15–20 mins at a concentration of 10 µl of beads/107 cells in 90 µl of MACS buffer (PBS+ 0.5% BSA+ 2mM EDTA). The cells were washed after labeling and re-suspended in the MACS buffer before proceeding for magnetic separation. The purity of cell separation was checked by flow cytometry. Only cell populations with purity > 98% were used in the experiments.
Antibody and cytokine treatments
FGK115, a CD40 agonist rat IgG2a mAb, was provided by Claudia Mauri (University College London, London, United Kingdom). The mab was purified from culture supernatants by affinity chromatography, using Hi-Trap Protein G column (GE Healthcare, AB). The mice were treated every other day with 100 µg/ml purified FGK115 mAb for three weeks. Anti-Thy1.1 mAb (clone 1A14) was purified from culture supernatants by Protein-G column chromatography (Amersham Biotech, Sweden), and 100 µg were injected intraperitoneally for depletion of donor T cells. Recombinant mouse IL-4 and purified anti-mouse IL-4 (clone 11B11) were purchased from eBiosciences (San Diego, CA). IL-4 was mixed at a 2:1 molar ratio (1:6 weight ratio) with anti-IL-4 mAb (11B11) to prepare IL-4/anti-IL-4 mAb complexes (IL-4C), which greatly increases the in vivo half-life and activity of IL-4 [14]. After 2 min at room temperature, complexes were diluted with 1% CD4KO serum to a concentration of 1µg/ml for injection. The complexes were always freshly prepared before use. The mice were treated thrice a week for a total of three weeks.
Detection of autoantibodies
Autoantibodies were assessed by ELISA, as previously described [11; 15]. Briefly, plates were coated with optimal concentration of autoantigens: 1) Chromatin, purified from chicken erythrocyte nuclei, was used at 5 µg/ml; 2) dsDNA from calf thymus DNA (Sigma-Aldrich, St. Louis, MO) was extracted with chloroform, precipitated by addition of ethanol, treated with S1 nuclease for 45 min at 37°C to remove single strand regions, and used at 3 µg/ml. Antigens were diluted in borate-buffered saline (BBS), added to polyvinyl micro-titer plates (Dynatech Laboratories, Alexandria, VA), and incubated overnight at 4°C. For the anti-dsDNA ELISA, plates were first coated with 1 µg/ml poly-L-lysine (Sigma-Aldrich, St. Louis, MO), before incubating with the autoantigen. The plates were washed with BBS and blocked with BBT (BBS, 0.4% Tween 80, 0.5% BSA, and 0.1% NaN3) for 1 h at room temperature. Serum samples, diluted 1/250 in BBT, were added in duplicate and incubated overnight at 4°C. Biotinylated goat anti–mouse IgG (pFc' specific; Jackson ImmunoResearch Laboratories, West Grove, PA) was added as secondary Ab. For reference, standard serum from a diseased MRL/lpr mouse with high titer autoantibodies was tested at serial two-fold dilutions from 1:250 to 1:128,000. The plates were washed and incubated for 1 h at room temperature with avidin-alkaline phosphatase (Zymed Laboratories, South San Francisco, CA). The plates were washed again, and para-nitrophenyl phosphate substrate (Sigma, St. Louis, MO), 1 mg/ml in 0.01 M diethanolamine, pH 9.8, was added. The plates were read at various time points with an automated ELISA reader (Molecular Devices, Sunnyvale, CA).
Evaluation of urine protein
Urine protein concentration was detected at 2- 4-wk intervals by using Uristix reagent strips (Bayer Corporation, Elkhart, IN). The strips were wetted with fresh urine and immediately scored according to color change on a scale of 0–4.
Immunofluorescence staining
The following conjugated Abs were purchased from BD PharMingen (San Diego, CA): APC anti-CD19 (1D3), PE anti-CD40 (3/23), FITC anti-class II (AF6-120.1), PE anti-B7-2 (GL1), PE anti-CD23 (B3B4) and PE anti-Fas (Jo2). Anti-FcγR (2.4G2), used for blocking, was grown in our laboratory. Cell surface staining was routinely performed with age- and sex-matched controls, as previously described [11; 16]. A total of 1.5 × 106 cells were blocked with 50 µl of 2.4G2 culture supernatant. The cells were then incubated with directly labeled Abs for 30 min and washed. Cells were fixed in PBS containing 1% paraformaldehyde and analyzed on a BD Biosciences FACScan (Mountain View, CA). Relative fluorescence intensity was plotted on a logarithmic scale using Flow-Jo software.
Statistical analysis
Statistical analysis was performed using Student’s t test. A value of p < 0.05 was considered to be significant.
RESULTS
Kinetics of CD4 T-cell help needed during B-cell development
Our previous work showed that CD4 T cells are required during the development and maturation of B cells in order to elicit an autoimmune response after induction of cGVH [11]. This led us to investigate further the timing of the CD4 T cell help. We utilized the irradiation-auto-reconstitution model established by Allman et al [12; 13]. CD4KO mice were subjected to a sub-lethal dose of irradiation (5 Gy). Ten million CD4 T cells, purified from B6 mice by using anti-CD4 magnetic beads, were transferred intravenously at different time points to irradiated recipients. One group of recipients received CD4 T cells on day 1 following irradiation, so that the initial wave of newly generated splenic B cells would mature in the presence of these syngeneic CD4 T cells. A second group of irradiated recipients received CD4 T cells at day 7 post-irradiation, because during the radiation-induced auto reconstitution process some of the early immature splenic B cells differentiate into mature B cells within 5–7 days. Thus, at that point the peripheral B cell pool would consists of both mature and early immature B cells. Another experimental group received CD4 T cells at day 21 post-radiation, when the peripheral reconstitution process is presumed to have concluded. A control group of recipients received no syngeneic CD4 T cells. cGVH was induced in all four groups at day 22 by transferring allogeneic bm12 CD4 T cells. Figure 1 shows the serum anti-dsDNA and anti-chromatin autoantibodies in the different groups of CD4KO recipients following induction of cGVH. Only recipients that received syngeneic CD4 T cells at day 1 showed significant autoantibody levels compared to the negative control group. Our data thus indicate that the response to cGVH is dependent on the stage at which the developing B cells had been exposed to syngeneic CD4 T cells. Only immature B cells whose entire development and maturation has occurred in the presence of syngeneic CD4 T cells are capable of responding fully to allo-stimulation. When CD4 T cells are transferred to a mixed pool of mature and immature B cells (recipients at day 7) or to a completely mature B-cell population which had never been exposed to CD4 T cells (day 21 recipients), the elicited autoantibody response is greatly diminished.
Fig. 1. CD4 T cells during early B cell ontogeny permit B cells to respond to allo-stimulus.
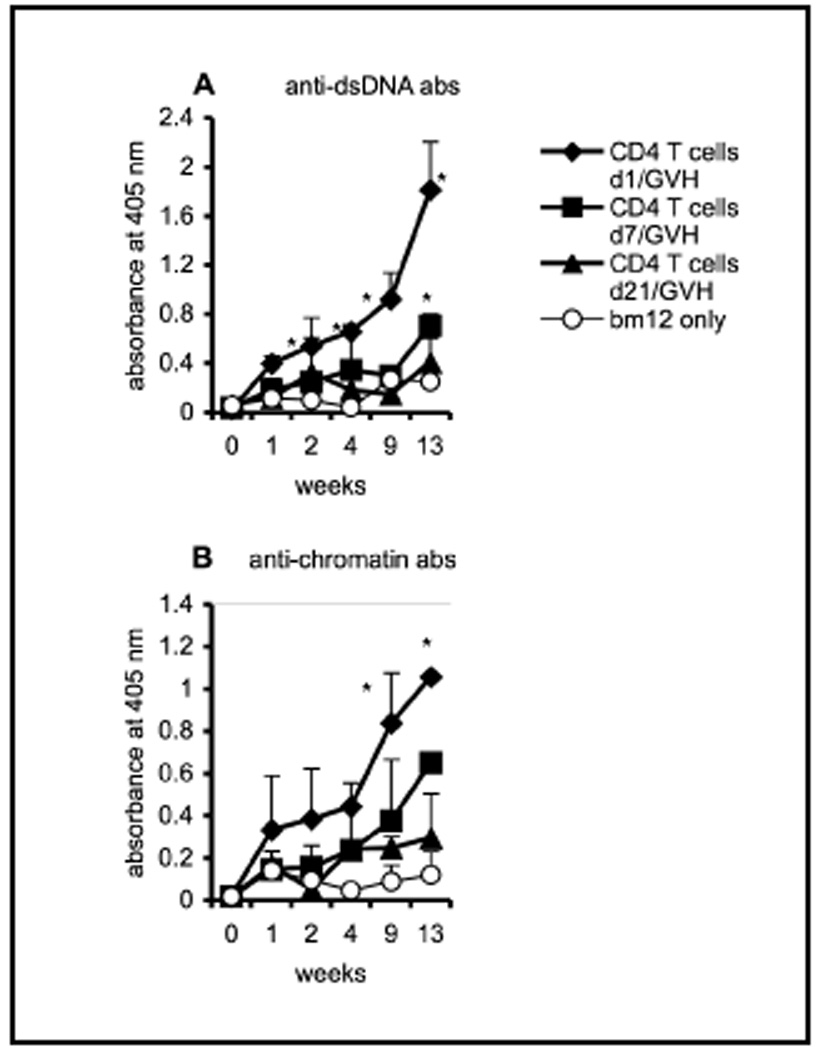
Ten million CD4 T cells, purified from B6 mice, were transferred intravenously into different groups of irradiated (5 Gy) CD4KO recipients (n=5) at days 1 (filled diamonds), 7 (filled squares) or 21 (filled triangles). The negative control group (open circles) received no syngeneic donor CD4 T cells. cGVH was induced in all four groups by transferring 40 × 106 bm12 allogeneic CD4 T cells on day 22 post-irradiation. Each group consisted of 4 mice, except the negative control group (n=3). Serum autoantibodies, (A) anti-dsDNA and (B) anti-chromatin antibodies, were evaluated by ELISA. *, p < 0.05 compared to the negative control group that received only bm12 T cells. The data are representative of two independent experiments. The error bars represent mean ±SE of the samples.
To define better the time frame needed for the action of CD4 T cells during the early ontogeny of B cells in equipping B cells to respond to allo-stimulation, we modified the irradiation-autoreconstitution model so that we could remove the CD4 T cells at various time points. Ten million CD4 T cells purified from B6.Thy1.1 mice were transferred to irradiated CD4KO recipients on day 0. The donor CD4 T cells were then depleted with a single injection of 100 µg of anti-Thy1.1 mAb (1A14) on days 1, 7, 14 or 21 post irradiation and cell transfer, and cGVH was established in all groups at day 22 by transferring CD4 T cells from bm12 mice. Using B6.Thy1.1 mice as I-A compatible donors guaranteed that the mAb treatment would exclusively deplete the donor CD4 T cells and not affect the allogeneic bm12 cells that express Thy1.2 allele. As shown in Figure 2, CD4 T cells needed to be present during the entire 21 days of B cell auto-reconstitution and maturation process in order to prepare recipients to mount a full cGVH reaction, as measured by subsequent anti-DNA and anti-chromatin antibodies and proteinuria. Depleting the transferred syngeneic T cells at day 1 greatly impaired the cGVH response. Depletion at day 7 also strongly suppressed the cGVH response, while depletion at day 14 did not affect the autoantibody levels, although it did attenuate the proteinuria. Depletion at day 21 had no discernable effect compared to mice not depleted of syngeneic CD4 T cells. Our data, therefore, demonstrate that presence of MHC syngeneic CD4 T cells is necessary to “nurture” B cells during their ontogeny and development in order to enable B cells to respond to an allo-stimulus, yet they play no role in the subsequent cGVH itself.
Fig. 2. CD4 T cells are needed throughout the B cell autoreconstitution and maturation process.
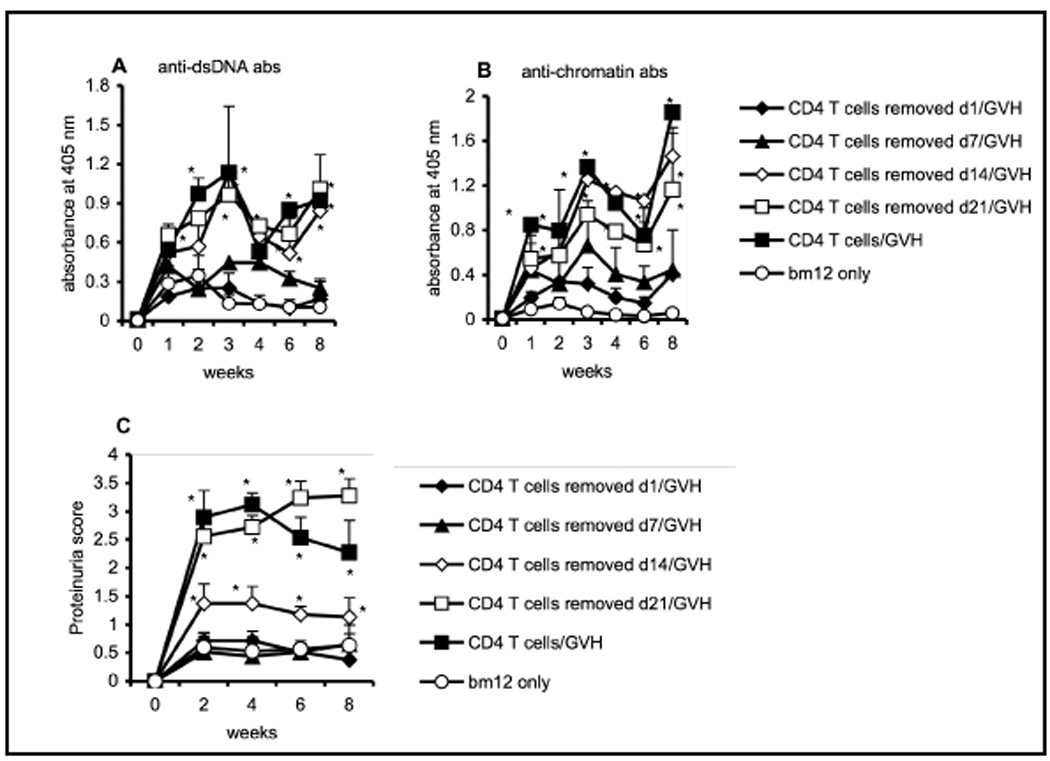
Ten million CD4 T cells, purified from B6.Thy1.1 mice, were transferred intravenously into different groups (n=5) of irradiated (5 Gy) CD4KO recipients. The donor syngeneic T cells were then depleted with 100 µg per mouse of anti-Thy1.1 mAb (1A14) at various time points post-irradiation and cell transfer: day 1 (filled diamonds); day 7 (filled triangles); day 14 (open diamonds) or day 21 (open squares). The positive control group (filled squares) received donor CD4 T cells, but the cells were not depleted with mAb. The negative control group (open circles) received no donor CD4 T cells. All the groups were challenged with 40–50 × 106 bm12 T cells at day 22 post-irradiation. (A) anti-dsDNA and (B) anti-chromatin antibodies. (C) proteinuria. *, p < 0.05 compared to the negative control group. The data are representative of two separate experiments.
Many of the critical interactions between CD4 T cells and B cells are known to be contact-mediated, usually involving MHC + peptide as well as various co-receptors [17]. We, therefore, wanted to investigate whether specific cognate interactions between donor CD4 T cells and the newly formed immature peripheral B cells in CD4KO mice were required for the B cells to respond subsequently to allo-stimulus. In order to address this question, CD4KO recipients were again subjected to sub-lethal radiation (5 Gy) to eliminate their peripheral B cell population. Then, 5 × 106 million purified CD4 T cells from AND-TCR tg mice (backcrossed onto RAG-1−/− mice) were transferred to the irradiated recipients. AND transgenic T cells were positively selected on a H-2b background [18] and the absence of rag ensured that no endogenous T-cell receptor rearrangement occurred. The bm12 T cells were transferred at day 22 post-irradiation. Our data in Figure 3 show that adoptively transferred tg CD4 T cells were just as effective as wild-type B6 CD4 T cells in assisting the ability of B cells to secrete (A) anti-dsDNA and (B) anti-chromatin autoantibodies in cGVH syndrome. These data suggest that the antigenic specificity of the “nurturing” CD4 T cells is irrelevant.
Fig. 3. Cognate interactions between CD4 T cells and B cells are not required to nurture B cells to react to an allo-stimulus.
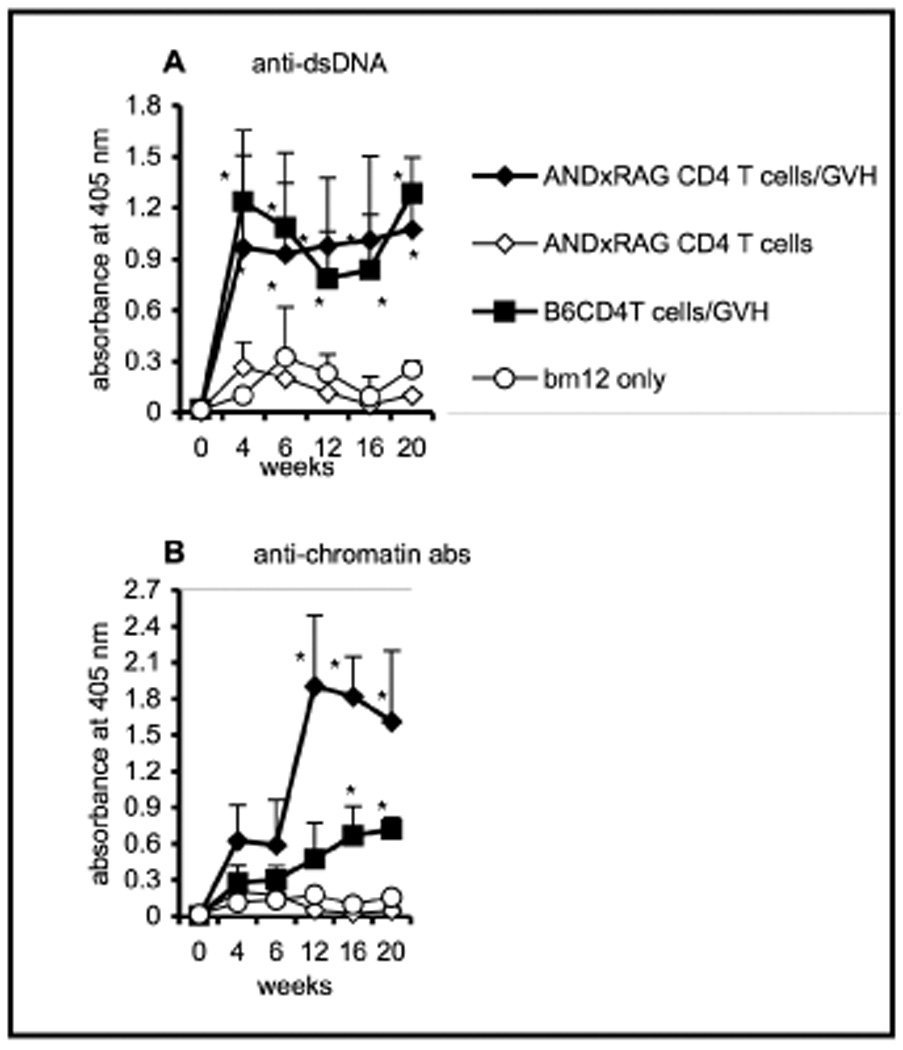
Purified CD4 T cells (5 × 106) from AND x RAG mice were transferred i.v. to irradiated (5 Gy) CD4KO recipients (n=5). The experimental group received AND x RAG CD4 T cells and bm12 T cells (filled diamonds). Control groups received only AND x RAG CD4 T cells and no bm12 cells (open diamonds); only bm12 cells and no AND x RAG CD4 T cells (open circles); or five million B6 CD4 T cells and bm12 T cells (filled squares). (A) anti-dsDNA and (B) anti-chromatin antibodies. *, p < 0.05 compared to the control group that received only bm12 cells. The data are representative of two experiments. Error bars represent SEM of the samples.
Role of T-cell cytokines in the nurturing of B cells
Our data using AND transgenic T cells indicate that, although CD4 T cells need to be present during the course of B cell development, the “nurturing” need not require interaction of a diverse population of antigen-specific T cells with the developing B cells. This suggested that the T-cell “nurturing” process might be mediated through cytokines. Such a mechanism would not be surprising, since T cell cytokines are known to have a profound impact on B-cell development and to influence the outcome of immune responses [19]. To test the requirement for cytokines in nurturing, we used B6 T cells that were genetically deficient in several cytokines in the B-cell autoreconstitution system. CD4KO recipients were irradiated (5 Gy), and CD4 T cells purified from various cytokine-knockout mice were transferred on the following day. cGVH was established on day 22 by injecting bm12 CD4 T cells. As shown in Figure 4, B cells from CD4KO recipients that received donor CD4 T cells from IFN-γKO, IL-10KO and IL-6KO mice mounted vigorous cGVH responses with elevated autoantibody titers, indicating that these cytokines were not essential to render B cells receptive to allo-signals. In contrast, IL-4 deficient T cells were completely ineffective in “nurturing” B cells and making them reactive to cGVH stimulation. Taken together, our data demonstrate IL-4 to be indispensable for B cells to acquire the ability to respond to cGVH reactions.
Fig. 4. The role of certain cytokines in nurturing developing B cells to respond to the cGVH.
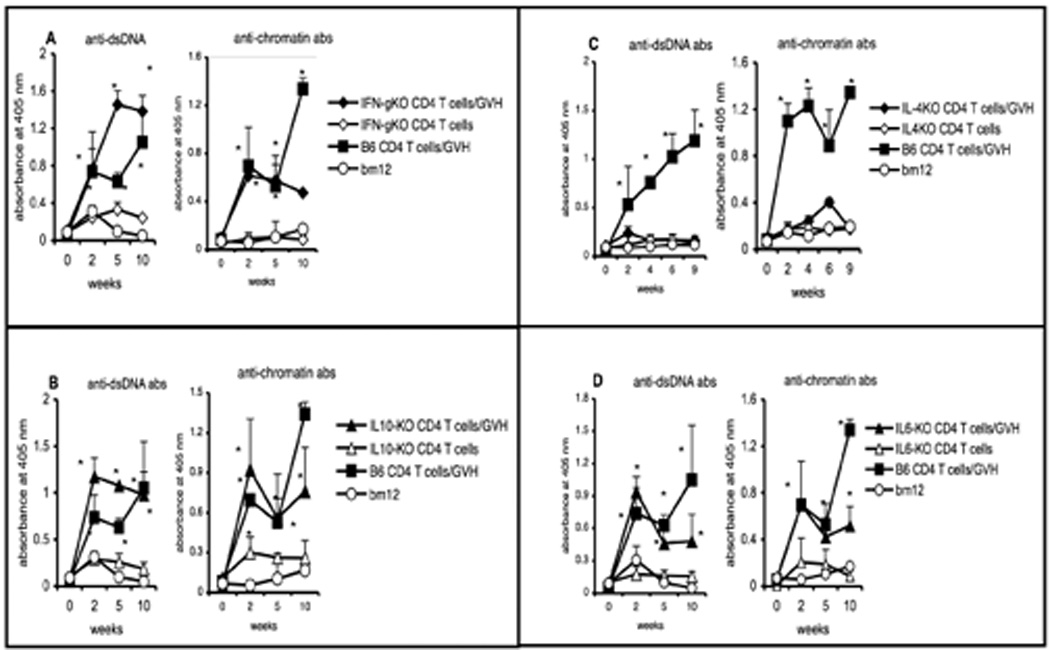
Purified CD4 T cells (5 × 106) from various cytokine-KO B6 mice were transferred i.v. into irradiated (5 Gy) CD4KO recipients (n=5). cGVH was induced by transferring 40 × 106 bm12 T cells on day 22 post-irradiation (filled symbols). Control groups received CD4 T cells from the different KO mice but no bm12 cells (open symbols). The positive control group received WT B6 CD4 T cells and bm12 T cells (filled squares) and the negative control group received only bm12 T cells (open circles). Anti-dsDNA and anti-chromatin antibodies are shown for recipients with donor T cells from (A) IFN-γKO (diamonds) mice; (B) IL-10KO (triangles) mice; (C) IL-4KO (diamonds) mice. (D) IL-6KO (triangles) mice. *, p < 0.05 compared to the control group that received only bm12 cells. The data presented in panels A, B and D were obtained from a single ELISA assay. The data are representative of two experiments. Error bars represent SEM of the samples.
rIL-4 can reconstitute B cell function in vivo and elicit cGVH response
IL-4 has been known to augment B-cell immune responses and the survival [20] and maturation of autoreactive B cells [21]. Since our previous data also suggested IL-4 as an important component in nurturing B cells to become reactive to an allo-stimulus, we investigated whether large doses of this cytokine could substitute for CD4 T cells. Irradiated (5 Gy) CD4KO recipients were treated with recombinant IL-4 (rIL-4) at a dose of 1 µg per mouse, given in combination with anti-IL-4 mAb, starting from day 1 post irradiation, three times a week for three weeks. cGVH was induced by transferring bm12 CD4 T cells on day 22 post-radiation. Interestingly, most of our experimental recipients perished in less than a week, presumably due to very intense cGVH reactions. The control recipients that received rIL-4 treatment, but no allo T cells, survived with no evidence of ill effects. In fact, even 2-wks after stopping rIL-4 treatment, transfer of bm12 CD4 T cells to these ‘control’ mice induced high levels of autoantibodies (data not shown). We therefore repeated the rIL-4 treatment experiment with a 10-day hiatus between the last dose of IL-4 and the transfer of bm12 T cells. Our results (Figure 5) showed that B cells from this rIL-4 treatment group produced anti-DNA and anti-chromatin autoantibodies. At 2-wks post cGVH induction, the IgG subclass distribution of total serum IgG and of anti-DNA antibodies was also comparable between recipients that received rIL-4 and those that received syngenic CD4 T cells (Supplemental Figures 1 and 2). However, the antibody levels declined more rapidly in the IL-4 treated group compared to the positive control group that received B6 CD4 T cells. Nevertheless, the course of renal involvement, as indicated by proteinuria scores, was similar in the two groups that developed autoantibodies. Along with the positive control mice, the IL-4-treated mice also developed dermatitis and skin lesions that are characteristic manifestations of cGVH reactions. Interestingly, the IL-4-treated mice also showed patches of skin de-pigmentation. At the end of 12-wks, the mice were sacrificed and their splenic B cells were analyzed by FACS. Activation markers such as CD86, MHC II, CD40 and Fas and developmental markers such CD23 were up-regulated on gated B cells in the experimental group compared to the negative control group in accordance to the phenotypic changes associated with cGVH seen in our previous studies [11; 16]. Taken together, these data indicate that administration of rIL-4 can substitute for CD4 T cell help and can confer on B cells the ability to respond in cGVH reactions.
Fig. 5. Treatment with recombinant IL-4 during B-cell auto-reconstitution permits a cGVH response in CD4KO mice.
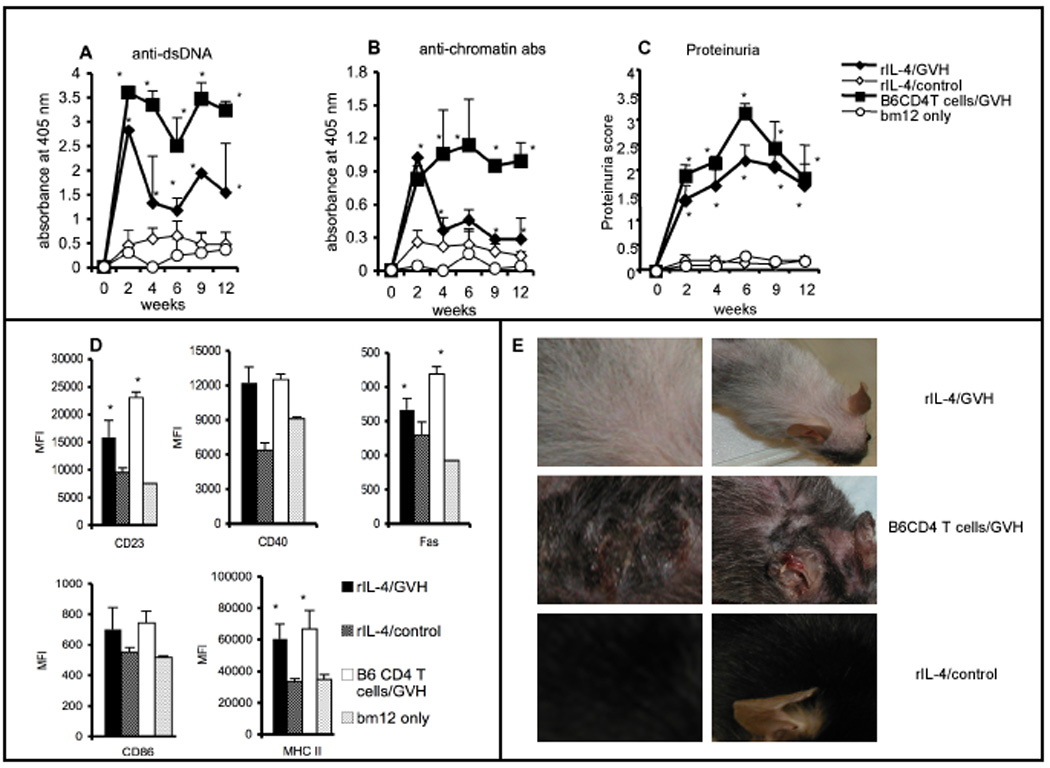
CD4KO mice were irradiated (5 Gy) to remove peripheral B cells. They were treated with 1 µg per mouse of rIL-4 thrice a week for three weeks. cGVH was induced by transferring 40 × 106 bm12 T cells on day 32 post-radiation (10 days after the last IL-4 treatment). The experimental group (n=7; filled diamonds) received rIL-4 treatment and bm12 T cells. The positive control group (n=4; filled squares) received B6 CD4 T cells, instead of rIL-4, plus bm12 T cells, while the negative control group (n=3; open circles) received only bm12 cells and no rIL-4. Another control group (n=5; open diamonds) received only rIL-4 but no bm12 cells. (A) anti-dsDNA; (B) anti-chromatin antibodies; (C) proteinuria score. (D) Mice were sacrificed at 12-wk, and CD19+-gated spleen cells were analyzed by flow cytometry for activation and developmental markers. (E) Skin de-pigmentation and lesions seen in experimental groups. The left panel shows a high resolution image and the right panel shows a low resolution image. *, p < 0.05, compared to the negative control group that received only bm12 cells. Error bars represent SEM of samples.
Treatment with agonist anti-CD40 mAb induces cGVH response in CD4KO mice
CD40, a member of TNF receptor family, is a potent regulator of lymphocyte function. The interaction of CD40 on B cells with its ligand (CD40L) on T cells provides a B-cell co-stimulatory signal that induces proliferation, Ig production with class switching, germinal center formation and B-cell memory [22; 23; 24]. In some situations, stimulation through CD40 alone can mimic the effects of T-B interactions. We therefore tested whether an agonist anti-CD40 mAb could nurture B cells during their development. Starting from day 1, after 5 Gy irradiation, CD4KO mice were treated every other day with 100 µg of an agonistic anti-CD40 mAb (FGK-115, a rat IgG2a Ab). The treatment continued for 21 days, and cGVH was induced on day 22 with bm12 T cells. As shown in Figure 6, anti-CD40-treated CD4KO mice demonstrated a robust cGVH reaction, with high titers of anti-DNA and anti-chromatin autoantibodies, as well as significant increases in urine protein excretion. In addition, they had extensive skin lesions, splenomegaly, and weight loss (Figure 6D and data not shown). The severity of the cGVH reaction required the experiment to be terminated at 6 wks, at which time the splenic B cells in the anti-CD40 + bm12 T cell group showed up-regulated B cell activation markers such as CD86, Fas and MHC II (data not shown), consistent with phenotypic changes normally displayed in cGVH. Taken together, our data demonstrate that cross-linking CD40 on B cells by using anti-CD40 agonistic mAb provides surrogate CD4 T cell nurturing to B cells in CD4KO mice and confers upon them the ability to respond to allo-stimulus and become autoreactive.
Fig. 6. Treatment with agonistic anti-CD40 mAb substitutes for CD4 T cells during B cell ontogeny.
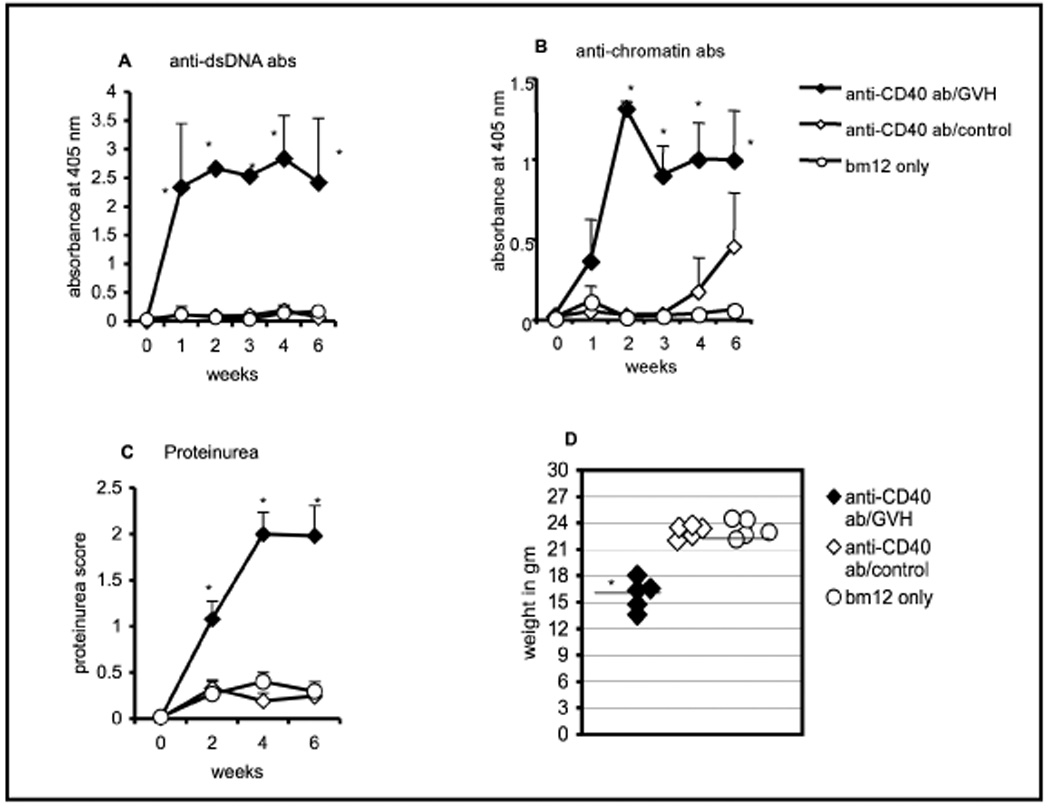
CD4KO mice were irradiated (5 Gy). The experimental group (n=5; filled diamonds) received 100 µg/ml of anti-CD40 mAb every other day for 3 wks before inducing cGVH with bm12 T cells at day 22. The negative control groups either received no mAb treatment and only bm12 T cells (n=3; open circles) or only mAb but no bm12 T cells (n=5; open diamonds). (A) Serum anti-dsDNA (B) anti-chromatin antibodies (C) proteinuria were measured at 2-wk intervals. (D) Body weights at 6-wks after cGVH was induced. *, p < 0.05 comparing the experimental group with the negative control group with only bm12 T cells. The data represent two independent experiments. Error bars represent SEM of the samples.
DISCUSSION
The autoimmune cGVH syndrome is due to cognate interactions of host B cells with allo-reactive donor T cells [2; 3]. Although such a model does not postulate an essential role for the recipient’s endogenous T cells, our previous work has shown that host CD4 T cells were essential [10; 11]. B cells from CD4KO mice failed to generate a cGVH response, even in the presence of syngeneic CD4 T cells. However, if syngeneic T cells are present during the ontogeny of B cells, they become capable of responding to the cGVH reactions [11]. In this study we further investigated how endogenous CD4 T cells "nurture" developing B cells and confer upon them the ability to react to cGVH stimulus. Our results show that this "nurturing" process does not require antigen-specific cognate interactions between CD4 T cells and B cells, but does require IL-4. The CD4 T cell nurturing may also be accomplished by large doses of recombinant IL-4 and or agonistic anti-CD40 mAb.
Our previous studies showed that cGVH could be induced only when B cells matured in the presence of CD4 T cells over 21 days [11]. However, the studies did not define the precise timing of the CD4 help. Our present studies further elucidate this issue. We found that CD4 T cells are needed very early during B cell maturation. When syngenic CD4 T cells were transferred to a heterogeneous pool of immature or mature B cells seven days after sub-lethal irradiation of CD4KO mice, the B cell response to cGVH was greatly impaired. Since we induce cGVH in all the recipients at the same time, it is possible to argue that the disparity in B cell response in different groups of recipients is mainly due to difference in duration of T-B interactions, and does not reflect the need for CD4 T cells in early B-cell development. To address this, we transferred Thy-1 congenic donor B6 T cells on day 1 and then, depleted the CD4 T cells at different time points. This approach provided various windows during which CD4 T cells would be present during peripheral B cell maturation. Our data demonstrated clearly that eradicating CD4 T cells at seven days severely attenuates the ability of B cells to react to allogeneic stimuli and become autoreactive, while removing them at 14 days had a much smaller effect. Therefore, the presence of CD4 T cells through much of the maturation of peripheral B cells is imperative for B cells to become autoreactive under cGVH stimulus.
Activation of B cells usually requires cognate interactions with CD4 T cells through multiple molecular interactions, the most important being the specific interactions of MHC II with the TCR, with the additional interaction of the CD4 co-receptor [25]. Studies have also shown that CD4-mediated help available for B cells responding to allo-antigens involves both a non-cognate interaction that activates B cells and a cognate interaction that is required for differentiation and IgG alloantibody production [26]. To explore these possibilities in our model, we used B6 donor CD4 T cells from AND x RAG −/− mice, in which all T cells bore the same TCR and endogenous TCR rearrangements was prevented. Impressively, these tg CD4 T cells provided very robust nurturing to B cells in their response to allo-stimulation, and the mice produced high titers of autoantibodies after induction of cGVH. Thus, the nurturing of B cells during their development does not require cognate recognition of the B cells by a polyclonal repertoire of CD4 T cells. On the other hand, the fact that the AND TCR is positively selected in the presence of I-Ab suggests that it must recognize this molecule, probably in the presence of some autologous peptides, and probably with low affinity, since it is not subsequently negatively selected. It is possible then that this level of recognition may play a role in B cell nurturing.
Our conception of the potential mechanisms for the B-cell nurturing must take into account our results with substituting soluble molecules for CD4 T cells. It is known that T-cell derived cytokines profoundly affect the pathogenesis of autoimmune diseases, such as lupus. Although IFN-γ, IL-6 and IL-10 are known to play pathogeneic role in development of lupus, our studies showed that these cytokines did not have any "nurturing" effect on autoreactive B cells and their absence also did not affect B cell response to cGVH stimulus. On the other hand, IL-4 seems to play a pivotal role in mediating B cell development and their response to allo-signal.
IL-4 is a multifunctional cytokine that is mainly secreted by CD4+ T cells. IL-4 has multiple effects on the antibody responses of B cells and most of these are mediated through the Stat6 pathway. IL-4 up-regulates the expression of several cell surface markers that are involved in T cell mediated B cell stimulation, such as MHC II, CD40, CD23, mIgM and the IL-4R. It also regulates isotype switching to IgG1 and IgE in B cells, promotes migration of circulating B cells to the spleen, inhibits spontaneous apoptosis of cultured B cells and decreases B-cell susceptibility to Fas-mediated killing during cognate interactions with CD4 T cells. Therefore, IL-4 plays a key role in providing T-cell derived help to B cells. IL-4 is also known to influence B-cell functions indirectly through its effects on T cells, mast cells, macrophages and dendritic cells [27]. Because IL-4 is known to stimulate B cells, it is not surprising that it has a role in mediating lupus like conditions. MRL/lpr mice treated with IL-4 antagonists had lower anti-dsDNA titers and associated renal disease. When MRL/lpr mice were back-crossed to IL-4−/− mice, they developed significantly reduced lymphadenopathy and end-organ disease. Treatment with anti-IL-4 mAb before the onset of lupus abrogated IgG anti-dsDNA antibody production in lupus-prone NZB/W F1 mice [28; 29]. Even in an inducible lupus model, Deocharan et al., showed that absence of IL-4 resulted in decreased autoantibody response [30]. All these reports highlight the importance of IL-4 in B cell activation. It is therefore not surprising that our data support the possibility that IL-4 is one of the key T-cell mediated cytokines that is required to nurture B cells and make them receptive to allo-signal. CD4KO recipients that received donor CD4 T cells from IL-4−/− mice, produced no autoantibodies upon induction of cGVH. In addition, treatment with large amounts rIL-4 could substitute for CD4 T cell help and nurture the B cells and predispose them to become autoreactive. It is possible that this effect of IL-4 is mediated by reducing expression of inhibitory receptors on B cells [31] and allowing the immature B cells to mature to a developmental stage that is more resistance to cell death and increasing the survival of autoreactive B cells in the spleen [20]. Interestingly, our data in fact showed that rIL-4 treatment generated more robust anti-dsDNA antibody responses than anti-chromatin responses.
It is known that pathogenic autoantibodies in lupus-prone mice generally belong to the IgG2a and IgG3 subclasses and these are generally suppressed by type 2 cytokines such as IL-4 [32]. However, in one study it was shown that over-expression of IL-4 by B cells of (NZW x C57BL/6.Yaa)F1 mice resulted in lower titers of IgG2a and in an increase in IgG1 sub-classes of anti-dsDNA antibodies [33]. Also, in some inducible lupus models (mercuric-chloride induced, pristane induced) the autoantibodies produced are mostly of IgG1 isotypes and are dependent on IL-4 [30; 34]. However, our data showed that rIL-4 treatment did not necessarily promote IgG1 subclasses and deviate the antibody response to type 2; rather all IgG subclasses, including IgG2c, IgG2b and IgG3 were significantly elevated. It is possible that upon activation by allo-T cells, the B cell autoreactivity is so intense that it drives the antibody response predominantly to Th1-type. It is interesting to note that the autoantibody levels in rIL-4 treated recipients abated significantly by 4 weeks after induction of cGVH, unlike the positive control recipients that were treated with syngeneic CD4 T cells. One possible explanation is that the syngeneic CD4 T cells persist in the recipients (data not shown) and can subsequently continue to nurture newly emerging immature B cells. On the other hand, the rIL-4 treatment was stopped before induction of cGVH, and it would not remain in circulation for more than 3 days [14]. Therefore, autoantibody responses in recipients with rIL-4 treatment gradually diminished.
Various studies have shown that IL-4 also affects lupus nephritis. NZM.2410 mice that over-express IL-4 in vivo develop severe glomerulosclerosis and renal disease [35]. Our studies also showed that IL-4 treated recipients had remarkably high urine protein concentrations. It is possible to argue that the autoantibody production and proteinuria scores in our IL-4 treated recipients have been in part augmented by effects of the IL-4 beyond the nurturing effect on B cells. It is also interesting to note that even though autoantibody titers subsided in rIL-4 treated recipients, the proteinuria level continued to remain high. Recent studies from Shu Man Fu's group using the (SWR x NZB) F1 mouse model, have demonstrated that lupus nephritis can persist even in the absence of anti-dsDNA antibodies [36]. In another study, it was shown that STAT-4 deficient NZM2410 mice developed renal disease despite decreases in anti-dsDNA antibody titers [35]. We do not know whether the progressive renal disease in our model is in part driven by autoantibody specificities other than anti-DNA or anti-chromatin or whether ongoing antibody-independent inflammatory processes are initiated in the damaged glomeruli.
One intriguing finding of our study was that several weeks after induction of cGVH in rIL-4 treated CD4KO recipients, many of the mice developed patches of skin de-pigmentation and vitiligo like conditions. This was not observed in controls that received only rIL-4 or recipients that received syngeneic CD4 T cells, followed by allo-T cells. Our previous studies in cGVH models have documented severe skin lesions with progression of the disease [10; 11]. We did observe skin lesions in our recipients with donor syngeneic T cells, but no skin discoloration. Vitiligo has been frequently reported in association with autoimmune diseases such as lupus, thyroid disease, diabetes mellitus and alopecia areata [37]. Many pro-inflammatory cytokines such as IL-1, IL-2, IL-4 and IL-13 are considered key players mediating the disease [38]. This is the first time we have observed vitiligo like conditions in our cGVH mice after rIL-4 treatment. It is also interesting to note that when we tried to establish cGVH responses immediately after stopping IL-4 treatment, most of our recipients perished within a week. This was not due to a direct toxic effect of IL-4, since the control recipients that received IL-4 but no allo-T cells, survived well. It is known through in vivo studies that IL-4 treatment strongly stimulate CD8+ T cell proliferation. Also, IL-4 can act rapidly through Stat6-dependent pathway to induce NK and NKT cells to produce IFN-γ [39]. Work from Via's group have shown that IFN-g production and CD8+ T cell activation lead to up-regulation of Fas [40] and to an acute GVH response in the parent-into-F1 model of GVH [41]. Although there was no obvious source of MHC class I directed allo-reactivity in our model, we speculate that our recipients might have suffered from acute GVH instead of a chronic one when a strong allo-stimulus was given immediately after rIL-4 treatment.
Signaling through CD40-CD40L interaction is considered an important aspect of cognate contact-dependent help for antibody production. CD40 is a 48kDa transmembrane glycoprotein cell surface receptor that shares sequence homology with TNF receptor family and is expressed by a variety of cells including B cells, macrophages, dendritic cells and endothelial cells. The ligand for CD40, CD154 (CD40L) is transiently expressed on activated T cells, mainly CD4 subsets. Engagement of CD40 on B cells with its ligand (CD40L) on T cells provides a B cell co-stimulatory signal that induces B cell proliferation, Ig production, class switching, germinal center formation, affinity maturation and generation of long lived plasma cells [23; 24]. The importance of CD40-CD154 ligation in the development of autoimmune disease has been illustrated in several murine models of autoimmunity using blocking abs and knockout mice [42; 43]. Recent data from SLE patients and murine lupus models have demonstrated that lupus T cells have prolonged expression of CD40L, and this probably leads to excessive B cell activation [44; 45]. CD40 agonists have long been used to boost immune responses [46]. Our data also demonstrate that engagement of CD40 by using an agonistic CD40 mab could effectively nurture B cells in CD4KO mice and prime them for allo-reactivity, in the absence of syngeneic CD4 T cells. Interestingly, upon induction of cGVH in recipients that received agonist CD40 mab, the disease severity was exacerbated. The mice had severe skin lesions (data not shown), elevated protein concentration in their urine and high rate of mortality. We postulate that heightened engagement of CD40 provides very robust nurturing effect to the B cells, and this is reflected in the magnified cGVH response in the recipients. This situation is perhaps analogous to the response we saw to pharmacologic doses of IL-4.
Although it is well documented that T-B cell interactions are needed to activate mature B cells in a TD response, our data further document an essential role for T-B interactions in order to nurture B cells and make them receptive to allo-stimuli. Our data elucidate that this T cell effect is required not only during the initial stages of B cell development, but also needs to be sustained over 2–3 weeks, ie., throughout the maturation of peripheral B cells. This process of nurturing is achieved in a polyclonal fashion and is not dependent on specific receptor interactions. It can also be effectively achieved through engagement of CD40-CD40L. Our data also highlight the importance of cytokines such as IL-4 in this nurturing process. We propose that a low affinity T-B interaction, analogous to positive selection of T cells as discussed above, would allow local interactions through CD40 and a local high concentration of IL-4, to signal B cells to become receptive to an eventual allo-stimulus. Thus, in the absence of T cells, massive amounts of IL-4 or a CD40 agonistic mab could provide the same signals.
The requirement for CD4 T cells during the early ontogeny of B cells in order to permit the B cells to respond to allo-stimulation reflects fundamental aspects of B-cell development. More recently, we have found that B cells from CD4KO mice are also incapable of responding to an exogenous T dependent antigen (Choudhury et al. manuscript in preparation). Since B cell development is an ongoing, life long process, this hitherto unrecognized T-B interaction may have implications beyond lupus and other autoimmune diseases. For example, these findings may also provide insights into role of CD4 T cells in HIV. HIV infection induces a wide array of B cell dysfunctions [47; 48]. Although the absence of immediate CD4 T cell help certainly must contribute to some of the immunodeficiency in these cases, our results suggest that the B cells themselves may become intrinsically defective, based on their ontogeny in a CD4 T cell deficient environment. HIV is also characterized by dysregulation of APC functions due to impaired CD40L function [49; 50]. It is therefore tempting to speculate that agonistic CD40 mab may abrogate some of the immune dysfunctions in this disease.
Supplementary Material
The experimental group (filled bar) received rIL-4 treatment followed by induction of cGVH. The positive control group (shaded bar) received B6CD4+ T cells, while the negative control group (open bar with vertical stripes) received only bm12 T cells. The other control group (open bar) was treated with rIL-4 only but received no bm12 T cells. Each bar represents the mean of ODs in each group, and the error bars represent the SEM of each group. N = 5 in each group.
See legend of Supplemental Figure 1. Each bar represents the mean of ODs in each group and the error bars represent the SEM of each group.
ACKNOWLEDGEMENTS
This work was supported by the NIH (R01-AR34156, R01-AI036206, R01-DE017590), the Lupus Research Institute, Alliance for Lupus Research, the United States Department of Veteran Affairs, the American Autoimmune Related Diseases Association and the Lupus Foundation of South Jersey. A. C. was supported by a postdoctoral fellowship from the Arthritis Foundation.
Abbreviations used in this paper
- SLE
systemic lupus erythematosus
- GVH
graft-vs-host
- cGVH
chronic GVH
- CD4KO
CD4 knockout mice
- bm12
B6.C-H2bm12 /KhEg mice
- B6
C57BL/6
- ab
antibodies
- autoab
autoantibodies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hahn BH, Tsao BP. Antibodies to DNA. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus. Baltimore: Williams & Wilkins; 1997. p. 407. [Google Scholar]
- 2.Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Autoantibodies in chronic graft versus host result from cognate T-B interactions. Journal of Experimental Medicine. 1990;171:503–517. doi: 10.1084/jem.171.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris SC, Cohen PL, Eisenberg RA. Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clinical Immunology & Immunopathology. 1990;57:263–273. doi: 10.1016/0090-1229(90)90040-w. [DOI] [PubMed] [Google Scholar]
- 4.Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Allotype-specific immunoregulation of autoantibody production by host B cells in chronic graft-versus host disease. Journal of Immunology. 1990;144:916–922. [PubMed] [Google Scholar]
- 5.Eisenberg RA. The Chronic Graft-versus-Host Model of Systemic Autoimmunity. In: Nemazee D, editor. B Cell Biology in Autoimmunity. Basel: Karger; 2002. pp. 228–243. [DOI] [PubMed] [Google Scholar]
- 6.Hansen TH, Tse HY. Insights into immune-response gene function using an Ia mutant mouse strain. Crit Rev Immunol. 1987;7:169–191. [PubMed] [Google Scholar]
- 7.La Cava A. Lupus and T cells. Lupus. 2009;18:196–201. doi: 10.1177/0961203308098191. [DOI] [PubMed] [Google Scholar]
- 8.Gleichmann E, Van Elven EH, Van der Veen JP. A systemic lupus erythematosus (SLE)-like disease in mice induced by abnormal T-B cell cooperation. Preferential formation of autoantibodies characteristic of SLE. Eur J Immunol. 1982;12:152–159. doi: 10.1002/eji.1830120210. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez M, Merino R, Gonzalez AL, Merino J. The ability of B cells to participate in allogeneic cognate T-B cell interactions in vitro depends on the presence of CD4+ T cells during their development. J Immunol. 1995;155:1091–1100. [PubMed] [Google Scholar]
- 10.Chen F, Maldonado MA, Madaio M, Eisenberg RA. The role of host (endogenous) T cells in chronic graft-versus-host autoimmune disease. J Immunol. 1998;161:5880–5885. [PubMed] [Google Scholar]
- 11.Choudhury A, Maldonado MA, Cohen PL, Eisenberg RA. The Role of Host CD4 T Cells in the Pathogenesis of the Chronic Graft-versus-Host Model of Systemic Lupus Erythematosus. J Immunol. 2005;174:7600–7609. doi: 10.4049/jimmunol.174.12.7600. [DOI] [PubMed] [Google Scholar]
- 12.Allman DM, Ferguson SE, Cancro MP. Peripheral B cell maturation. I. Immature peripheral B cells in adults are heat-stable antigenhi and exhibit unique signaling characteristics. J Immunol. 1992;149:2533–2540. [PubMed] [Google Scholar]
- 13.Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation. II. Heat-stable antigen(hi) splenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J Immunol. 1993;151:4431–4444. [PubMed] [Google Scholar]
- 14.Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, Maliszewski CR. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151:1235–1244. [PubMed] [Google Scholar]
- 15.Eisenberg R, Choudhury A. The anti-DNA knock-in model of systemic autoimmunity induced by the chronic graft-vs-host reaction. Methods Mol Med. 2004;102:273–284. doi: 10.1385/1-59259-805-6:273. [DOI] [PubMed] [Google Scholar]
- 16.Choudhury A, Cohen PL, Eisenberg RA. Mature B cells preferentially lose tolerance in the chronic graft-versus-host disease model of systemic lupus erythematosus. J Immunol. 2007;179:5564–5570. doi: 10.4049/jimmunol.179.8.5564. [DOI] [PubMed] [Google Scholar]
- 17.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 18.Kaye J, Hsu ML, Sauron ME, Jameson SC, Gascoigne NR, Hedrick SM. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- 19.Croft M, Swain SL. B cell response to fresh effector T helper cells. Role of cognate T-B interaction and the cytokines IL-2, IL-4, and IL-6. J Immunol. 1991;146:4055–4064. [PubMed] [Google Scholar]
- 20.Mori M, Morris SC, Orekhova T, Marinaro M, Giannini E, Finkelman FD. IL-4 promotes the migration of circulating B cells to the spleen and increases splenic B cell survival. J Immunol. 2000;164:5704–5712. doi: 10.4049/jimmunol.164.11.5704. [DOI] [PubMed] [Google Scholar]
- 21.Morris SC, Dragula NL, Finkelman FD. IL-4 promotes Stat6-dependent survival of autoreactive B cells in vivo without inducing autoantibody production. J Immunol. 2002;169:1696–1704. doi: 10.4049/jimmunol.169.4.1696. [DOI] [PubMed] [Google Scholar]
- 22.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 24.Laman JD, Claassen E, Noelle RJ. Functions of CD40 and its ligand, gp39 (CD40L) Crit Rev Immunol. 1996;16:59–108. doi: 10.1615/critrevimmunol.v16.i1.40. [DOI] [PubMed] [Google Scholar]
- 25.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–990. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele DJ, Laufer TM, Smiley ST, Ando Y, Grusby MJ, Glimcher LH, Auchincloss H., Jr Two levels of help for B cell alloantibody production. J Exp Med. 1996;183:699–703. doi: 10.1084/jem.183.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 28.Singh RR. IL-4 and many roads to lupuslike autoimmunity. Clin Immunol. 2003;108:73–79. doi: 10.1016/s1521-6616(03)00145-1. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima A, Hirose S, Yagita H, Okumura K. Roles of IL-4 and IL-12 in the development of lupus in NZB/W F1 mice. J Immunol. 1997;158:1466–1472. [PubMed] [Google Scholar]
- 30.Deocharan B, Marambio P, Edelman M, Putterman C. Differential effects of interleukin-4 in peptide induced autoimmunity. Clin Immunol. 2003;108:80–88. doi: 10.1016/s1521-6616(03)00096-2. [DOI] [PubMed] [Google Scholar]
- 31.Rudge EU, Cutler AJ, Pritchard NR, Smith KG. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and Fc gamma RII-mediated B cell suppression. J Exp Med. 2002;195:1079–1085. doi: 10.1084/jem.20011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi S, Fossati L, Iwamoto M, Merino R, Motta R, Kobayakawa T, Izui S. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest. 1996;97:1597–1604. doi: 10.1172/JCI118584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santiago ML, Fossati L, Jacquet C, Muller W, Izui S, Reininger L. Interleukin-4 protects against a genetically linked lupus-like autoimmune syndrome. J Exp Med. 1997;185:65–70. doi: 10.1084/jem.185.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubicka-Muranyi M, Kremer J, Rottmann N, Lubben B, Albers R, Bloksma N, Luhrmann R, Gleichmann E. Murine systemic autoimmune disease induced by mercuric chloride: T helper cells reacting to self proteins. Int Arch Allergy Immunol. 1996;109:11–20. doi: 10.1159/000237226. [DOI] [PubMed] [Google Scholar]
- 35.Singh RR, Saxena V, Zang S, Li L, Finkelman FD, Witte DP, Jacob CO. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol. 2003;170:4818–4825. doi: 10.4049/jimmunol.170.9.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshmukh US, Bagavant H, Fu SM. Role of anti-DNA antibodies in the pathogenesis of lupus nephritis. Autoimmun Rev. 2006;5:414–418. doi: 10.1016/j.autrev.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Kemp EH, Waterman EA, Waterman AP. Autoimmune aspects of vitiligo. Autoimmunity. 2001;34:65–77. doi: 10.3109/08916930108994127. [DOI] [PubMed] [Google Scholar]
- 38.Yu HS, Chang KL, Yu CL, Li HF, Wu MT, Wu CS. Alterations in IL-6, IL-8, GM-CSF, TNF-alpha, and IFN-gamma release by peripheral mononuclear cells in patients with active vitiligo. J Invest Dermatol. 1997;108:527–529. doi: 10.1111/1523-1747.ep12289743. [DOI] [PubMed] [Google Scholar]
- 39.Morris SC, Orekhova T, Meadows MJ, Heidorn SM, Yang J, Finkelman FD. IL-4 induces in vivo production of IFN-gamma by NK and NKT cells. J Immunol. 2006;176:5299–5305. doi: 10.4049/jimmunol.176.9.5299. [DOI] [PubMed] [Google Scholar]
- 40.Shustov A, Nguyen P, Finkelman F, Elkon KB, Via CS. Differential expression of Fas and Fas ligand in acute and chronic graft-versus-host disease: up-regulation of Fas and Fas ligand requires CD8+ T cell activation and IFN-gamma production. J Immunol. 1998;161:2848–2855. [PubMed] [Google Scholar]
- 41.Puliaev R, Nguyen P, Finkelman FD, Via CS. Differential requirement for IFN-gamma in CTL maturation in acute murine graft-versus-host disease. J Immunol. 2004;173:910–919. doi: 10.4049/jimmunol.173.2.910. [DOI] [PubMed] [Google Scholar]
- 42.Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol. 1995;154:1470–1480. [PubMed] [Google Scholar]
- 43.Wang X, Huang W, Mihara M, Sinha J, Davidson A. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol. 2002;168:2046–2053. doi: 10.4049/jimmunol.168.4.2046. [DOI] [PubMed] [Google Scholar]
- 44.Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97:2063–2073. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98:826–837. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mauri C, Mars LT, Londei M. Therapeutic activity of agonistic monoclonal antibodies against CD40 in a chronic autoimmune inflammatory process. Nat Med. 2000;6:673–679. doi: 10.1038/76251. [DOI] [PubMed] [Google Scholar]
- 47.Moir S, Ogwaro KM, Malaspina A, Vasquez J, Donoghue ET, Hallahan CW, Liu S, Ehler LA, Planta MA, Kottilil S, Chun TW, Fauci AS. Perturbations in B cell responsiveness to CD4+ T cell help in HIV-infected individuals. Proc Natl Acad Sci U S A. 2003;100:6057–6062. doi: 10.1073/pnas.0730819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol. 2009;9:235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chougnet C. Role of CD40 ligand dysregulation in HIV-associated dysfunction of antigen-presenting cells. J Leukoc Biol. 2003;74:702–709. doi: 10.1189/jlb.0403171. [DOI] [PubMed] [Google Scholar]
- 50.Zhang R, Fichtenbaum CJ, Hildeman DA, Lifson JD, Chougnet C. CD40 ligand dysregulation in HIV infection: HIV glycoprotein 120 inhibits signaling cascades upstream of CD40 ligand transcription. J Immunol. 2004;172:2678–2686. doi: 10.4049/jimmunol.172.4.2678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The experimental group (filled bar) received rIL-4 treatment followed by induction of cGVH. The positive control group (shaded bar) received B6CD4+ T cells, while the negative control group (open bar with vertical stripes) received only bm12 T cells. The other control group (open bar) was treated with rIL-4 only but received no bm12 T cells. Each bar represents the mean of ODs in each group, and the error bars represent the SEM of each group. N = 5 in each group.
See legend of Supplemental Figure 1. Each bar represents the mean of ODs in each group and the error bars represent the SEM of each group.