

Abstract
A micro-reactor was applied to produce ortho-substituted [18F]fluoroarenes from the reactions of cyclotron-produced [18F]fluoride ion (t1/2 = 109.7 min) with diaryliodonium salts. The micro-reactor provided a very convenient means for running sequential reactions rapidly with small amounts of reagents under well-controlled conditions, thereby allowing reaction kinetics to be followed and Arrhenius activation energies (Ea) to be measured. Prepared symmetrical iodonium chlorides (Ar2I+Cl−) rapidly (< 8 min) gave moderate (Ar = 2-MeOC6H4, 51%) to high (Ar = Ph or 2-MeC6H4, 85%) decay-corrected radiochemical yields (RCYs) of a single radioactive product (Ar18F). Reaction velocity with respect to Ar group was 2-MeOC6H4 < Ph < 2-MeC6H4. Activation energies were in the range 18–28 kcal/mol. Prepared unsymmetrical salts (e.g., 2-RC6H4I+2’-R’C6H4X−; X = Cl or OTs) also rapidly gave two products (2-RC6H418F and 2-R’C6H418F) in generally high total RCYs (79–93%). Selectivity for product [18F]fluoroarene was controlled by the nature of the ortho substituents. The power of ortho substituents to impart an ortho effect was in the following order, 2,6-di-Me > 2,4,6-tri-Me > Br > Me > Et ∼ iPr >> H > OMe. For (2-methyphenyl)(phenyl)iodonium chloride, the time-course of reaction product selectivity was constant and consistent with the operation of the Curtin-Hammett Principle. These results will aid in the design of diaryliodonium salt precursors to 18F-labeled tracers for molecular imaging.
Keywords: fluorine-18, micro-reactor, kinetics, [18F]fluoroarene, iodonium salt
1. Introduction
Fluorine-18 is a short-lived (t1/2 = 109.7 min) positron-emitter that finds increasing application for labeling tracers for molecular imaging in animal and human subjects with positron emission tomography (PET).1 For many of these applications, the fluorine-18 must be at high specific radioactivity, meaning that dilution of the radiotracer with non-radioactive tracer must be low i.e., the ratio of 18F to 19F compound must be high. In practice, this ratio is of the order of 1 (18F): 200 (19F), and corresponds to specific radioactivities of the order of 8 Ci/µmol. In PET imaging studies, this level of specific radioactivity is usually adequate for avoiding toxic or pharmacological effects, and also for establishing true tracer conditions in which a biochemical target (e.g., a neurotransmitter receptor) or pathway to be imaged does not become saturated and obscured with the co-administered non-radioactive tracer. For fluorine-18 to be at high specific radioactivity, it must usually be produced as the single-atom species, [18F]fluoride ion. This is usually achieved by the 18O(p,n)18F reaction on 18O-enriched water, using a moderate-energy proton beam from a compact cyclotron.2 This is a highly productive nuclear reaction, capable of generating multi-Curie levels of fluorine-18.
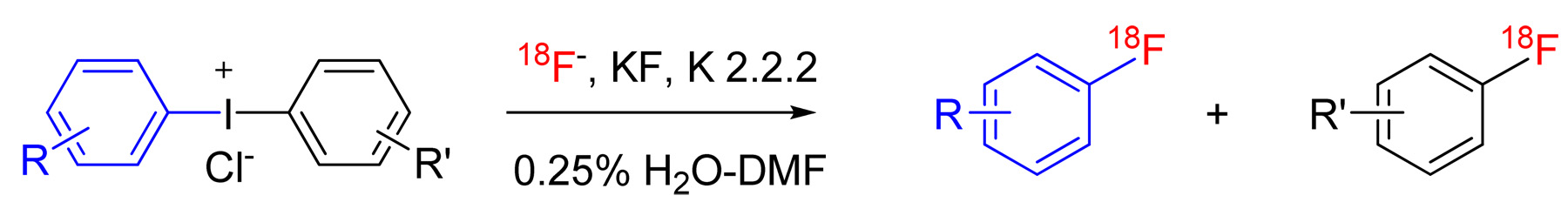
A major challenge in PET radiotracer development is to find efficient methods for rapidly incorporating cyclotron-produced [18F]fluoride ion into organic molecules. This may be achieved at aliphatic sites and in electron-deficient arenes by nucleophilic substitution reactions.1 A greater challenge is the incorporation of [18F]fluoride ion into electron-rich arenes, for which classical aromatic nucleophilic substitution is generally a disfavored process, giving low or negligible yields. The reactions of diaryliodonium salts with [18F]fluoride ion3 (Scheme 1) do not share this limitation and may be applied to labeling both electron-rich and electron-deficient arenes with fluorine-18. These reactions have therefore gained considerable interest.4, 5, 6 Notwithstanding, these reactions exhibit unusual features and their detailed mechanisms are not yet well understood. As part of our ongoing investigations into the scope and nature of these reactions for PET radiotracer development, we wished to explore the influence of substituents, particularly ortho substituents, on reaction rates, energetics and product selectivity under varied but well-controlled conditions. Here, we first show the utility of a micro-reactor apparatus for this radiochemical purpose and secondly further insights into the mechanism and outcomes of the radiofluorination reaction. These findings will assist in future PET radiotracer design and production.
Scheme 1.

Reactions of diaryliodonium salts with [18F]fluoride Ion
2. Results and Discussion
2.1 Chemistry
Two main methods were used to prepare substituted diaryliodonium tosylates, namely reaction of Koser’s reagent ([hydroxy(tosyloxy)]iodobenzene; PhI(OH)OTs) or an ortho-substituted derivative (RC6H4I(OH)OTs) with an arylboronic acid7 or tri-(alkyl)stannylarene8 (Scheme 2). The derivatives of Koser’s reagent were prepared by treating the appropriate iodoarene diacetate with p-tosic acid.9 The reactions of Koser’s reagent and derivatives with (tri-(alkyl)stannyl)arenes exhibited excellent regioselectivity and gave moderate (38%) to high yields (90%). Reactions with arylboronic acids were regioselective for acids bearing an o-methyl substituent but not for arylboronic acids bearing an o-methoxy substituent. Yields were low (20%) to moderate (49%). Direct reactions with mesitylene were effective for making (mesityl)aryliodonium salts, since with the methyl substitution pattern, only one positional isomer is possible. Diaryliodonium tosylates were readily converted into their corresponding halides in moderate (27%) to high yields (88%) by metathesis reactions. In each case, metathesis was readily confirmed by the absence of signals for tosylate in the 1H-NMR spectrum.
Scheme 2.

Routes used for the syntheses of diaryliodonium salts in this study
2.2. Radiofluorination Reactions within a Micro-reactor
The potential advantages of micro-reactor technology for PET radiochemistry are increasingly recognized. 10,11 In this study, all radiofluorination reactions were performed in a commercially available micro-reactor apparatus (Nanotek; Advion). The use of this apparatus offers several advantages over traditional methods of studying radiofluorination reactions, which are usually performed singly with substantial amounts of non-radioactive precursor (e.g., milligram-quantities of precursor for labeling).1,6 As we have recently shown,11 by use of the micro-reactor apparatus, a series of reactions can be performed rapidly with very low fixed quantities of reagents, under precisely controlled conditions of temperature, time and reaction stoichiometry. The whole micro-reactor is heated to a controlled temperature, which can be well above the boiling point of most organic solvents due to the elevated pressure of the system. Because of the small internal volume of the micro-reactor (15.7 µL for 2-m length; 31.4 µL for 4-m length), entering reactant solutions rapidly mix under laminar flow and reach the set reactor temperature. Reaction time simply equates to the residence time of reactants within the reactor, which is determined by the flow rates of the reactants. These flow rates were precisely determined with mechanically-driven syringes. Reaction stoichiometry was controlled by setting the concentration of reactants in the loaded solutions and also their flow rates into the reactor.
In this study we performed reactions with [18F]fluoride ion in either a no-carrier-added state (NCA) (i.e., at high specific radioactivity) or a carrier-added (CA) state (at low specific activity). [18F]Fluoride ion was dried in the presence of the cryptand (Kryptofix 2.2.2; K 2.2.2), plus base (K2CO3) to maintain a NCA state or in the presence of a known amount of potassium fluoride-K 2.2.2 to produce CA reagent. The generated [18F]fluoride ion complex was then dissolved in the solvent for reaction. A solution of the diaryliodonium salt was prepared in the same solvent. These reagent solutions were loaded into their respective storage loops in the micro-reactor apparatus and fed into the reactor at set flow rates (Scheme 3).
Scheme 3.

Set-up of microfluidic apparatus for the study of radiofluorination reactions
Reactions were quenched by diluting the output of the reactor in water-acetonitrile (1: 1 by vol.) at room temperature. Recovery of radioactivity from the reactor was 83 ± 19%, (mean ± SD, n = 13) and 79 ± 16% (mean ± SD, n = 20) for CA and NCA reactions respectively. There was no discernible dependence of these values on the nature of the iodonium salt used in the reaction. Control experiments showed that all the retained radioactivity was polar in reverse phase HPLC analysis, consistent with identity as [18F]fluoride ion. Quenched reaction mixtures were analyzed by reverse phase radio-HPLC. All reactions gave a simple radioactive product distribution. Thus, chromatograms showed only unchanged [18F]fluoride ion (at or near the solvent front), followed either by one peak for a single [18F]fluoroarene (from a symmetrical diaryliodonium salt) or two peaks for two [18F]fluoroarenes (from an unsymmetrical salt). Recovery of radioactive injectate was checked by collection of all radioactive eluate fractions and separate measurement in a calibrated well-counter. Decay-corrected radiochemical yields (RCYs) were calculated from the radio-chromatograms.
Initially, we tested the feasibility of using the micro-reactor apparatus for radiofluorination of a diaryliodonium salt, the reaction of [18F]fluoride ion with diphenyliodonium chloride. Reactions were performed with NCA [18F]fluoride ion in DMF or CA [18F]fluoride ion in DMF-0.25% water at set temperatures (Figure 1). At 90 °C NCA [18F]fluoride ion gave higher RCY than CA [18F]fluoride ion. CA reactions performed at higher temperatures gave higher RCYs. Remarkably, an 85% RCY of [18F]fluorobenzene was produced from [18F]fluoride ion in < 6.5 min at 110 °C. Clearly the presence of a low concentration of water was well tolerated in these CA reactions.
Figure 1.

Time-course of [18F]fluorobenzene RCY from reactions of [18F]fluoride ion with diphenyliodonium chloride (5 mM) in DMF-0.25% water in the presence of 0.5 mM KF-K 2.2.2 at 90, 105 and 110 °C (CA), or in DMF in the presence of K2CO3-K 2.2.2 at 90 °C (NCA).
2.3. Investigation of the Ortho-Effect
An intriguing feature of the reaction of diaryliodonium salts with nucleophiles is the influence of ortho substitutents, which have been reported to direct an incoming nucleophile to attack the same ring as the substituent.12 Here we wished to study this effect more closely for the reactions of [18F]fluoride ion with ortho-substituted diaryliodonium salts. We therefore extended the application of the micro-reactor to the study of the NCA radiofluorination of four (2-methylphenyl)(phenyl)iodonium salts in different solvents. Each reaction was studied at various temperatures up to 200 °C for reaction times up to 8 min. The highest observed RCYs of [18F]fluoroarenes are listed in Table 1 with the corresponding reaction conditions. Each salt gave higher RCY in DMF than in acetonitrile containing a low proportion (1.5%) of water, or for one salt, the tosylate, in anhydrous acetonitrile. The bromide and tosylate of (2-methylphenyl)(phenyl)iodonium gave almost quantitative incorporation of [18F]fluoride ion into [18F]fluoroarenes in DMF, while incorporations were significantly lower for the iodide and chloride. Ermert et al.13 have previously observed the similar reactivity of diaryliodonium tosylate and bromides with [18F]fluoride ion. In all our reactions, [18F]2-fluorotoluene was the major radioactive product and [18F]fluorobenzene the minor radioactive product. The selectivity for producing [18F]2-fluorotoluene was quite similar across reactions, ranging from 2.00 to 3.00, thereby verifying that a methyl substituent confers an ortho-effect in these reactions.
Table 1.
NCA Radiofluorination of (2-Methylphenyl)(phenyl)iodonium Salts in Different Solvents
| Entry | Anion | Solvent | Temp (°C) |
Time (s) |
RCY of [18F]fluoroarenes* (%) |
Selectivity for main product |
||
|---|---|---|---|---|---|---|---|---|
| Total | [18F]2-F-toluene | [18F]F-benzene | ||||||
| 1 | Cl− | MeCN-1.5% H2O | 150 | 417 | 46 | 32 | 14 | 2.29 |
| 2 | DMF | ” | 490 | 77 | 53 | 24 | 2.21 | |
| 3 | Br− | MeCN-1.5% H2O | ” | 262 | 26 | 18 | 8 | 2.25 |
| 4 | DMF | 130 | ” | 99 | 67 | 32 | 2.09 | |
| 5 | I− | MeCN-1.5% H2O | 160 | ” | 12 | 8 | 4 | 2.00 |
| 6 | DMF | 130 | 354 | 48 | 33 | 15 | 2.54 | |
| 7 | OTs− | MeCN | 140 | 536 | 12 | 9 | 3 | 3.00 |
| 8 | MeCN-1.5% H2O | 150 | 322 | 50 | 34 | 16 | 2.13 | |
| 9 | DMF | 130 | 474 | 98 | 66 | 32 | 2.06 | |
Highest observed RCYs (the temperature and times are the conditions giving these yields).
In order to further understand the ortho-effect, studies were extended to a larger range of diaryliodonium salts bearing a range of ortho substituents. The radiofluorination of each salt was studied under either CA or NCA conditions in dry DMF or wet DMF over various temperatures and times. Maximal observed RCYs, and the corresponding reaction conditions, are recorded in Table 2. The total RCYs of [18F]fluoroarenes were impressively high, ranging from 51 to 95%. Yields were lower for salts bearing an o-methoxy group, with the symmetrical di-o-methoxy substituted salt giving lowest yield. For unsymmetrical salts, product selectivities ranged from 1.20 to 18.8 (Table 2). Surprisingly, an o-methoxy group failed to confer an ortho-effect, since 2-[18F]fluoroanisole was always the least preferred product (Table 2, entries 4 and 5). From all the product selectivity values listed in Table 2, it may be deduced that the power of ortho substituents to impart an ortho-effect was in the following order: 2,6-di-Me > 2,4,6-tri-Me > Br > Me > Et ∼ iPr > H > OMe. The selectivity values will be useful in the rational design of diaryliodonium salt precursors to 18F-labeled tracers.
Table 2.
Radiochemical Yields of [18F]Fluoroarenes from the Radiofluorination of o-Substituted Diaryliodonium Salts.
| Entrya | Diaryliodonium salt (ArI+Ar’X) |
Temp. (°C) |
Time (s) |
RCY of [18F]fluoroarene (%) |
Selectivity for main product |
GTs1-GTs2 (kcal/mol) |
|||
|---|---|---|---|---|---|---|---|---|---|
| Ar | Ar’ | Total | Ar18F | Ar’18F | |||||
| 1 | Ph | Ph | 110 | 377 | 85 | 85 | - | - | - |
| 2 | 2-MeC6H4 | 2-MeC6H4 | 140 | ” | 83 | 83 | - | - | - |
| 3 | 2-MeC6H4 | Ph | 110 | ” | 82 | 57 | 25 | 2.28 | 0.63 |
| 4 | Ph | 2-MeOC6H4 | 140 | ” | 66 | 60 | 6.5 | 9.22 | 1.8 |
| 5 | 2-MeC6H4 | 2-MeOC6H4 | ” | ” | 79 | 75 | 4 | 18.8 | 2.4 |
| 6 | 2-MeOC6H4 | 2-MeOC6H4 | ” | ” | 51 | 51 | - | - | - |
| 7 | 2-BrC6H4 | 2-MeC6H4 | 170 | 471 | 86 | 48 | 38 | 1.26 | 0.20 |
| 8 | 2-MeC6H4 | 2-EtC6H4 | ” | ” | 95 | 52 | 43 | 1.21 | 0.17 |
| 9 | 2-MeC6H4 | 2-iPrC6H4 | ” | ” | 88 | 48 | 40 | 1.20 | 0.16 |
| 10 | 2,6-di-MeC6H3 | 2-MeC6H4 | ” | ” | 74 | 62 | 11 | 5.56 | 1.5 |
| 11 | 2,4,6-tri-MeC6H2 | 2-MeC6H4 | 130 | ” | 92 | 59 | 33 | 1.78 | 0.46 |
| 12 | 2,4,6-tri-MeC6H2 | Ph | 150 | ” | 82 | 63 | 19 | 3.24 | 0.99 |
| 13 | 2,6-di-MeC6H3 | 2,4,6-tri-MeC6H2 | 190 | ” | 82 | 61 | 21 | 3.00 | 1.0 |
| 14 | 2-BrC6H4 | Ph | ” | ” | 93 | 68 | 25 | 2.72 | 0.92 |
Entries 1–6 were performed with CA [18F]fluoride ion and chloride salts in DMF-0.25% water. Entries 7–11 and 14 were performed with NCA [18F]fluoride ion and chlorides in DMF. Entries 12 and 13 were performed with NCA [18F]fluoride ion and tosylates in DMF.
The ortho-effect has frequently been ascribed to the steric influence of the o-substituent on the conformation of the iodonium salt, leading to a preference for the ortho-substituted aryl ring to rest in the equatorial plane of a trigonal bipyramidal structure with therefore easy access for attack by an apically-associated nucleophile. Previous studies on the ortho-effect have only investigated o-alkyl substituents.12 Our results with o-methoxy-substituted diaryliodonium salts show that the ortho-effect is not purely related to substituent bulk. Rather than substituent bulk, selectivity may be enhanced by the presence of one or more ortho hydrophobic groups (e.g., methyl) which create a lipophilic microenvironment in which the incoming [18F]fluoride ion can act as a powerful nucleophile, by first loosely binding to the hypervalent iodine atom and then attacking the locally lipophilic ortho-substituted ring. Opposing (as for OMe) or reinforcing (as for Br) electronic influences also appear to be important in determining product selectivity.
The reliability of the radiofluorination reactions of diphenyliodonium salts have been claimed to be enhanced by addition of a free-radical scavenger, such as TEMPO.14 In this study, we found that radiofluorination reactions were consistent and reliable in the absence of a free-radical scavenger. In fact, in a pilot experiment, we found that TEMPO had no beneficial effect. In general, iodonium salts are well known to show photosensitivity 15 and indeed much research has been directed to their use as photoinitiators. 16 The micro-reactor device used in this study stores and uses the iodonium salt reagent in the dark, and this may have contributed to the reliability of the reactions by avoiding radical generation. The tolerance of these reactions for a low concentration of water is also striking and is accord with a previous claim.17 This tolerance indicates the generally high reactivity of the iodonium salts for reaction with fluoride ion that is in a moderately hydrated and therefore moderately nucleophilic state.
2.4. Kinetics and Energetics of Radiofluorination Reactions
Examples of the determination of the Arrhenius activation energies (Ea) for reactions of [18F]fluoride ion are rare, being confined to isotopic exchange with polyfluoroarenes.18 The absence of such data may partly derive from the practical limitations of working with a short-lived radionuclide from a cyclotron-source. The micro-reactor apparatus afforded an opportunity to overcome some of these limitations, since well-controlled reaction conditions could be established for rapid sequential reactions from the same ‘stocks’ of radioactive and non-radioactive reagents. In order to gain kinetic data and derive Ea values, we decided to perform reactions with CA [18F]fluoride ion (0.5 mM) with a 10-fold molar excess of diaryliodonium salt (5 mM) in the presence of K+-K.2.2.2. Fluoride ion nucleophilicity is well known to depend on the state of ion hydration.1,19 Therefore, we decided to include a small concentration of water (0.25% v/v) in the reaction medium (DMF) to control the hydration of the [18F]fluoride ion at a fixed carrier concentration and hence its nucleophilicity. Six diaryliodonium chlorides were studied in this manner. Reactions were performed at various set temperatures for several reaction times up to 377 s.
Figure 2 shows an example of acquired kinetic data, that from the reaction of CA [18F]fluoride ion with bis-(o-methylphenyl)iodonium chloride. The kinetic data were used to estimate initial reaction velocities which were then used in Arrhenius plots to estimate activation energies (e.g., Figure 3). The Ea values for the reactions of several diaryliodonium chlorides with CA [18F]fluoride ion in DMF-0.25% water were estimated to be in the range 18–28 kcal/mol. Activation energies appeared to increase as first one substituent, either an o-methyl or o-methoxy group, and then a second was introduced into the opposite ring of the diaryliodonium salt (Table 3). The symmetrical o-methoxy-substituted salt was estimated to have the highest Ea value. Estimates of Ea values for other reactions of diaryliodonium salts with nucleophiles have been reported, and they fall in the range 19–37 kcal/mol. For example, the decomposition of various substituted diphenyliodonium halides (halide = Cl−, Br− or I−) in DMF occurred with Ea values in the range 25.5–36.7 kcal/mol.20 Reactions of diphenyliodonium salts with phenoxide ions in dioxane-water (1: 1, v/v) to give diphenyl ethers have an Ea value of 25.9 kcal/mol.21
Figure 2.

Kinetics of the radiofluorination of bis-(o-methylphenyl)iodonium chloride in DMF-0.25% H2O to give [18F]2-fluorotoluene at different temperatures.
Figure 3.

Arrhenius plot for the reaction of [18F]fluoride ion with bis-(2-methylphenyl)iodonium chloride (data derived from that in Figure 2). The solid line is the best fit determined by linear regression and the dotted lines are the 90% confidence limits (see Experimental).
Table 3.
Arrhenius Activation Energies (Ea) for the Reactions of Diaryliodonium Chlorides with CA [18F]Fluoride Ion in DMF-0.25% H2O (in the presence of K+-K 2.2.2).
 | |||
|---|---|---|---|
| Diaryliodonium chloride |
Ea (kcal/mol) |
||
| R | R’ | R | R’ |
| H | H | 20.9 ± 1.6 | 20.9 ± 1.6 |
| H | 2-MeO | 22.5 ± 2.9 | n.d. |
| H | 2-Me | 23.5 ± 1.6 | 18.3 ± 0.9 |
| 2-Me | 2-MeO | 25.1 ± 2.6 | n.d. |
| 2-Me | 2-Me | 27.5 ± 2.0 | 27.5 ± 2.0 |
| 2-MeO | 2-MeO | 28.0 ± 1.6 | 28.0 ± 1.6 |
2.5. Selectivities of Reactions – Compliance with the Curtin-Hammett Principle and Mechanistic Considerations
During the accumulation of the data summarized in Table 2, we were impressed that product selectivities appeared constant for any particular diaryliodonium salt precursor (small variations occurred more with solvent than with anion; see Examples reported in Table 1). This suggested that reactions might be taking place in accord with the Curtin-Hammett Principle.22 This principle states that the relative amounts of products formed from two critical conformations of substrate are completely independent of the relative populations of the conformations and depend only upon the difference in the free energy of the transition states, provided that the rates of reaction are much slower than the rates of conformational interconversion. The appearance of products in a constant ratio during the reaction course under set conditions constitutes a test of whether a process that involves one substrate to give two products complies with the Curtin-Hammett Principle. 23 Thus, we set up an experiment with CA [18F]fluoride ion and (o-methylphenyl)(phenyl)iodonium chloride as reactants in equimolar proportions in DMF at 110 °C, and measured the RCYs of the two products with time (Figure 4). The ratio of products (2.21) was found to be constant over the measured time course (up to 30% total substrate conversion) in accord with the Curtin-Hammett Principle. The product selectivity was also measured at other temperatures and was essentially invariant. Scrutiny of other data accumulated in this study revealed very little variation in product selectivity over the time course of reactions.
Figure 4.

Time-course of the decay-corrected radiochemical yield (RCY) of [18F]2-fluorotoluene (△) and [18F]fluorobenzene (▼), and of the product selectivity (●), from the reaction of CA [18F]fluoride ion with (2-methylphenyl)(phenyl)iodonium chloride at 110 °C in DMF-0.25% H2O, in the presence of K+-K 2.2.2. Reagents and K+-K 2.2.2 were each initially at 2.5 mM. The dashed line is the line of linear regression for the product selectivity data.
Monomeric iodonium cations are expected to be highly fluxional and able to interconvert through conformations in which the ligands rapidly interchange equatorial and axial positions relative to the anion, which is secondarily bonded to the pivotal hypervalent iodine atom.24 It may be envisaged that when the anion is an incoming nucleophile it will be able to attack only the carbon atom in the equatorial ring from an apical position. Thus, we propose that the radiofluorination of an unsymmetrical diaryliodonium salt involves attack of [18F]fluoride ion onto either of two rapidly inter-converting conformers, yielding two transition states that each give a single radiofluorinated product (Scheme 4). Assuming all the radiofluorination reactions follow the Curtin-Hammett Principle, the ratio of radiofluorinated products then depends solely on the difference in the free energies of the two transition states. This difference can be calculated from the product selectivity value (P1/P2) according to the equation P1/P2 = e−(GTs1–GTs2)/RT, where GTs1 and GTs2 are the free energies of the transition states 1 and 2, respectively. The values for GTs1–GTs2 for the reactions studied here range from 0.16 to 2.4 kcal/mol (Table 2), and are therefore relatively small compared to the activation energies for the same reactions (Table 3).
Scheme 4.

Suggested outline mechanism for the radiofluorination of an unsymmetrical diaryliodonium salt, through transition states TS1 and TS2 to give products P1 and P2, respectively.
3. Summary
This study demonstrates that micro-reactor technology can be excellent for the rigorous and efficient study of radiofluorination reactions under well-controlled conditions using small amounts of material. Furthermore, the reactions of CA and NCA [18F]fluoride ion with readily synthesized ortho-substituted diaryliodonium salts can produce [18F]fluoroarenes rapidly and in very high RCYs, even for salts bearing electron-donating alkyl or methoxy groups. The powers of individual ortho substituents to impart the ortho-effect were ranked and showed that this effect is not due solely to substituent bulk. The activation energies determined for some of these reactions are within the range estimated for similar reactions of diaryliodonium salts. Finally, we found evidence that the reactions of [18F]fluoride ion with unsymmetrical diaryliodonium salts comply with the Curtin-Hammett Principle. These results will aid in our design of diaryliodonium salts as substrates for producing previously inaccessible 18F-labeled tracers for molecular imaging.
4. Experimental Section
Materials, general methods and details of diaryliodonium salts syntheses are documented in Supplementary Information.
4. 1. Production of [18F]Fluoride Ion Reagents
4.1.1. No-carrier-added
Cyclotron-produced no-carrier-added [18F]fluoride ion (10–200 mCi) in [18O]water (98 atom %, 225–350 µL) was first adsorbed onto a small anion resin (i.e., MP-1 or QMA) cartridge within the CE module of a NanoTek apparatus (Advion; Louisville, TN), and then released with a solution of K2CO3 (0.8 mg; 5 nmol) plus K 2.2.2 (4.5 mg; 11 nmol) in MeCN-H2O (9: 1 v/v; 150 µL) into a 5-mL V-vial. The solution was dried by two cycles of azeotropic evaporation with acetonitrile (0.45 mL) at 100 °C.
4.1.2. Carrier-added
Cyclotron-produced no-carrier-added [18F]fluoride ion (120–200 mCi) in [18O]water (225–350 µL) was dried25 by four cycles of azeotropic evaporation with acetonitrile (0.65 mL for each addition) at 90 W × 2 min in a microwave cavity (type-521; Resonance Instruments Inc., Skokie, IL) housed within a modified Synthia26 module. KF (1 mM) and K 2.2.2 (1 mM) in 0.5 % H2O/DMF (v/v; 400 µL) were added to the prepared dry 18F−-K 2.2.2-K+ complex and mixed in a 5-mL V-vial. The solution was then loaded into the storage loop (255 µL) of the microfluidic apparatus.
4.2. Configuration and Operation of the Micro-reactor Apparatus
The setup of the LF module used in this work is depicted in Scheme 3. The micro-reactor is composed of coiled silica glass tubing (e.g., length, 2 m; internal diameter, 100 µm; internal volume, 15.7 µL), housed in a brass ring filled with high-temperature resistant poly(silicone). The reactor could be heated and thermostatted to a set temperature. The reactor may be fed with reagents from storage loops at set flow rates, by the action of mechanically-driven syringes. The reactor may operate up to pressures of 300 psi, which is monitored.
The apparatus was cleaned by flushing tubes and reactors with acetonitrile at the beginning of each experimental session and again at the end. Reagent solutions were loaded into their respective storage loops from different source vials. To perform one reaction, 10–20 µL of solution from each loop were infused (via valve connection from the respective syringe pump → E → D → C) into the micro-reactor at a set flow rate and temperature. A bolus of acetonitrile was then infused from syringe pump 1 (via valve connection from syringe pump 1 → B) to sweep all reaction mixture out of the micro-reactor and into the collection vial via the distribution valve. The volume of the sweeping solvent just exceeded the aggregate internal volume of the micro-reactor and subsequent transfer tubing. Once the source vials were in place, all operations were programmed to run in sequence automatically and remotely.
4.3. Radiofluorination Reactions
Dry 18F−-K 2.2.2-K+ complex (100–150 mCi) was dissolved in reaction solvent (typically, DMF-0.5% H2O, DMF or MeCN; 255 µL). A solution of diaryliodonium salt (10 mM) was prepared in matching solvent. Each of the two solutions (255 µL) was loaded into a separate storage loop of the microfluidic apparatus. For reactions, each solution (10–15 µL) was infused simultaneously into either the 2 m- or 4 m-length coiled silica glass tube micro-reactor of the apparatus at a set flow rate in the range 5–100 µL/min and pre-set temperature. The micro-reactor output was quenched with MeCN-H2O (1: 1 v/v; 1 mL). The radical scavenger, TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl), was added to some reactions.14 Amounts of precursor, temperature and flow rates were varied. Decay-corrected radiochemical yields (RCYs) of [18F]fluoroarenes were measured by reverse phase radio-HPLC on a Luna C18 column (250 × 4.6 mm i.d., 10 µm; Phenomenex, Torrance, CA) eluted at 1.75 mL/min with a gradient of MeCN-H2O (60: 40, v/v) with the percentage of MeCN increased linearly from 60 to 90 over 7 min.
4.4. Kinetics and Energetics of Radiofluorination Reactions
Reactions were run between a large molar excess of diaryiodonium salt, relative to fluoride ion, in DMF containing 0.5% water, in the presence of carrier potassium fluoride and K 2.2.2. Reaction times were controlled by the rates at which [18F]fluoride/KF/K 2.2.2 solution and the diaryliodonium chloride solution were passed through the micro-reactor. Reactor outputs were quenched by dilution with acetonitrile-water (1: 1 v/v) at room temperature. Reactions were run at a minimum of six different temperatures each separated by at least 5 °C for several reaction times ranging up to 377 s. RCYs were determined by radio-HPLC analysis of reaction products, by comparing the integral of the radio-chromatogram for each labeled product with that of unchanged [18F]fluoride ion. Full recovery of radioactivity from the HPLC system was verified by measurement of all eluted radioactivity in a dose calibrator. With the program Prism 3.0, RCYs below 20% were plotted against time (including zero RCY at zero time) and each curve fitted to a first order polynomial. The initial apparent velocities of reaction (Voapp, expressed as % RCY/min) were calculated as the initial slopes of the fitted curves (i.e., as the coefficient of the second term in the equation of the polynomial fit). lnVoapp was plotted against the inverse of the reaction temperature (1/T °K−1), and the data fitted to a linear curve with Prism 3.0, which also provided curves for 90% confidence limits. Arrhenius activation energies (Ea) were calculated from the fitted slopes, which equal – Ea/RT (R = gas constant).
4.5. Test of Curtin-Hammett Principle
Reactions between CA [18F]fluoride ion (2.5 mM) and (2-methylphenyl)(phenyl)iodonium chloride (2.5 mM) in DMF-0.25% water, in the presence of K+-K 2.2.2 (2.5 mM) were performed in the microfluidic apparatus at 110 °C for various times up to 377 s. The radiochemical yields of the two radioactive products, [18F]2-fluorotoluene and [18F]fluorobenzene, were measured and the product selectivity for major product, as the ratio of these two products, was calculated for each reaction time.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program (project # Z01-MH-002793) of the National Institutes of Health (National Institute of Mental Health), and also by the NIH Intramural Research Program through the Center for Information Technology. We are grateful to the NIH Clinical PET Center for the production of fluorine-18.
Footnotes
Supporting Information Available: Full experimental and characterization data for the new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cai LS, Lu SY, Pike VW. Eur. J. Org. Chem. 2008;17:2853–2873. [Google Scholar]
- 2.(a) Guillaume M, Luxen A, Nebeling B, Argentini M, Clark JC, Pike VW. Appl. Radiat. Isot. 1992;42:749–762. [Google Scholar]; (b) Qaim SM, Clark JC, Crouzel C, Guillaume M, Helmeke HJ, Nebeling B, Pike VW, Stöcklin G. In: Radiopharmaceuticals for Positron Emission Tomography. Stöcklin G, Pike VW, editors. Dordrecht, Netherlands: Kluwer Academic Publishers; 1993. pp. 1–43. [Google Scholar]
- 3.Pike VW, Aigbirhio FI. J. Chem. Soc. Chem. Commun. 1995;21:2215–2216. [Google Scholar]
- 4.(a) Shah A, Pike VW, Widdowson DA. J. Chem. Soc. Perkin Trans. 1998;1:2043–2046. [Google Scholar]; (b) Gail R, Hocke C, Coenen HH. J. Label. Compd. Radiopharm. 1997;40:50–52. [Google Scholar]; (c) Zhang MR, Kumata K, Suzuki K. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]; (d) Pike VW, Aigbirhio FI. J. Label. Compd. Radiopharm. 1995;37:120–122. [Google Scholar]
- 5.Martín-Santamaría S, Carroll MA, Carroll CM, Carter CD, Pike VW, Rzepa HS, Widdowson DA. Chem. Commun. 2000:649–650. [Google Scholar]
- 6.Ross L, Ermert J, Hocke C, Coenen HH. J. Am. Chem. Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 7.Carroll MA, Pike VW, Widdowson DA. Tetrahedron Lett. 2000;41:5393–5396. [Google Scholar]
- 8.Pike VW, Butt F, Shah A, Widdowson DA. J. Chem. Soc., Perkin Trans 1. 1999:245–248. [Google Scholar]
- 9.(a) Neiland G, Karele B. J. Org. Chem. USSR (Engl. Transl.) 1970:889. [Google Scholar]; Zh. Org. Khim. 1970;6:885. [Google Scholar]; (b) Koser GF, Wettach RH. J. Org. Chem. 1977;42:1476–1478. [Google Scholar]
- 10.(a) Lu SY, Watts P, Chin FT, Hong J, Musachio JL, Briard E, Pike VW. Lab on Chip. 2004;4:523–525. doi: 10.1039/b407938h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee CC, Sui GD, Elizarov A, Shu CYJ, Shin YS, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng HR. Science. 2005;310:1793–1796. doi: 10.1126/science.1118919. [DOI] [PubMed] [Google Scholar]; (c) Gillies JM, Prenant C, Chimon GN, Smethurst GJ, Perrie W, Hamblett I, Dekker B, Zweit J. Appl. Radiat. Isot. 2006;64:325–332. doi: 10.1016/j.apradiso.2005.08.007. [DOI] [PubMed] [Google Scholar]; (d) Steel CJ, O’Brien AT, Luthra SK, Brady F. J. Label. Compd. Radiopharm. 2007;50:308–311. [Google Scholar]; (e) Lu SY, Pike VW. In: PET Chemistry – The Driving Force in Molecular Imaging. Schubiger PA, Lehmann L, Friebe M, editors. Heidelberg: Springer-Verlag; 2007. pp. 271–287. [Google Scholar]; (f) Audrain H. Angew. Chem. Int. Ed. 2007;46:1772–1775. doi: 10.1002/anie.200603509. [DOI] [PubMed] [Google Scholar]; (g) Elizarov AM. Lab on Chip. 2009;9:1326–1333. doi: 10.1039/b820299k. [DOI] [PubMed] [Google Scholar]
- 11.Lu S, Giamis AM, Pike VW. Curr. Radiopharmaceuticals. 2009;2:49–55. doi: 10.2174/1874471010902010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Yamada Y, Kashima K, Okawara M. Bull. Chem. Soc. Japan. 1974;47:3179–3180. [Google Scholar]; (b) Lancer KM, Wiegand GH. J. Org. Chem. 1976;41:3360–3364. [Google Scholar]; (c) Yamada Y, Okawara M. Bull. Chem. Soc. Japan. 1972;45:1860–1863. [Google Scholar]
- 13.Ermert J, Hocke C, Ludwig T, Gail R, Coenen HH. J. Label. Compd. Radiopharm. 2004;47:429–441. [Google Scholar]
- 14.(a) Wadsworth HJ, Widdowson DA, Wilson E, Carroll MA. WO 2005/061415. 2005 [Google Scholar]; (b) Carroll MA, Nairne J, Smith G, Widdowson DA. J. Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 15.Irving H, Reid RW. J. Chem. Soc. 1960:2078–2081. [Google Scholar]
- 16.(a) Hacker NP, Leff DV, Dektar JL. J. Org. Chem. 1991;56:2280–2282. [Google Scholar]; (b) Yagci Y, Yilmaz F, Kiralp S, Toppare L. Macromol. Chem. Phys. 2005;206:1178–1182. [Google Scholar]; (c) He JH, Mendoza VS. J. Polym. Sci. Part A - Polym. Chem. 1996;34:2809–2816. [Google Scholar]; (d) He JH, Zhang JC. J. Polym. Sci. Part A - Polym. Chem. 1999;37:4521–4527. [Google Scholar]; (e) He JH, Zhang JC.J. Photochem. Photobiol 1997107, 249–252. [Google Scholar]; (f) Padon KS, Scranton AB. J. Polym. Sci. Part A - Polym. Chem. 2000;38:2057–2066. [Google Scholar]; (g) Merritt EA, Oloffsson B. Angew. Chem. Int. Ed. 2009;48:2–21. [Google Scholar]
- 17.Wadsworth HJ, Devenish T. WO 2005/097713 A1. 2005 [Google Scholar]
- 18.Cacace F, Speranza M, Wolf AP, Macgregor RR. J. Fluorine Chem. 1982;21:145–158. [Google Scholar]
- 19.Landini D, Maia A, Rampoldi A. J. Org. Chem. 1989;54:328–332. [Google Scholar]
- 20.Beringer FM, Mausner M. J. Am. Chem. Soc. 1958;80:4535–4536. [Google Scholar]
- 21.Beringer FM, Gindler EM. J. Am. Chem. Soc. 1955;77:3203–3207. [Google Scholar]
- 22.Seeman JI. Chem. Rev. 1983;83:83–134. [Google Scholar]
- 23.Eliel EL, Wilen SH, Mander LN. Stereochemistry of Carbon Compounds. New York: Wiley Interscience; 1994. Ch. 10; p. 652. [Google Scholar]
- 24.Carroll MA, Martín-Santamaría S, Pike VW, Rzepa HS, Widdowson DA. J. Chem. Soc. Perkin Trans 2. 1999:2707–2714. [Google Scholar]
- 25.Lazarova N, Siméon FG, Musachio JL, Lu SY, Pike VW. J. Label. Compd. Radiopharm. 2007;50:463–465. [Google Scholar]
- 26.Bjurling P, Reineck R, Westerburg G, Gee AD, Sutcliffe J, Långström B. In: Proc. Sixth Workshop on Targetry and Target Chem. Link JM, Ruth TJ, editors. Vancouver: TRIUMF; 1995. pp. 282–284. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.