

Summary
Several human neurological disorders have been associated with various mutations affecting mitochondrial enzymes involved in cellular ATP production. One of these mutations, T9176C in the mitochondrial DNA (mtDNA), changes a highly conserved leucine residue into proline at position 217 of the mitochondrially encoded Atp6p (or a) subunit of the F1FO-ATP synthase. The consequences of this mutation on the mitochondrial ATP synthase are still poorly defined. To gain insight into the primary pathogenic mechanisms induced by T9176C, we have investigated the consequences of this mutation on the ATP synthase of yeast where Atp6p is also encoded by the mtDNA. In vitro, yeast atp6-T9176C mitochondria showed a 30% decrease in the rate of ATP synthesis. When forcing the F1FO complex to work in the reverse mode, i.e. F1-catalyzed hydrolysis of ATP coupled to proton transport out of the mitochondrial matrix, the mutant showed a normal proton-pumping activity and this activity was fully sensitive to oligomycin, an inhibitor of the ATP synthase proton channel. However, under conditions of maximal ATP hydrolytic activity, using non-osmotically protected mitochondria, the mutant ATPase activity was less efficiently inhibited by oligomycin (60% inhibition versus 85% for the wild type control). BN-PAGE analyses revealed that atp6-T9176C yeast accumulated rather good levels of fully assembled ATP synthase complexes. However, a number of subcomplexes (F1, Atp9p-ring, unassembled α-F1 subunits) could be detected as well, presumably because of a decreased stability of Atp6p within the ATP synthase. Although the oxidative phosphorylation capacity was reduced in atp6-T9176C yeast, the number of ATP molecules synthesized per electron transferred to oxygen was similar compared with wild type yeast. It can therefore be inferred that the coupling efficiency within the ATP synthase was mostly unaffected and that the T9176C mutation did not increase the proton permeability of the mitochondrial inner membrane.
Introduction
Most of the ATP in human cells is produced by an F1FO-type ATP synthase or complex V located in the mitochondrial inner membrane [1]. This enzyme utilizes the energy of the electrochemical proton gradient established by the mitochondrial electron transport chain to drive the synthesis of ATP from ADP and inorganic phosphate. Pronounced defects in the ATP synthase can be responsible for various very deleterious disorders in humans, especially in the highly energy-demanding neuromuscular system [2–4]. Such disorders like NARP (neuropathy ataxia retinitis pigmentosa) and MILS (maternally-inherited Leigh’s syndrome) have been associated to several mutations in the mitochondrial ATP6 gene encoding subunit a of the ATP synthase (referred to as Atp6p in yeast). This subunit contains most of the residues involved in the proton translocating activity of the FO domain of the ATP synthase [5]. Proton movements mediated by subunit a lead to the rotation of a ring of c subunits (referred to as Atp9p in yeast) that contacts subunit a in the membrane. The resulting mechanical energy is then used to induce conformational changes at the level of the catalytic sites in the F1 extra-membrane domain of the enzyme that favor the synthesis of ATP and its subsequent release into the mitochondrial matrix.
One of the known pathogenic ATP6 mutations, T9176C, leads to the replacement of a highly conserved leucine into proline at amino acid position 217 of subunit a [6, 7]. Despite unequivocal genetic evidence for the pathogenicity of this mutation, its consequences on mitochondrial ATP synthase are still poorly defined. We recently reported the construction and properties of yeast models of other pathogenic mutations found in the human ATP6 gene: T9176G [8], T8993G [9] and T8993C [10]. The yeast and human ATP synthases proved to be similarly affected by these mutations, which is not very surprising due to the high conservation of the mitochondrial ATP synthase in terms of structure and function [4]. To gain insight into the primary pathogenic mechanisms of T9176C, we have investigated the consequences of this mutation on the yeast ATP synthase.
Experimental Procedures
Yeast strains and media
The S. cerevisiae strains and their genotypes are listed in Table 1. The preparation of rich media containing glucose, glycerol, or galactose carbon sources, and synthetic complete drop-out media supplemented with adenine to 40 mg/L, was as described [11]. Where indicated, ethanol (2%) was included with glycerol (3%) to prepare enriched plates with two non-fermentable substrates (EG). Oligomycin was added to media as indicated in the legend to Figure 1.
Table 1.
Genotypes and sources of yeast strains
| Strain | Nuclear genotype | mtDNA | Source |
|---|---|---|---|
| DFS160 | MATα leu2Δ ura3-52 ade2-101 arg8::URA3 kar1-1 | ρ0 | [39] |
| NB40-3C | MATa lys2 leu2-3,112 ura3-52 his3Δ HinDIII arg8::hisG | ρ+cox2-62 | [39] |
| SDC30 | Matα, kar1, ade2, ura3, leu2, Δ arg8::URA3 | ρ−ATP6 COX2 | [11] |
| MR6 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+ WT | [11] |
| MR10 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ−atp6::ARG8m | [11] |
| RKY37 | MATα leu2Δ ura3-52 ade2-101 arg8::URA3 kar1-1 | ρ−atp6-L247P | this study |
| RKY38 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+atp6-L247P | this study |
| RKY20 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+atp6-L183P | [10] |
| RKY25 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+atp6-L247R | [8] |
| RKY48 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+atp6-Δ leader | [20] |
| W303-1A | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 | ρ + | a |
| W303ΔATP11 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 ATP11::HIS3 | ρ+ | [40] |
| Atp11(Δ40–75)p | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 ATP11::HIS3 [URA3-pG13/Δ40–75s] | ρ+ | [22] |
| Atp11(Δ40–111)p | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 ATP11::HIS3 [URA3-pG13/Δ40–111s] | ρ+ | [22] |
| Atp11(R183)p | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 ATP11::HIS3 [URA3-pG13/R183m] | ρ+ | [22] |
| Atp11(A300)p | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 ATP11::HIS3 [URA3-pG13/A300m] | ρ+ | [22] |
Rothstein
Fig. 1. The T9176C mutation (atp6-L247P) does not compromise the growth of yeast on respiratory substrates.
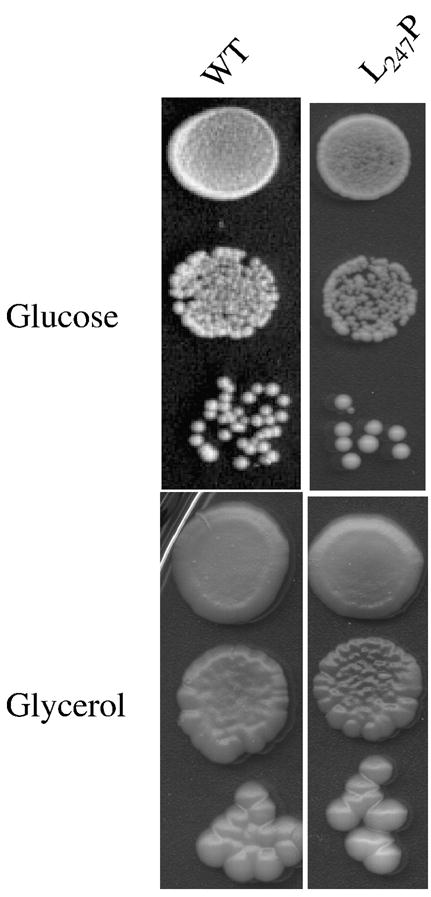
Panel A: Freshly grown cells of wild type yeast (MR6) and atp6-L247P mutant (RKY38) were serially diluted and 5 μl of each dilution were spotted onto YPGA (glucose) and N3 (glycerol) plates. The plates were incubated at 28°C and photographed after three (YPGA) or six (N3) days.
Construction of atp6–L247P mutant strains
The mutagenesis was performed on an EcoRI-BamHI C-terminal fragment of the yeast ATP6 locus cloned in pUC19 (plasmid pSDC9, [11]). This fragment contains the 38 codons of the 3′ end of ATP6 gene (codons 233–260). Using the QuikChange XL Site-directed Mutagenesis Kit from Stratagene, the TTA codon 247 of ATP6 gene was changed into the proline CCA codon with the following primers: 5′ GGATATGTCTGGGCTATTCCAACAGCATCATATTTA, and 5′ TAAATATGATGCTGTTGGAATAGCCCAGACATATCC (the mutated bases are in bold). The mutated fragment was cleaved from the plasmid by EcoRI/SapI digestion and cloned into pSDC14 using the same sites, to create pRK8 plasmid, which encodes a whole ATP6 gene containing the L247P mutation. The pRK8 plasmid contains also the yeast mitochondrial COX2 gene as a marker for mitochondrial transformation [9]. The pRK8 plasmid was introduced by co-transformation with the nuclear selectable LEU2 plasmid pFL46 into the ρo strain DFS160 by microprojectile bombardment using a biolistic PDS-1000/He particle delivery system (Bio-Rad) as described [12]. Mitochondrial transformants were identified among the Leu+ nuclear transformants by their ability to produce respiring clones when mated to the nonrespiring NB40-3C strain bearing a deletion in the mitochondrial COX2 gene. One mitochondrial transformant (synthetic ρ− RKY37) was crossed to the atp6::ARG8m deletion strain MR10 (Table 1, see also ref. [11]). The crosses produced cytoductants (called RKY38) harboring the MR10 nucleus and mtDNA in which the ARG8m ORF had been replaced with the atp6-L247P gene. The RKY38 clones were identified by virtue of their inability to grow in the absence of an external source of arginine. Sequencing of the mutated atp6 locus in RKY38 revealed no change other than L247P.
Miscellaneous procedures
Determination of ρ−/ρ° cells in yeast cultures, mitochondrial respiratory and ATP synthesis/hydrolysis activities, mitochondrial membrane potential measurements, cytochrome spectral analyses, SDS- and BN-PAGE, and pulse labelling of mtDNA encoded proteins were performed as described in [11] and references therein.
Results
Genetic stability and respiratory growth of yeast mutant atp6-L247P
The leucine residue 217 of human subunit a that is changed into proline by the T9176C mutation corresponds to the leucine residue 247 of the yeast homologous protein Atp6p [4]. We have converted the TTA triplet encoding this residue into the CCA codon for proline (see Materials and Methods). In view of earlier findings that mutations of the ATP synthase often promote instability of the mitochondrial genome in yeast in the form of ρ− clones issued from large deletions in the mtDNA [13], two independent atp6-L247P isolates of the mutant RKY38 (Table 1) were tested for the percentage of ρ− cells in cultures grown under non-selective conditions, in rich glucose or galactose medium. These cultures contained only 2% secondary ρ− cells like those of the corresponding wild type strain (MR6), indicating that the leucine to proline change did not adversely affect the stability of yeast mtDNA. The atp6-L247P mutant grew nearly identically to the wild type on a non-fermentable substrate (glycerol) both at 28°C, the optimal temperature for growing yeast (Fig. 1), and at 36°C (not shown). This does not imply that the atp6-L247P mutation had no deleterious effects on the ATP synthase. Instead, it is necessary to decrease the rate of ATP synthesis by ~85% to observe an obvious growth defect on non-fermentable substrates ([14] and see below).
Consequences of the atp6-L183P mutation on several activities related to respiration and oxidative phosphorylation
a) Oxygen consumption
Mitochondria isolated from the atp6-L247P mutant and from the corresponding wild type strain MR6 were assayed for oxygen consumption. With NADH as an electron donor, the basal (state 4) oxygen consumption rate was about the same for the two strains (Table 2). The respiratory rates were stimulated in the presence of excess ADP (state 3 or phosphorylating conditions), but the effect was more pronounced in wild type mitochondria (2.2-fold) compared to the mutant sample (1.6-fold). A qualitatively similar result was observed when maximal respiratory rates were measured in the presence of the uncoupler, m-chlorophenylhydrazone (CCCP), in which case oxygen consumption increased 3.9-fold and 2.9-fold, respectively, in wild type and mutant mitochondria (Table 2). Notably, while the rates of stimulated oxygen consumption were lower in the mutant versus wild type mitochondria, the magnitude of the effect produced by CCCP was ~1.3-fold greater relative to ADP in both samples. This finding suggests that the atp6-L247P mutation caused a decrease in the content of respiratory enzymes rather than a modification of the passive proton permeability of the inner mitochondrial membrane.
Table 2.
Influence of the atp6-L247P mutation on yeast mitochondrial respiration, ATP hydrolytic, and ATP synthesis activities.
| Strain | Respiration rates nmol O.min−1.mg−1 | ATPase activity nmol Pi.min−1.mg−1 | ATP synthesis rate nmol Pi.min−1.mg−1 | |||||
|---|---|---|---|---|---|---|---|---|
| NADH | NADH +ADP | NADH +CCCP | Asc/TMPD + CCCP | − oligo | + oligo | − oligo | + oligo | |
| MR6 | 279±17 | 613±12 | 1081±107 | 2013±290 | 4.474±0.222 | 0.665±0.108 | 637±18 | 15±8 |
| RKY38 | 274±23 | 428±26 | 798±45 | 1313±121 | 3.324±0.405 | 1.292±0.229 | 475±18 | 23±0.3 |
Mitochondria were isolated from wild type strain MR6 (WT) and mutant atp6-L247P (RKY38), grown for 5–6 generations in YPGALA medium (rich galactose) at 28°C. Additions were 0.15 mg/ml proteins, 4 mM NADH, 150 μM ADP, 12.5 mM ascorbate (Asc), 1.4 mM N,N,N,N,-tetramethyl-p-phenylenediamine (TMPD), 4 μM CCCP, 3 μg/ml oligomycin (+O). The MR6 and RKY38 both cultures contained 2% ρ−/ρ° cells. The values reported are averages of triplicate assays ± standard deviation. Respiratory and ATP synthesis activities were measured using freshly isolated mitochondria in iso-osmotic buffer (Mannitol 0.6M, EGTA 0.3 mM, Tris/Maleate 10 mM) at pH 6.8. For the ATPase assays, previously frozen mitochondria (kept at −80°C) were thawed and the reaction was analyzed in a hypo-osmotic solution (KCl 0.2M, MgCl2 10 mM, Tris/HCl 10 mM) at pH 8.4.
Our previous investigations of other yeast models of NARP/MILS diseases (atp6-L183P [10], atp6-L183R [9] and atp6-L247R [8]), and of an ATP6-deletion strain (Δatp6) [11], revealed a deficit for cytochrome c oxidase (complex IV) in each of the mitochondrial samples and correlated the magnitude of this effect with the degree of respiratory deficiency measured for each mutant strain (see Discussion). A similar relationship between the level of complex IV and respiratory function was demonstrated also for the atp6-L247P mutant in the current work. Measurements of oxygen consumption, in the presence of substrates that donate electrons directly to complex IV (ascorbate/TMPD), showed that this activity was reduced in the mutant 35% with respect to wild type (Table 2). Consistent with this finding, dithionite-reduced spectra recorded from whole cells showed that cytochromes a + a3 of complex IV were decreased ~30% in the mutant compared with wild type, while cytochromes c1 and b of complex III were in normal amounts (data not shown). Finally, the amount of immunologically reactive complex IV protein in mutant mitochondria was substantially below the wild type level (see Figure 2A for a typical result representative of three independent experiments).
Fig. 2. SDS- and BN-PAGE analyses of mitochondrial proteins from atp6-L247P mutant.

Panels A, B: BN-PAGE analyses of mitochondrial proteins from the yeast T9176C (atp6-L247P). For the analysis of complexes III and IV (panel A) the mitochondria (100 μg) were solubilized with 10 grams of digitonin per gram of proteins while 2 grams of digitonin per gram of proteins were used for the analysis of the ATP synthase (Panel B). After their electrophoretic separation the digitonin-extracted proteins (50 μg) were transferred to a nitrocellulose membrane and hybrized with the indicated antibodies. For the blots shown in panel B, between two hybridizations, the membrane was stripped to completely remove the previously hybridized antibodies. Panel C, SDS-PAGE analysis. Total mitochondrial proteins (10 μg) of atp6-L247P mutant and wild type strain MR6 were separated in SDS-PAGE, transferred onto a nitrocellulose membrane and probed with the indicated antibodies. Data are representative of at least 3 experiments.
b) Mitochondrial ATP synthesis/hydrolysis
The consequences of the atp6-L247P mutation were analyzed further by measuring the rate of mitochondrial ATP synthesis with NADH as a respiratory substrate. This activity was reduced in the mutant by ~30% compared with the wild type (Table 2), which was similar in scale to the difference observed between these samples with respect to rates of oxygen consumption that were measured under identical conditions (see above). Hence, the efficiency of oxidative phosphorylation, as defined by the number of ATP molecules formed per electron transferred to oxygen, appeared to be largely unaffected by the atp6-L247P mutation. The rate of ATP hydroysis measured in mitochondria from the atp6-L247P mutant was ~75 % the wild type level (Table 2). Of particular interest, oligomycin inhibited the ATPase activity by 85% in the wild type sample, but only by 60% in the mutant sample.
c) ATP-driven translocation of protons across the inner mitochondrial membrane
The proton-pumping activities of wild type and atp6-L247P mutant F1FO were measured in samples of whole mitochondria using a fluorescent dye, Rhodamine 123, to monitor changes in the membrane potential (ΔΨ) (Fig. 3); the dye accumulates inside the mitochondrial matrix, where its fluorescence is quenched, in response to an established ΔΨ [15]. Before testing for ATP-driven proton translocation, the mitochondria were energized with ethanol in order to effect the release of the natural inhibitory peptide (IF1) that binds F1FO in the resting state and prevents ATP hydrolysis (Fig. 3, EtOH). The imposed ΔΨ was then collapsed with KCN, and before rebinding of IF1 could occur (see ref. [16]). ATP was added and counter-exchanged for ADP in the matrix compartment. Comparable levels of proton pumping coupled to ATP hydrolysis were manifested in the wild type and mutant mitochondria by a large and sustained fluorescence quenching that was fully reversed upon addition of oligomycin (Fig. 3, oligo).
Fig. 3. Energization of mitochondria.

Energization of the mitochondrial inner membrane was monitored by rhodamine 123 fluorescence quenching with intact mitochondria from wild type yeast (MR6) and atp6-L247P mutant (RKY38). The additions were 0.5 μg/ml rhodamine 123, 0.15 mg/ml mitochondrial proteins (Mito), 10 μl of ethanol (EtOH), 6 μg/ml oligomycin (oligo), 0.2 mM potassium cyanide (KCN), 1 mM ATP, and 3 μM CCCP. Data are representative of at least 3 experiments.
Assembly/stability of the ATP synthase in the atp6-L247P mutant
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was used to examine the influence of the atp6-L247P mutation on the assembly and stability of the ATP synthase. Following electrophoretic separation, digitonin-extracted mitochondrial proteins were transferred to nitrocellulose and hybridized, successively, with antibodies against four subunits representative of different ATP synthase domains, the proton channel (Atp6p, Atp9p), the peripheral stalk (Atp7p) and the F1 unit (Atp1p or subunit α). The antibodies against Atp6p and Atp7p detected two immunological signals at comparable levels in mitochondria from the wild type and atp6-L247P strains, corresponding to fully assembled F1FO dimers and monomers (Figure 2B). However, the two samples were distinguished with antibodies against Atp9p and subunit α(Atp1p), which detected evidence of an Atp9p-ring, free F1, and incomplete α subunit assemblies in the mutant sample only (Fig. 2B). Pulse-chase analysis of mitochondrial translation products in isolated mitochondria revealed that Atp6p was synthesized with similar efficiencies in the atp6-L247P and wild type organelles and that there was no difference between the two in the turnover rates for newly synthesized mitochondrial gene products. Consistent with these findings, Western blots of SDS-PAGE gels showed equivalent steady-state levels of Atp6p, Atp9p and subunit α in the wild type and mutant samples (Fig. 2C). Taken together, these data indicated only a modest effect of the atp6-L247P mutation on ATP synthase biogenesis and stability.
The atp6-L47P mutant is more sensitive to oligomycin in vivo than the wild type
A previous report [14] indicated that the ATP synthase is far from being limiting for yeast proliferation on non-fermentable substrates (e.g. glycerol); rather the activity of the enzyme needs to be decreased by ~85% before non-fermentative growth is inhibited. Hence, it is not surprising that yeast models of atp6-L247P and atp6-L183P mutations (Table 1) showed ~wild type growth on glycerol plates (Fig. 1A), despite a 30% and 40% deficit, respectively, in mitochondrial ATP production (Table 2, [10]). However, since the atp6-L183P and atp6-L247P mutants are compromised for oxidative phosphorylation, we reasoned that the viability of these strains on glycerol might be more sensitive to oligomycin than wild type yeast. This prediction was validated in experiments that showed the atp6-L247P mutant ceased to grow on glycerol in the presence of an oligomycin concentration (0.25 μg/ml) that did not interfere significantly with wild type propagation on this medium (Fig. 5A). The atp6-L183P mutant showed a similar phenotype on oligomycin-supplemented glycerol plates (data not shown).
Fig. 5. The T9176C mutation (atp6-L247P) renders the growth of yeast on respiratory substrates more sensitive to oligomycin.

Panel A: Freshly grown cells of wild type yeast (MR6), atp6-L247P mutant (RKY38), and a strain lacking the leader peptide of Atp6p (RKY48, atp6-Δleader) [20] were serially diluted and 5 μl of each dilution spotted onto glucose (YPGA) and glycerol (N3) plates, and onto glycerol plates containing the indicated concentrations of oligomycin (Oligo). The plates were incubated at 28°C and photographed after 4 days. Panel B: Growth of yeast atp11 mutants on EG ± oligomycin. Wild type and atp11 mutant strains described in [22] were grown overnight on rich glucose plates (2% glucose, 2% bactopeptone, 1% yeast extract), replica-plated to non-fermentable media (3% glycerol, 2% ethanol, 2% bactopeptone, 1% yeast extract) that was either untreated (EG) or supplemented with oligomycin to 2% (EGO), and incubated at 30°C for 48 h. From top to bottom, the strains analyzed were W303-1A, W303ΔATP11, and W303ΔATP11 transformants harboring plasmids for Atp11(R183)p, Atp11(Δ40–111)p, Atp11(Δ40–75)p, or Atp11(A300)p. The amount of oligomycin-sensitive ATPase activity measured in mitochondria isolated from each strain is shown on the left. For stylistic reasons, two separate regions of the same photograph were fused (white dotted line) to create the digital image of the EG and EGO plates shown in the figure.
Interpretation of these results was complicated by the fact that several mutations in Atp6p were shown to render the yeast ATP synthase more resistant to oligomycin [17, 18]. Perhaps the atp6-L183P and atp6-L247P mutations affected the binding of oligomycin in a way that enhanced, rather than reduced, sensitivity; e.g. by decreasing the dissociation constant of the drug. Hence, it was deemed useful to measure oligomycin sensitivity in vivo for additional yeast mutants that showed a 50–80% deficit in mitochondrial ATP sythase activity, and which were known to be unaffected in the oligomycin binding pocket. One such mutant lacked the leader peptide of Atp6p (strain RKY48, Table 1) that encompasses the N-terminal 10 amino acids of the full-length gene product, and which is cleaved during the assembly of Atp6p into FO [19]. While not essential, this peptide enhanced the association of Atp6p with the Atp9 (subunit c)-ring [20]. The leaderless mutant (RKY48), which showed a 50% reduction in ATP production, grew normally on plain glycerol plates but was more sensitive than the wild type to oligomycin (Fig. 5A). We also examined atp11 mutants in this work. Although Atp11p is not part of the F1FO complex, this protein is required for assembly of the β-subunit into the F1 sector and its absence renders yeast respiratory defective [21]. The mutants investigated in this work were disrupted at the ATP11 locus (Δatp11), produced N-terminal or C- terminal deleted forms of Atp11p from episomal plasmids, and displayed mitochondrial ATP synthase activity above the threshold (15%) that is required to sustain growth on non-fermentable carbons [22]. None of these atp11 mutants grew on ethanol-glycerol plates that were supplemented with 0.2% oligomycin while the wild type parental strain was unaffected in this respect (Fig. 5B). These results indicate that an increased sensitivity in vivo to oligomycin can be caused by mutations that alter ATP synthase levels without affecting the properties of the oligomycin-binding pocket itself.
DISCUSSION
The present study describes a yeast model that has investigated the consequences imposed by the T9176C mutation of human mtDNA that was identified in NARP/MILS patients. This mutation leads to replacement of a highly conserved leucine residue into proline within the Atp6p (subunit a) of the ATP synthase (at position 217 in humans, 247 in yeast). Current models predict this residue is located within an α-helical trans-membrane segment that is in close proximity to the Atp9p (or subunit c)-ring near residues (c-D61 and a-R159), which are known to be critical for FO activity [23]. Introducing an α-helix breaker proline residue is this region could conceivably affect the interactions of Atp6p with Atp9p and/or the functioning of the Atp9p-ring/Atp6p system.
In yeast, the T9176C mutation (RKY38 strain, Table 1) caused a moderate but significant deficit (~30%) in mitochondrial ATP production. At first glance, this reduction seemed to be caused, at least in part, by defects in the assembly/stability of the ATP synthase. Indeed, BN-analyses of atp6-L247P mitochondrial extracts revealed the presence, in addition to fully assembled ATP synthase, of several sub-complexes (Atp9p-ring, F1, and F1α-subunit derivatives) that were virtually absent in the wild type. The same types of sub-complexes were observed previously in an ATP6-deletion strain (Δatp6, [11]), in a mutant lacking the leader peptide of Atp6p [20], and in yeast models of other NARP/MILS mutations (T9176G (atp6-L247R) [8] and T8993C (atp6-L183P) [10]). Prior work showed that large assemblies lacking Atp6p, but containing most of the other subunits of the ATP synthase, formed in these mutants, but that these assemblies were fragile and dissociated easily under the conditions of BN-PAGE analysis [8, 10, 11]. Furthermore, in each case, the amounts of assembled ATP synthase correlated with the steady state levels of Atp6p, presumably because Atp6p proteins have been shown to be rapidly eliminated from the cells when they fail to assemble [24–27]. As Atp6p was efficiently synthesized in the atp6-L247P mutant (Fig. 3), it can be inferred that it is the incorporation or stability of Atp6p within the ATP synthase that is compromised in this mutant. However, there was no obvious difference in the steady-state level of Atp6p in atp6-L247P yeast compared with the wild type (Fig. 2C). It is possible that, in vivo, Atp6p is incorporated in the ATP synthase of the atp6-L247P mutant, but the association of the mutant protein is unusually labile such that the complex dissociates partially when the proteins are resolved in blue native gels.
The observed decrease in the capacity of oligomycin to inhibit the mitochondrial ATPase activity in atp6-L247P yeast (60% versus 85% for the wild type) provides further evidence that the enzyme is, in fact, defective in the mutant strain. We propose that the difference in the efficacy of drug inhibition does not reflect a gain of resistance by the mutant, since ATP synthesis was normally inhibited by oligomycin in mitochondria from the atp6-L247P strain (Table 2). Instead, we suggest that the coupling between the F1 and FO sectors is less efficient when the mutated ATP synthase works in the reverse mode. In this respect, it must be noted that the ATPase assays are performed with mitochondria in a hypotonic solution that is buffered at a non-physiological pH (8.4), and in the presence of a large excess of ATP. Under these conditions, the turnover of the F1FO complex is approximately ten times that of the enzyme when it synthesizes ATP. Considering the decreased stability of the F1FO complex of atp6-L247P yeast, as revealed in BN-PAGE analyses (see above), it is possible that in the extreme conditions of the ATPase assays, a sub-population of the mutant complex dissociates and this provides free F1-ATPase particles in the mitochondrial matrix that are no longer sensitive to the effect of oligomycin interaction with FO. Instead under the conditions of the ATP-driven proton translocation assays (Fig. 3), more of the ATP synthase remains intact such that the magnitude of oligomycin-sensitive coupled activity is comparable to what was observed in wild type mitochondria.
The atp6-L247P mutant showed a decreased electron transfer capacity that was correlated with to a lower content in complex IV. We propose that this is a secondary consequence of the defects in the ATP synthase, as a similar relationship between respiratory activity and complex IV levels was observed previously in other yeast models of NARP/MILS mutations (atp6-L183P [10], atp6-L183R [9] and atp6-L247R [8]) and in Δatp6 yeast [11]. Our current data, considered together with what has been reported earlier, reveals an interesting phenomenon. Notwithstanding the small number of mutations analyzed, there is a linear correlation between the extent to which ATP synthase activity is reduced with the prominence of a complex IV deficit (Fig. 6). These findings are relevant with respect to published studies [8–11, 28] that suggested complex IV biogenesis might be controlled by the level of active ATP synthase, perhaps as a means to adjust the rate of electron transfer to the ATP needs of the cell or to modulate the mitochondrial membrane potential when glycolytic ATP has to be imported into the organelle (i.e. anaerobic conditions) [29]. To date, there is no evidence to absolutely refute the hypothesis that the amount of complex IV might be regulated by the rate of ATP synthesis in human cells, as we propose might occur in yeast. However, transient regulatory responses to the intramitochondrial ATP/ADP ratio have been postulated to regulate cytochrome c oxidase activity in mammals through adenine nucleotide binding to specific sites on the enzyme (for a review see [30]). Hence, it appears that at the very least, evolution has preserved complex IV as an important target for the co-regulation of the electron transfer and ATP synthesis activities in mitochondria.
Fig. 6. The rate of ATP synthesis is linearly correlated with the cytochrome c oxidase activity in yeast atp6 mutants.
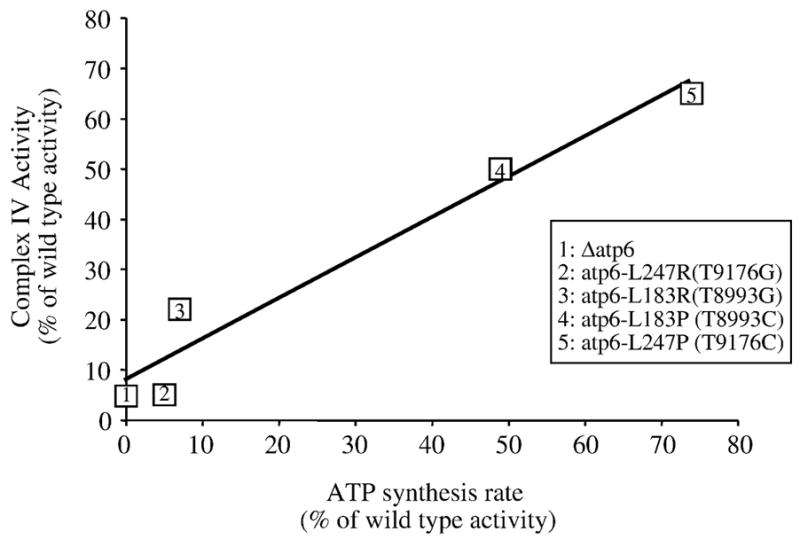
The diagram shows a linear correlation between the ATP synthesis rate (expressed as % of the wild type activity) and cytochrome c oxidase activity (expressed as % of the wild type activity) in Δatp6, atp6-L183P (T8993C), atp6-L183R (T8993G), atp6-L247P (T9176C) and atp6-L247R (T9176G) strains.
Despite a 30% deficit in mitochondrial ATP production, the yeast atp6-L247P and atp6-L183P mutants grew like the wild type on non-fermentable substrates, but ceased to proliferate by respiration if limiting amounts of oligomycin was included in the medium. While Atp6p is known to contribute to the binding of oligomycin [17], we provide evidence that the increased sensitivity of these mutants to oligomycin need not reflect a structural change in the oligomycin binding pocket itself. The data presented in Figs. 5A and 5B show that yeast deficient for mitochondrial ATP synthase, due to mutations unrelated to oligomycin binding (atp6-Δleader, atp11 variants), were more sensitive to oligomycin relative to wild type strains. On this basis, we propose that the respiratory capacity of atp6-L247P and atp6-L183P mutants is unusually sensitive to oligomycin in vivo because such strains are already partially compromised for ATP synthesis. These findings are interesting in view of a previous study that showed evidence for an oligomycin-induced genetic drift in favor of wild type mtDNA in populations of humans cells heteroplasmic for the T8993G mutation [31]. The mechanism by which this genetic drift occurs is poorly understood. Our observations support the possibility that cells segregating less mutant mtDNA will be less vulnerable to oligomycin because of an increase in their oxidative phosphorylation capacity. Our findings are also interesting in view of the poor correlations between genotype and phenotype expression for some of the ATP6 pathogenic mutations, especially T9176C. Some of the reported cases of this mutation were characterized by a relatively mild phenotype [7] while others had an acute, severe clinical course with fulminant Leigh syndrome and a sudden unexpected death [6]. Since the amount of mutated DNA was similar in the different analyzed probands, it would appear that additional determinants may be responsible for the differences in phenotypic presentation. The synthetic lethality that is produced when ATP synthase deficient yeast are exposed to levels of oligomycin that are below what is necessary to prevent wild type growth on non-fermentable substrates illustrates this concept.
In conclusion, notwithstanding the caveats in the extrapolation of data from yeast to humans, we argue that the four ATP6 pathogenic mutations created to model the human syndromes of NARP/MILS (T8993G [9], T8993C [10], T9176G [8], and T9176C (this study)), have similar impacts on the ATP synthase of yeast and human origins. This idea is supported by the fact amongst these mutations, the two that have been shown to correlate with particularly severe forms of the disease (T8993G, T9176G) were also much more detrimental in yeast compared with T8993C and T9176C. Defective assembly/stability of Atp6p was provided as the basis for the major deficit in ATP production imposed by the T9176G mutation both in yeast [8] and in human cells [32, 33]. In vivo, the T8993G mutation had essentially no impact on the assembly/stability of yeast ATP synthase [9], similarly to what has been reported in a thorough study of T8993G cybrids [34]. However, other studies repeatedly demonstrated the presence of partial ATP synthase assemblies in human tissues [35] or cell lines [36] containing the T8993G mutation. T8993C cybrids have been reported not to exhibit a defect in F1FO biogenesis [34]. However this finding is at odds with another study that revealed the presence of free F1 in mitochondria of fresh muscle biopsies from T8993C patients [37]. The latter observation mirrors what was reported for this mutation in a yeast model [10]. The T9176C mutation, which is the principal focus of the current paper, was not characterized in earlier reports, to explain the reduction in ATP production in biopsies of fresh muscle [38]. Very recently, defects identical to those reported here in yeast, were found in fibroblasts from patients who are homoplasmic for the T9176C mutation (V. Proccacio et al., in preparation). These similarities between yeast and humans likely reflect a strong conservation of the Atp9p-ring/Atp6p structure, especially in the region of Atp6p where the pathogenic mutations are located [2, 4].
Fig. 4. Influence of the atp6-L247P mutation on mitochondiral protein synthesis.
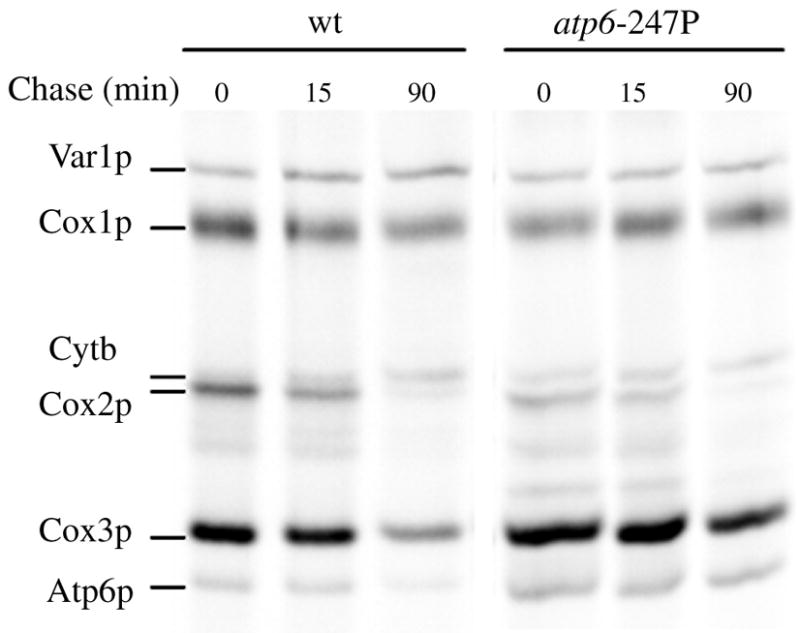
Proteins encoded by the mtDNA were in vivo labeled with [35S]-(methionine + cysteine) for 20 min. at 28°C in the presence of cycloheximide to inhibit cytosolic protein synthesis. Turnover of newly synthesized proteins was evaluated by the addition of excess cold (methionine + cysteine) and incubation for the indicated periods of time. After the labeling reactions, total protein extracts were prepared from the cells (0.2 OD at 650 nm) and loaded on a 12.5% polyacrylamide-4 M urea gel containing 25% glycerol. After electrophoresis the gel was dried and radioactive proteins visualized with a phosphoImager.
Acknowledgments
We are grateful to J. Velours for the generous gift of antibodies. R.K was supported by postdoctoral fellowships from the French Ministry of Research and the Agence Nationale de la Recherche (ANR); N.E was supported by the ANR, and E.C. by post-doctoral fellowships from the ANR and the Association Française contre les Myopathies (AFM). This work was supported by grants from the AFM, the GIS Maladies rares, and ANR to M.B. and J.-P.dR; from the Conseil de la Région Aquitaine to J.-P.dR, from the National Institute of Healh (NIH) to S.H.A. (grant GM48157); from the University Hospital of Angers, the University of Angers, the Association contre les Maladies Mitochondriales (AMMi), Retina France, Ouvrir les Yeux (OLY), and Union Nationale des Aveugles et Déficients Visuels (UNADEV) to V.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 2.Schon EA, Santra S, Pallotti F, Girvin ME. Pathogenesis of primary defects in mitochondrial ATP synthesis. Semin Cell Dev Biol. 2001;12:441–448. doi: 10.1006/scdb.2001.0281. [DOI] [PubMed] [Google Scholar]
- 3.Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006;1757:1400–1405. doi: 10.1016/j.bbabio.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Kucharczyk R, Zick M, Bietenhader M, Rak M, Couplan E, Blondel M, Caubet SD, di Rago JP. Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim Biophys Acta. 2009;1793:186–199. doi: 10.1016/j.bbamcr.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Fillingame RH, Angevine CM, Dmitriev OY. Mechanics of coupling proton movements to c-ring rotation in ATP synthase. FEBS Lett. 2003;555:29–34. doi: 10.1016/s0014-5793(03)01101-3. [DOI] [PubMed] [Google Scholar]
- 6.Dionisi-Vici C, Seneca S, Zeviani M, Fariello G, Rimoldi M, Bertini E, De Meirleir L. Fulminant Leigh syndrome and sudden unexpected death in a family with the T9176C mutation of the mitochondrial ATPase 6 gene. J Inherit Metab Dis. 1998;21:2–8. doi: 10.1023/a:1005397227996. [DOI] [PubMed] [Google Scholar]
- 7.Thyagarajan D, Shanske S, Vazquez-Memije M, De Vivo D, DiMauro S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann Neurol. 1995;38:468–472. doi: 10.1002/ana.410380321. [DOI] [PubMed] [Google Scholar]
- 8.Kucharczyk R, Salin B, di Rago JP. Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum Mol Genet. 2009;18:2889–2898. doi: 10.1093/hmg/ddp226. [DOI] [PubMed] [Google Scholar]
- 9.Rak M, Tetaud E, Duvezin-Caubet S, Ezkurdia N, Bietenhader M, Rytka J, di Rago JP. A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene. J Biol Chem. 2007;282:34039–34047. doi: 10.1074/jbc.M703053200. [DOI] [PubMed] [Google Scholar]
- 10.Kucharczyk R, Rak M, di Rago JP. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim Biophys Acta. 2009;1793:817–824. doi: 10.1016/j.bbamcr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Rak M, Tetaud E, Godard F, Sagot I, Salin B, Duvezin-Caubet S, Slonimski PP, Rytka J, di Rago JP. Yeast cells lacking the mitochondrial gene encoding the ATP synthase subunit 6 exhibit a selective loss of complex IV and unusual mitochondrial morphology. J Biol Chem. 2007;282:10853–10864. doi: 10.1074/jbc.M608692200. [DOI] [PubMed] [Google Scholar]
- 12.Bonnefoy N, Fox TD. Genetic transformation of Saccharomyces cerevisiae mitochondria. Methods in cell biology. 2001;65:381–396. doi: 10.1016/s0091-679x(01)65022-2. [DOI] [PubMed] [Google Scholar]
- 13.Contamine V, Picard M. Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiol Mol Biol Rev. 2000;64:281–315. doi: 10.1128/mmbr.64.2.281-315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukhopadhyay A, Uh M, Mueller DM. Level of ATP synthase activity required for yeast Saccharomyces cerevisiae to grow on glycerol media. FEBS Lett. 1994;343:160–164. doi: 10.1016/0014-5793(94)80310-2. [DOI] [PubMed] [Google Scholar]
- 15.Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- 16.Venard R, Brethes D, Giraud MF, Vaillier J, Velours J, Haraux F. Investigation of the role and mechanism of IF1 and STF1 proteins, twin inhibitory peptides which interact with the yeast mitochondrial ATP synthase. Biochemistry. 2003;42:7626–7636. doi: 10.1021/bi034394t. [DOI] [PubMed] [Google Scholar]
- 17.John UP, Nagley P. Amino acid substitutions in mitochondrial ATPase subunit 6 of Saccharomyces cerevisiae leading to oligomycin resistance. FEBS Lett. 1986;207:79–83. doi: 10.1016/0014-5793(86)80016-3. [DOI] [PubMed] [Google Scholar]
- 18.Ray MK, Connerton IF, Griffiths DE. DNA sequence analysis of the Olir2-76 and Ossr1-92 alleles of the Oli-2 region of the yeast Saccharomyces cerevisiae. Analysis of related amino-acid substitutions and protein-antibiotic interaction. Biochim Biophys Acta. 1988;951:213–219. doi: 10.1016/0167-4781(88)90042-5. [DOI] [PubMed] [Google Scholar]
- 19.Michon T, Galante M, Velours J. NH2-terminal sequence of the isolated yeast ATP synthase subunit 6 reveals post-translational cleavage. Eur J Biochem. 1988;172:621–625. doi: 10.1111/j.1432-1033.1988.tb13934.x. [DOI] [PubMed] [Google Scholar]
- 20.Zeng X, Kucharczyk R, di Rago JP, Tzagoloff A. The leader peptide of yeast Atp6p is required for efficient interaction with the Atp9p ring of the mitochondrial ATPase. J Biol Chem. 2007;282:36167–36176. doi: 10.1074/jbc.M705436200. [DOI] [PubMed] [Google Scholar]
- 21.Wang ZG, Ackerman SH. The assembly factor Atp11p binds to the beta-subunit of the mitochondrial F(1)-ATPase. J Biol Chem. 2000;275:5767–5772. doi: 10.1074/jbc.275.8.5767. [DOI] [PubMed] [Google Scholar]
- 22.Wang ZG, Ackerman SH. Identification of functional domains in Atp11p. Protein required for assembly of the mitochondrial F1-ATPase in yeast. J Biol Chem. 1996;271:4887–4894. doi: 10.1074/jbc.271.9.4887. [DOI] [PubMed] [Google Scholar]
- 23.Moore KJ, Fillingame RH. Structural interactions between transmembrane helices 4 and 5 of subunit a and the subunit c ring of Escherichia coli ATP synthase. J Biol Chem. 2008;283:31726–31735. doi: 10.1074/jbc.M803848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ackerman SH, Tzagoloff A. Function, structure, and biogenesis of mitochondrial ATP synthase. Prog Nucleic Acid Res Mol Biol. 2005;80:95–133. doi: 10.1016/S0079-6603(05)80003-0. [DOI] [PubMed] [Google Scholar]
- 25.Zeng X, Neupert W, Tzagoloff A. The metalloprotease encoded by ATP23 has a dual function in processing and assembly of subunit 6 of mitochondrial ATPase. Mol Biol Cell. 2007;18:617–626. doi: 10.1091/mbc.E06-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzagoloff A, Barrientos A, Neupert W, Herrmann JM. Atp10p assists assembly of Atp6p into the F0 unit of the yeast mitochondrial ATPase. J Biol Chem. 2004;279:19775–19780. doi: 10.1074/jbc.M401506200. [DOI] [PubMed] [Google Scholar]
- 27.Lefebvre-Legendre L, Vaillier J, Benabdelhak H, Velours J, Slonimski PP, di Rago JP. Identification of a nuclear gene (FMC1) required for the assembly/stability of yeast mitochondrial F(1)-ATPase in heat stress conditions. J Biol Chem. 2001;276:6789–6796. doi: 10.1074/jbc.M009557200. [DOI] [PubMed] [Google Scholar]
- 28.Barrientos A, Gouget K, Horn D, Soto IC, Fontanesi F. Suppression mechanisms of COX assembly defects in yeast and human: insights into the COX assembly process. Biochim Biophys Acta. 2009;1793:97–107. doi: 10.1016/j.bbamcr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefebvre-Legendre L, Balguerie A, Duvezin-Caubet S, Giraud MF, Slonimski PP, Di Rago JP. F1-catalysed ATP hydrolysis is required for mitochondrial biogenesis in Saccharomyces cerevisiae growing under conditions where it cannot respire. Mol Microbiol. 2003;47:1329–1339. doi: 10.1046/j.1365-2958.2003.03371.x. [DOI] [PubMed] [Google Scholar]
- 30.Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 31.Manfredi G, Gupta N, Vazquez-Memije ME, Sadlock JE, Spinazzola A, De Vivo DC, Schon EA. Oligomycin induces a decrease in the cellular content of a pathogenic mutation in the human mitochondrial ATPase 6 gene. J Biol Chem. 1999;274:9386–9391. doi: 10.1074/jbc.274.14.9386. [DOI] [PubMed] [Google Scholar]
- 32.Carrozzo R, Wittig I, Santorelli FM, Bertini E, Hofmann S, Brandt U, Schagger H. Subcomplexes of human ATP synthase mark mitochondrial biosynthesis disorders. Ann Neurol. 2006;59:265–275. doi: 10.1002/ana.20729. [DOI] [PubMed] [Google Scholar]
- 33.Carrozzo R, Tessa A, Vazquez-Memije ME, Piemonte F, Patrono C, Malandrini A, Dionisi-Vici C, Vilarinho L, Villanova M, Schagger H, Federico A, Bertini E, Santorelli FM. The T9176G mtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology. 2001;56:687–690. doi: 10.1212/wnl.56.5.687. [DOI] [PubMed] [Google Scholar]
- 34.Cortes-Hernandez P, Vazquez-Memije ME, Garcia JJ. ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J Biol Chem. 2007;282:1051–1058. doi: 10.1074/jbc.M606828200. [DOI] [PubMed] [Google Scholar]
- 35.Houstek J, Klement P, Hermanska J, Houstkova H, Hansikova H, Van den Bogert C, Zeman J. Altered properties of mitochondrial ATP-synthase in patients with a T-->G mutation in the ATPase 6 (subunit a) gene at position 8993 of mtDNA. Biochim Biophys Acta. 1995;1271:349–357. doi: 10.1016/0925-4439(95)00063-a. [DOI] [PubMed] [Google Scholar]
- 36.Nijtmans LG, Henderson NS, Attardi G, Holt IJ. Impaired ATP synthase assembly associated with a mutation in the human ATP synthase subunit 6 gene. J Biol Chem. 2001;276:6755–6762. doi: 10.1074/jbc.M008114200. [DOI] [PubMed] [Google Scholar]
- 37.Morava E, Rodenburg RJ, Hol F, de Vries M, Janssen A, van den Heuvel L, Nijtmans L, Smeitink J. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A. 2006;140:863–868. doi: 10.1002/ajmg.a.31194. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs LJ, de Coo IF, Nijland JG, Galjaard RJ, Los FJ, Schoonderwoerd K, Niermeijer MF, Geraedts JP, Scholte HR, Smeets HJ. Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Mol Hum Reprod. 2005;11:223–228. doi: 10.1093/molehr/gah152. [DOI] [PubMed] [Google Scholar]
- 39.Steele DF, Butler CA, Fox TD. Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc Natl Acad Sci U S A. 1996;93:5253–5257. doi: 10.1073/pnas.93.11.5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ackerman SH, Martin J, Tzagoloff A. Characterization of ATP11 and detection of the encoded protein in mitochondria of Saccharomyces cerevisiae. J Biol Chem. 1992;267:7386–7394. [PubMed] [Google Scholar]