

Abstract
We elucidate the role of late Na+ current (INaL) for diastolic intracellular Ca2+ (DCa) accumulation in chronic heart failure (HF). HF was induced in 19 dogs by multiple coronary artery microembolizations; 6 normal dogs served as control. Ca2+ transients were recorded in field-paced (0.25 or 1.5 Hz) fluo-4-loaded ventricular myocytes (VM). INaL and action potentials were recorded by patch-clamp. Failing VM, but not normal VM, exhibited (1) prolonged action potentials and Ca2+ transients at 0.25 Hz, (2) substantial DCa accumulation at 1.5 Hz, and (3) spontaneous Ca2+ releases, which occurred after 1.5 Hz stimulation trains in ~31% cases. Selective INaL blocker ranolazine (10 μM) or the prototypical Na+ channel blocker tetrodotoxin (2 μM) reversibly improved function of failing VM. The DCa accumulation and the beneficial effect of INaL blockade were reproduced in silico using an excitation-contraction coupling model. We conclude that INaL contributes to diastolic Ca2+ accumulation and spontaneous Ca2+ release in HF.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-010-0092-0) contains supplementary material, which is available to authorized users.
Keywords: Action potential remodeling, Ca2+ handling, Heart failure, Na+ current, Na+/Ca2+ exchange
Introduction
Congestive heart failure (HF) is associated with severe abnormalities in both cardiac rhythm and contractile function. While Ca2+ directly triggers contractions in cardiac myocytes, both cell Ca2+ content and dynamics are modulated by Na+ ions [1]. Abnormal Na+ handling and Na+/Ca2+ interplay in HF are being extensively investigated in order to develop new approaches to the treatment of HF [2–4]. While the majority of Na+ channels open and inactivate during the action potential (AP) upstroke, some of them reopen during the AP plateau, carrying the so-called late or persistent Na+ current (INaL) [5]. We found INaL in cardiomyocytes of both normal and failing human hearts [6], and further studies showed that INaL increases in HF, both in patients and in animal models [5, 7–9]. Augmented INaL plays an important role in impaired repolarization of cardiomyocytes of failing hearts [6, 7, 9–13].
Previous studies have suggested that during each beat a relatively small, but long lasting, INaL brings a substantial amount of Na+ into cardiomyocytes, comparable to that carried by a large but very brief transient Na+ current (INaT) [9]. The latter is reduced in chronic HF [8, 14, 15], whereas INaL is increased; hence, the relative contribution of INaL to the regulation of intracellular Na+ is expected to increase further in myocytes from failing hearts. Thus INaL, by altering AP and providing Na+ influx, might be implicated in Na+-related Ca2+ overload, especially in the context of up-regulated Na+/Ca2+ exchanger (NCX) in HF [16–18]; reduced sarcoplasmic reticulum (SR), Ca2+ ATPase (SERCA), or Ca2+-pump function; down-regulated Ca2+-release channel (ryanodine receptor, RyR); and increased SR Ca2+ leak [18–23]. The increased INaL by depolarizing the cell membrane and increasing late Na+ influx during AP is likely to facilitate a shift of the NCX operation in HF from the prime forward mode to the reverse mode, i.e., from Ca2+ efflux to Ca2+ entry [24], thereby contributing to cell Ca2+ accumulation in HF. Abnormal cell Ca2+ accumulation, in turn, worsens both contractility (via diastolic function) and rhythm (via spontaneous Ca2+ releases-triggered delayed after-depolarizations, DADs) [18, 25]. Therefore, inhibition of INaL could be a potential target to improve function of the failing heart, but this possibility remains largely unexplored. While some prior studies have indeed shown improved diastolic performance due to inhibition of INaL in different experimental conditions (including in vivo settings), those studies were mainly performed in a normal myocardium using pharmacological INaL enhancement with toxins or other Na+ channel agonists [26–28], as well as expressing LQT-3 syndrome-related mutant Na+ channels with retarded inactivation [29]. The properties of INaL in HF myocytes are distinctively different [9, 11, 12] from the non-inactivating INa produced by Na+ channel agonists and mutations in myocytes of normal hearts (NH), not to mention differences in Ca2+ handling. Thus, the role of an augmented INaL in HF remains to be established.
Using a combination of experimental and numerical modeling approaches, the present study tested the hypothesis that INaL is indeed a major contributor to the dynamic, Na+-dependent diastolic Ca2+ (DCa) accumulation in single ventricular myocytes isolated from failing hearts. We used a canine chronic HF model that causes numerous physiological deficiencies similar to those observed in patients with HF [30]. Isolated single ventricular myocytes from failing hearts exhibited abnormal Ca2+ handling, including substantial DCa accumulation at a moderate-high physiological pacing rate of 1.5 Hz, which is deleterious to left ventricular ejection fraction in HF patients [31, 32]. Inhibition of INaL greatly reduced DCa and substantially decreased the probability of spontaneous Ca2+ releases (SCaRs), a well-known initiation mechanism of DADs and triggered arrhythmia [18, 25]. Our numerical modeling confirmed that INaL provides a substantial contribution to diastolic cell Ca2+ accumulation observed experimentally. Thus, inhibition of INaL may suppress a significant portion of the Ca2+ accumulation and decrease occurrence of SCaRs in cardiomyocytes and, thereby, is likely to improve heart contractility and rhythm in HF.
Methods
This study conforms to the guide of the care and use of laboratory animals published by the NIH and was approved by the Institutional Animal Care and Use Committee of the Henry Ford Health System. Our experimental methods, data analysis, theoretical formulations, and calculations are given in the Electronic Supplementary Material (ESM). In brief, chronic HF in dogs was induced by multiple sequential coronary microembolizations. Ca2+ signals were measured in field-stimulated, fluo-4-loaded single ventricular myocytes using a photo-multiplier; electrophysiological recordings were performed using the patch-clamp technique. Ca2+ signals and APs were measured at 35°C and 1.8 mM [Ca2+]o, INaL was measured at room temperature (21–23°C).
We used ranolazine (RAN) and tetrodotoxin (TTX) as pharmacological tools. Whereas TTX blocks both INaT and INaL equally [9], RAN blocks INaL preferentially [13]. RAN is superior to other Class I or III drugs (RAN > amiodarone > flecainide > mexiletine > lidocaine) [13, 33–36] at blocking INaL over INaT. Furthermore, RAN at the concentration of 10 μM used in the present study (i.e., close to therapeutic range of 2–8 μM) has only minor effects on K+ currents [33], unlike amiodarone, which has major effects on K+ currents. Concentrations of RAN (10 μM) and TTX (2 μM) were chosen to produce a comparable INaL inhibition (~1.5 of IC50) based on our previous studies [9, 13].
Results are expressed as mean ± SEM (if not stated otherwise), and statistical significance was based on P < 0.05, ANOVA followed by Bonferroni’s post-hoc test. To evaluate the probability of the SCaRs we used nonparametric two-tailed Fisher’s exact test.
We simulated the effect of selective inhibition of INaL on DCa accumulation and APs in silico using a modified excitation-contraction (E-C) coupling model of failing canine ventricular myocytes developed previously by Winslow et al. [37] (see details in the ESM).
Results
Minor effects of ranolazine and TTX on intracellular Ca2+ in myocytes from normal heart
First we tested the effects of 10 μM RAN and 2 μM TTX in field-stimulated cardiomyocytes of normal dogs on the duration (90%) of Ca2+ transients (CaT90) and accumulation of DCa during a pulse train (see the ESM for definition) (Fig. 1). Both RAN and TTX only slightly reduced CaT90 at a low stimulation rate of 0.25 Hz, and these effects were not statistically different (Fig. 1c). At a high stimulation rate of 1.5 Hz, accumulation of DCa was negligible (a few percent) both in control and under RAN or TTX (Fig. 1b).
Fig. 1.
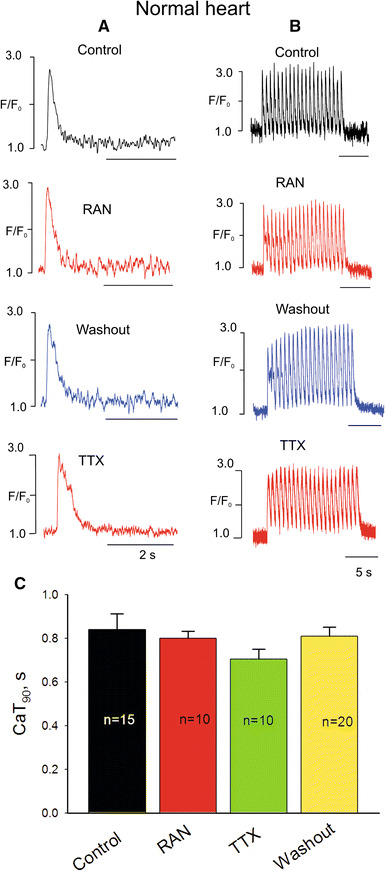
Representative examples of Ca2+ transient recordings in normal dog heart ventricular myocytes at low (a) and high (b) pacing rates before and after infusion of ranolazine (RAN) or tetrodotoxin (TTX). c Data summary (mean ± SEM, n = 10–20) for Ca2+ transient duration (CaT90)
Improvement of intracellular Ca2+ handling in myocytes from failing heart by TTX and ranolazine
Ca2+ transients were substantially different in field-stimulated myocytes of failing compared to normal heart. CaT90 of failing myocytes was significantly longer, and the CaT had a spike-dome configuration when myocytes were stimulated at a frequency of 0.25 Hz (Fig. 2). Some failing myocytes exhibited multiple oscillations on top of the “dome” (Fig. 2a, upper panel). Both RAN and TTX improved CaT, as they significantly and reversibly shortened CaT90 by suppressing the dome phase (Fig. 2c).
Fig. 2.
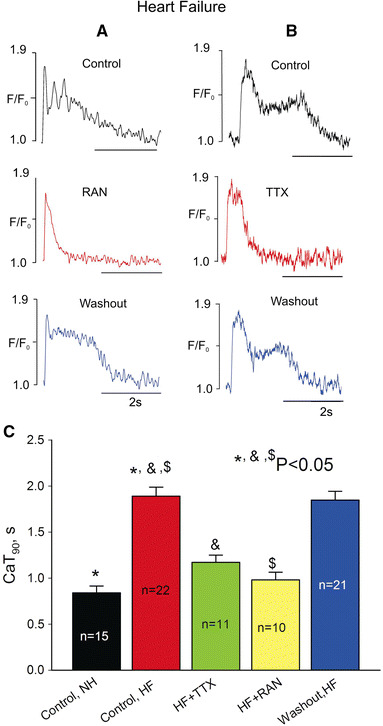
Ranolazine and TTX shorten Ca2+ transient duration (CaT90) in failing dog ventricular myocytes at low pacing rates (0.25 Hz). a, b Representative examples of Ca2+ transient recordings, c summary of the data (mean ± SEM, n = 10–22) for CaT90
Increasing the stimulation rate to 1.5 Hz markedly increased the DCa level during the pulse train (Fig. 3a, b, upper panels) and induced beat-to-beat CaT alternations in amplitude and duration in all HF myocytes studied (Fig. 3a, upper panel). Both RAN (Fig. 3a) and TTX (Fig. 3b) significantly and reversibly decreased DCa accumulation and eliminated these changes (Fig. 3c).
Fig. 3.
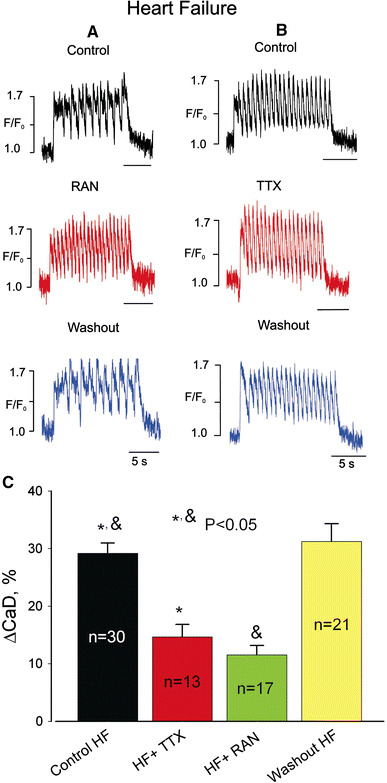
Ranolazine and TTX reduce diastolic Ca2+ elevation at high pacing rates (1.5 Hz) in failing dog ventricular myocytes. a, b Representative examples of Ca2+ transient recordings, c data summary for diastolic Ca2+ (CaD) changes (ΔCaD) during a pulse train. Data are mean ± SEM pooled from 13 to 30 cells
Next we determined whether failing myocytes generate SCaRs when subjected to 1.5 Hz trains of stimulation pulses. SCaRs occurred in ~31% of instances with a delay of 2.1 ± 0.2 s (n = 17) after the end of the last pulse of the 1.5 Hz train of pulses (Fig. 4a). SCaRs were absent in myocytes of NH. The SCaR amplitude was comparable to that of CaT within the pulse train (see below) and ranged from 163 to 251 nM (calculation of [Ca2+]i is given in the ESM). Both TTX and RAN greatly (~threefold) and reversibly reduced the probability of SCaR occurrence (Fig. 4).
Fig. 4.
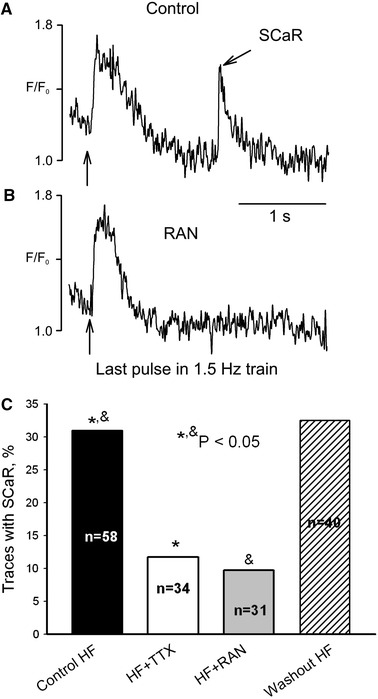
Ranolazine prevents spontaneous Ca2+ releases (SCaR) in myocytes from failing dog heart. Representative traces of Ca2+ signals just after the 1.5 Hz pulse train a in control and b in the presence of 10 μM ranolazine (RAN). Vertical arrows indicate the last pulse in the train. c RAN significantly and reversibly decreases the rate of SCaR. The probability of SCaR occurrence (bars) was evaluated as the percentage of traces that contained SCaR (similar to that shown in a). In control conditions 18 out of total 58 traces contained SCaR, in TTX it was present in 4 out of total 34 traces, in RAN it was present in 3 out of 31 traces, and after washout it was present in 13 out of total 40 traces. Statistical comparison was performed using two-tailed Fisher’s exact tests, and P < 0.05 was considered to be significant. Data were pooled from 31 to 58 cells
Does ranolazine affect SR Ca2+ content and NCX function?
The mechanism for the SCaRs, which are precursors for DADs, is the interplay between the SR Ca2+ load, cytoplasmic [Ca2+], and NCX (see for review [25]). Therefore, in order to further address the mechanism of the beneficial effects of RAN to decrease the probability of SCaR occurrence (Fig. 4), in the next experiments we determined SR Ca2+ content (or load) and NCX function by the caffeine-induced Ca2+-transients at a basal stimulation rate of 1.5 Hz in the presence and absence of the drug. The amplitude of the caffeine-induced Ca2+ transient was used as a measure for SR Ca2+ load [28], and the decay time constant was used as an estimate of NCX activity [38]. Figure 5a shows traces of the caffeine-induced Ca2+ transients in the absence (left panel) and presence of RAN (10 μM, right panel). Figure 5b and c shows summary data of the caffeine-induced transient amplitude (SR Ca2+ content) and decay time constant (NCX function). Our data show that the decay kinetics (Fig. 5c) after RAN application changes insignificantly. Although there was an apparent reduction in SR Ca2+ load (Fig. 5b), the statistical analysis does not reveal statistically significant difference in caffeine-induced Ca2+ transient amplitude (Fig. 5b). Therefore, we assumed that RAN affects neither SR Ca2+ load nor NCX function.
Fig. 5.
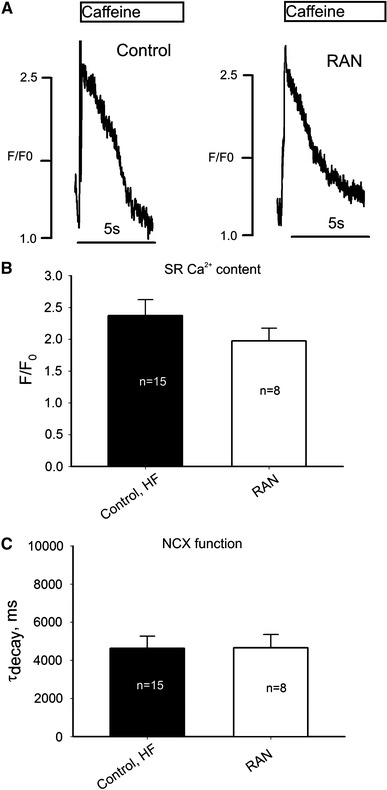
Ranolazine affects SR Ca2+ content or NCX function insignificantly in ventricular myocytes from failing dog hearts. a Original traces of Ca2+ transients during the exposure to caffeine (10 mM) in control (left panel) and in the presence of ranolazine (RAN, 10 μM, right panel). b Average data for SR Ca2+ load, c average data for Ca2+ decay as a measure of NCX function (single exponential fit). Statistical analysis does not reveal a statistical difference between control and RAN for these parameters (ANOVA). Data bars in b and c represent mean ± SEM pooled from 8 to 15 cells
Effects of ranolazine on INaL in myocytes from failing heart
In our previous study we have shown that RAN preferentially blocked INaL (IC50 = 6.46 μM, for HF) over INaT (IC50 = 294 μM, NH; IC50 = 244 μM, HF) [13]. The effect of the drug on INaL kinetics has not been previously studied and thus was examined in the present study. The decay of INaL was approximated by a double-exponential fit (Eq. 4 in the ESM; see examples in Fig. 5a) as previously suggested [12]. The fast component (τBM, tens of ms) represents burst mode (BM) and the slow component (τLSM, hundreds of ms) represents late scattered mode (LSM) of late Na+ channel openings [12]. The effect of RAN (10 μM) on INaL was complex and reversible: the density was reduced (Fig. 6b) and the drug accelerated decay kinetics of both INaL components (Fig. 6c, d).
Fig. 6.
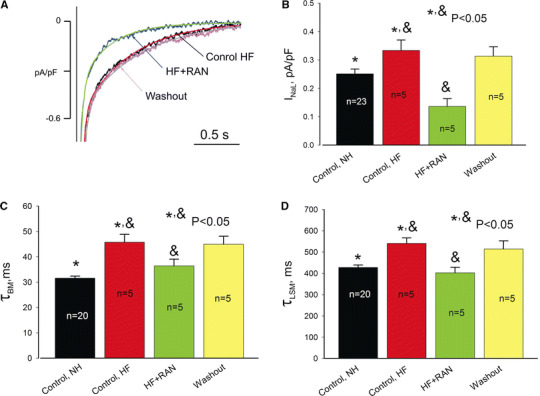
Effects of RAN (10 μM) on INaL in ventricular myocytes from canine failing hearts. a Representative traces along with double-exponential fit of the time course of INaL decay (solid lines). b Summary of the data on INaL density measured as a mean current within 200–220 ms after the depolarization onset, and decay kinetics (c, d) measured at −30 mV in control, in the presence of RAN, and during the washout. Data are mean ± SEM pooled from 5 to 23 cells. See details in text
Effects of ranolazine on action potentials in myocytes from failing heart
The AP shape and its duration, especially the variability of AP duration, play an important role in abnormalities of the E-C coupling in HF. We previously showed that AP duration of ventricular myocytes is prolonged (at low rates) and AP duration variability is increased in this canine HF model and in HF patients compared to NH, and the role of INaL in the AP duration abnormalities has been suggested [6, 7, 10]. Using the specific INaL inhibitor RAN allows further investigation of the role of INaL in AP regulation in HF. Accordingly, we determined the effects of RAN (10 μM) on AP duration and its variability at low and high pacing rates in myocytes from failing hearts. The results of these experiments are summarized in Fig. 7. At both stimulation rates RAN effectively and reversibly shortened the prolonged APs and abolished the beat-to-beat variability apparent as a large dispersion in the AP duration distribution histograms (Fig. 7c–h). For comparison the AP duration and its dispersion in normal dog ventricular myocytes recorded at 0.25 and 1.5 Hz were 337 ± 52 ms (mean ± SD, n = 8 cells) and 303 ± 40 ms (n = 9), respectively (data pooled from the previous publications [7, 9, 13]).
Fig. 7.
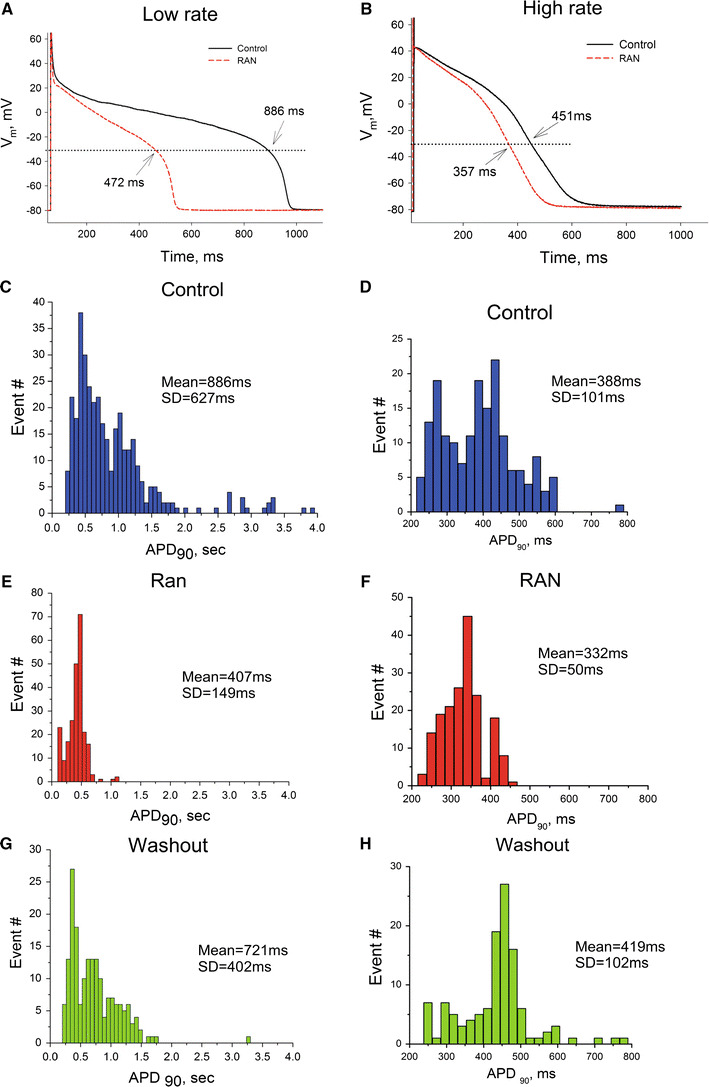
RAN reversibly shortens the AP duration (APD) and reduces the dispersion of APD in ventricular myocytes from canine failing hearts. a, b Representative AP traces at low and high pacing rates recorded at the end of a pulse train, respectively. Dotted line indicates −30 mV level; APD values at this potential are indicated at the traces. c–h Histograms of the distribution of the APD measured at 90% of repolarization (APD90). Bin sizes were 50 and 23 ms for c, e, g and for d, f, h, respectively. SD Standard deviation. APs were recorded in control (c, d), in the presence of 10 μM RAN (e, f), and after RAN washout (g, h). Data were pooled from 6 to 12 cells of four failing hearts
Numerical evaluation of INaL and late Na+ influx during action potential plateau and its importance for the diastolic Ca2+ accumulation in HF
Based on our experimental data, we performed several numerical estimates to answer three key important questions:
What is the time course for INaL and late Na+ influx (i.e., INaL integral) during AP plateau?
How much of this Na+ influx could be blocked by 10 μM of RAN?
Is late Na+ influx sufficient to account for a major portion of diastolic cell Ca2+ accumulation observed experimentally during the pulse train at 1.5 Hz?
In the first approximation we used experimentally recorded AP traces to simulate Na+ influx dynamics during AP plateau (above −30 mV) at low and high rates of stimulation before and after RAN (Fig. 8a, b) (see formulations in the ESM). Results of these AP-clamp simulations are as follows: Because the model includes RAN-induced steady-state inactivation (SSI) shifts found at the physiological resting potential [13] (see Table 1 in the ESM), it predicts greater inhibition of INaL and late Na+ influx by RAN in physiological conditions in comparison to the voltage-clamp data obtained from a very low holding potential (compare Figs. 6a, 8c). More specifically, RAN blocks about 77% of the late Na+ influx during the single AP (from 64.3 to 14.5 fC/pF at low rate and from 50.4 to 11.5 fC/pF at the high rate).
Fig. 8.
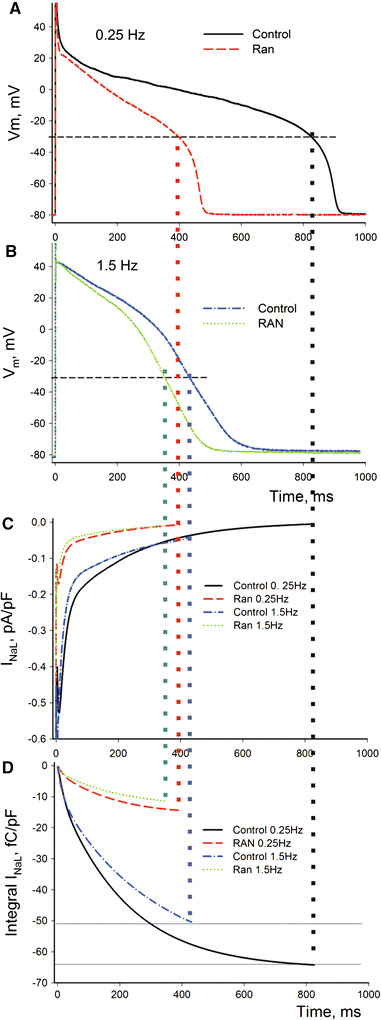
AP-clamp numerical model. A substantial inhibition of INaL and late Na+ influx (77%) during AP plateau by a therapeutic concentration (10 μM) of RAN. a, b Experimental AP recordings at low and high pacing rates in the absence or presence of RAN. c, d Predicted INaL and its integral (~late Na+ influx) during the corresponding AP shapes (Eq. 9 in the ESM)
A remarkable portion of late Na+ influx is likely exchanged by NCX for Ca2+ steady state during stimulation at 1.5 Hz (Fig. 3) for the following reasons: (1) NCX function is enhanced in HF [16–18], (2) NCX operates in the reverse mode during AP plateau in canine failing myocytes (see Fig. 1f in [37]) and extrudes 3 Na+ for influx of 1 Ca2+, and (3) INaL blockade by RAN substantially reduces DCa (Fig. 3) but affects neither SR Ca2+ loading, nor NCX function (Fig. 5). Based on this reasoning, our numerical estimate demonstrates that the late Na+ influx (~INaL integral) during the AP plateau might be indeed substantial and sufficient to increase cytosolic [Ca2+] by ~72 nM assuming its efflux by the reverse Na+/Ca2+ exchange at the steady state during a 1.5 Hz stimulation train (see the ESM for detailed formulations and calculations). While our AP clamp simulations, described above, include real AP shapes of myocytes in our specific HF model, they only consider INaL, NCX, and cell Ca2+ buffering. Thus, the above 72 nM is likely a higher estimate because this simple estimate does not include possible contributions of other Ca2+ and Na+ transport systems. Therefore, we further addressed the role on INaL in CaD accumulation in silico using our modified version of an established model for pacing-induced dog HF ventricular myocytes that includes all essential components of E-C coupling and Na+ homeostasis [37]. This pacing-induced HF model produces similar remodeling of AP and ionic currents, including the augmented INaL, found in patients with HF and in our dog HF model [6–9, 37, 39]. The details of the model modification to include INaL are given in the ESM. The results of our model simulations are shown in Fig. 9. We applied a train of 11 stimulation pulses in the model and recorded intracellular Ca2+ dynamics and APs in control and when INaL was set to zero. Our model simulations reproduced DCa accumulation (Fig. 9, upper panel) observed experimentally in failing myocytes (Fig. 3a, b, upper panels). Selective blockade of INaL in the model (either completely or partially, simulating RAN effect) shortened AP (Fig. 9, lower panel), and substantially reduced this simulated DCa accumulation similarly to that found in experiment (Figs. 7b–f, 3a, b, middle panels).
Fig. 9.
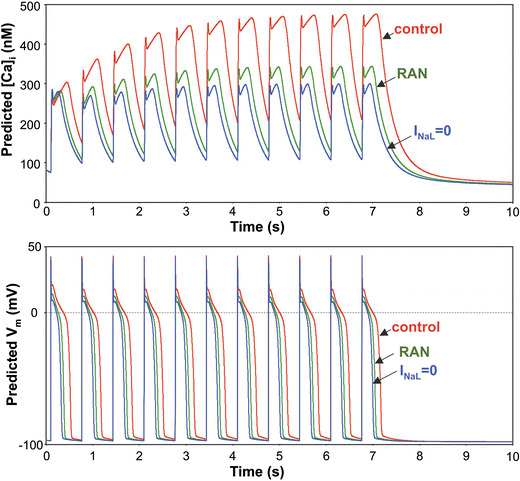
In silico demonstration of the role of the augmented INaL in AP shape and diastolic Ca2+ accumulation in canine failing ventricular myocytes. Numerically simulated dynamics of intracellular Ca2+ concentration ([Ca2+]i) (upper panel) in a train of 11 pulses applied with a rate of 1.5 Hz. Complete INaL elimination or reduction by RAN (10 μM) substantially reduces the diastolic Ca2+ accumulation and shortens AP duration (lower panel). Simulations were performed using a modified Winslow et al. E-C coupling model of failing canine ventricular myocytes [37] (see details in the ESM)
We next calculated the absolute cytosolic free Ca2+ dynamics during stimulation with the rate of 1.5 Hz from the respective Ca2+ signal traces represented originally by F/F0 values (ESM). The CaT amplitude was 210–325 nM in myocytes from the HF model, i.e., values similar to those reported previously for myocytes from pacing-induced models of HF in dogs [37] (~200 nM, see also Fig. 7e) or cats [40] (~340 nM). Finally, from our experimental traces (examples in Fig. 3a, b), we found that during 1.5 Hz pulse trains RAN indeed suppresses the major portion of DCa accumulation, which in absolute terms of [Ca2+] ranges from 62 to 98 nM. Thus, both of our numerical simulations closely predict this experimental estimate for substantial DCa accumulation suppression by INaL inhibition.
Discussion
Using a combination of experimental and numerical modeling studies, we demonstrate at the single cell level that in chronic HF augmented activity of the late Na+ channels exerts both direct electrophysiological effects and indirect effects on intracellular Ca2+ concentration in ventricular myocytes. As discussed in detail below, our findings thus suggest a novel cellular and molecular mechanism contributing to rate-dependent [31, 32] impaired diastolic function (i.e., poor relaxation) and DAD-mediated triggered activity, two major abnormalities associated with chronic HF [2, 18, 41, 42]. As we noted in the Introduction, the previous suggestions about the INaL-mediated mechanism in HF were based on results obtained in normal myocardium using Anemonia sulcata toxin, ATX-II, to produce persistent INa [27, 28]. The role of INaL in these latter studies could be exaggerated because ATX-II caused a fivefold increase in the persistent INa (Fig. 4a in [28]), whereas studies in failing human and dog hearts showed INaL amplitude increases of 30–50% [8, 9].
Specific mechanisms to improve Ca2+ handling and heart contractility by inhibition of late Na+ current
Diastolic Ca2+ accumulation
Previous studies in animal models of HF showed that partial blockade of either total Na+ current by saxitoxin or INaL by RAN improved contractility of myocytes from failing hearts [6, 13]. The results of the present study support the following specific mechanisms of late Na+ current contribution to abnormal Ca2+ handling and impaired contraction in chronic HF. Increased INaL in HF alters AP and simultaneously provides a substantial systolic late Na+ influx (during AP plateau). These two effects combined likely cause or modulate Ca2+ influx via the reverse mode Na+/Ca2+ exchange. This additional INaL-dependent systolic Ca2+ influx contributes to major abnormalities of cell Ca2+ handling, including DCa accumulation (compare upper panels in Figs. 1b and 3b), Ca2+ alternans (Fig. 3a, upper panel), and SCaR (Fig. 4). While the DCa accumulation and Ca2+ alternans can directly impair contractions of individual myocytes, increased rate of SCaRs can further worsen the diastolic cardiac muscle function. Indeed, heterogeneity of diastolic [Ca2+]i among cells within myocardial tissue caused by asynchronous SCaRs leads to heterogeneous myofilament activation, the summation of which produces a Ca2+-dependent component to diastolic tone [43].
The idea that INaL and its related Na+ influx cause a dynamic cell Ca2+ accumulation in HF is supported by the following results. We showed that partial blockade of INaL by RAN or TTX greatly improves Ca2+ handling in failing myocytes specifically; associated with inhibition of INaL, the DCa accumulation decreased (Fig. 3), alternans disappeared (Fig. 3a), and the probability of SCaRs decreased substantially (~threefold) (Fig. 4). This idea is also supported by recent studies in ventricular muscle strips isolated from end-stage failing human hearts [28] and in myocytes from dog failing hearts [13], which demonstrated that inhibition of INaL by RAN significantly reduces frequency-dependent increase in diastolic tension (i.e., diastolic dysfunction) by approximately 30% (in line with our results). Finally, inhibition of INaL and INaL-related Na+ influx in silico (both an AP clamp and a complete model of E-C coupling) substantially and selectively suppresses DCa accumulation of the myocytes of failing hearts (Figs. 7, 9).
Dynamic Ca2+ cycling abnormalities in failing heart; alternans and spontaneous Ca2+ releases
At a low stimulation rate of 0.25 Hz we also observed an abnormal tonic or dome component of CaT (compare upper panels in Figs. 1a, 2a, b) (first reported in heart ventricular muscle strips from patients with HF [44]). In addition we found some Ca2+ oscillations at low pacing rates (Fig. 2a upper panel) and beat-to-beat variations of Ca2+ transients at higher rates of stimulation (Fig. 3a, upper panel) that may be a sign of alternans of intracellular Ca2+ cycling [45]. TTX (middle panels in Fig. 2b) or RAN (middle panels in Fig. 2a) reversed these abnormalities.
The threshold for SCaR is lower in HF because of reduced SERCA function and increased SR Ca2+ leak [20, 21] in line with observation of SCaRs in our recordings in failing (Fig. 4) but not in normal myocytes (Fig. 1b). Diastolic SCaRs can initiate DADs via activation of the forward mode NCX inward current, and/or nonselective channels [18, 25]. Previous studies demonstrated that when SCaRs within a cardiomyocyte are sufficiently synchronized (e.g., multifocal Ca2+ waves), the resultant depolarization summates and can be sufficient to trigger a spontaneous AP [43]. The amplitude of SCaRs (F/F0 ~1.55) is close to that of CaT during 1.5 Hz pacing (Fig. 4a), which could be pro-arrhythmic, especially in the context of up-regulated NCX in HF [16–18]. Indeed, it has been documented that the threshold amplitude of SCaRs that is sufficient to evoke DADs is almost twofold lower for HF myocytes (~280 nM) compared to that in normal heart (~512 nM) [42]. This threshold is close to the amplitude of SCaRs reported here (~250 nM) (Fig. 4a). Increased Ca2+ entry is an established mechanism for SCaR from the SR [46]. Thus, the DCa accumulation represents a reasonable mechanism for SCaR after the pulse train in our chronic HF model. The fact that TTX and/or RAN inhibited SCaRs without significantly affecting SR load and NCX function (Fig. 5) supports this mechanism (Fig. 4b, c). On the other hand, small (albeit insignificant) decrease in SR Ca2+ load in the presence of RAN (Fig. 5b) does not completely rule out some involvement of SR load in SCaR occurrence, given the non-linear behavior of the SR (i.e., a relatively small change in load triggers the release) [47]. Also elucidation of possible contributions of changes in AP shape and duration in the improvement of Ca2+ handling, caused by the INaL reduction shown both experimentally and in silico (Figs. 7, 9), merits consideration in future studies.
Predictions in silico
Our model simulations provide quantitative evidence that INaL accounts for a major portion of diastolic cell Ca2+ accumulation in myocytes (stimulated at rate of 1.5 Hz) measured experimentally. A simplified numerical modeling of INaL during AP clamp shows that RAN (10 μM) almost completely (by 77%) inhibits late Na+ influx (Fig. 8d). At the same time we also show that Na+ influx via INaL can be indeed substantial. Taking into account that NCX operates in the reverse mode during AP in HF [24, 48] and NCX function is enhanced in HF [16–18], we reasoned that a large portion of Na+ influx is likely to be the cause of an increase in the dynamic exchanged by the reverse mode NCX for Ca2+. It is known, for example, that the reverse mode NCX-mediated Ca2+ influx is indeed substantial and may even result in the direct activation of contraction in HF [48]. We estimated that late Na+ influx is sufficient to cause a 72 nM increase in cytosolic [Ca2+] via this mechanism. We also tested the effect of INaL inhibition in in silico simulations of the E-C coupling process including all substantial mechanisms involved in the regulation of the Ca2+ and Na+ homeostasis in HF (Fig. 9). Our model simulations closely reproduce our experimental results and confirm the importance of INaL for both the AP duration and the DCa accumulation in failing myocardium.
Physiological significance, clinical relevance
In a recent clinical trial RAN significantly reduced arrhythmias in patients with non ST-segment elevation acute coronary syndrome [49] pointing to a potential clinical relevance of INaL. The beneficial effect of RAN in these patients can be thus explained, in part, by two mechanisms: (1) improvement of repolarization shown in previous studies [13, 33] and in this study (see Fig. 6) and (2) Ca2+ handling associated with a decreased probability of spontaneous releases (shown in this study, Fig. 4) and, hence, DADs and their triggered APs (see previous section). Partial inhibition of INaL by RAN, TTX, or saxitoxin greatly improves repolarization and decreases intrinsic beat-to-beat AP duration variability of failing cardiomyocytes [7, 9, 13] (Fig. 6). The cellular mechanisms of RAN effects proposed in this study may explain, at least in part, beneficial RAN effects previously reported in whole-animal studies by our group and others. In dogs and rabbits RAN reduced infarct size and Ca2+ overload in response to a regional ischemia-reperfusion [50, 51]. In dogs with coronary microembolization-induced HF, acute intravenous administration of RAN improved LV systolic function and LV mechanical efficiency without increasing myocardial oxygen consumption and without any increase in heart rate or reduction of systemic blood pressure [52].
INaL as a novel therapeutic target
The emerging paradigm with regard to Na+ channels in HF is that INaT is decreased [8, 14, 15] but simultaneously INaL is increased [5]. Blockers of INaT are pro-arrhythmic in HF because they will further slow conduction, thus worsening impulse conduction [53] and thereby facilitating development of re-entry. Hence, transient and late Na+ currents must be treated differently. The new type of “smart” drugs should preferentially block INaL over INaT [5, 54, 55]. The potential benefits (preventing Ca2+ overload and arrhythmias) of the preferential INaL blockade can be also expected in hypoxia and ischemia, in which INaL increase and Na+-induced Ca2+ overload are major features. In ischemia, accumulation of toxic metabolite lysophosphatidylcholine and reactive oxygen species dramatically increases INaL [56–58]. Indeed Na+ channels are critically involved in this process because their blockers or activators reduce or increase Ca2+ overload in these pathological conditions, respectively [59, 60].
In conclusion, we provide evidence that INaL and its systolic Na+ influx contribute to the dynamic DCa accumulation and spontaneous release in ventricular myocytes from dog model of chronic HF. Therefore, the results of the present study support the idea that selective blockade of this Na+ current represents a plausible strategy to treat Ca2+-related diastolic dysfunction and arrhythmia in HF.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was supported by grants from the NIH (HL074328, A. Undrovinas), American Heart Association (0350472Z, A. Undrovinas), Gilead Palo Alto Inc. (A. Undrovinas), and by the Intramural Research Program of the NIH, National Institute on Aging (V. A. Maltsev).
Conflict of interest statement
Dr. A. Undrovinas reports research support from Gilead Palo Alto, Inc., and Dr. L. Belardinelli is an employee of Gilead Palo Alto Inc. All other authors report no conflicts.
Footnotes
N. A. Undrovinas and V. A. Maltsev contributed equally to this work.
References
- 1.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 2.Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, et al. Relationship between Na+–Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 3.Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.CIR.0000023042.50192.F4. [DOI] [PubMed] [Google Scholar]
- 4.Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–299. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe? Prog Biophys Mol Biol. 2008;96:421–451. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 7.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S, Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Mol Life Sci. 1998;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 12.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 17.Flesch M, Schwinger RH, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, et al. Evidence for functional relevance of an enhanced expression of the Na(+)-Ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- 18.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 19.Brillantes AM, Allen P, Takahashi T, Izumo S, Marks AR. Differences in cardiac calcium release channel (ryanodine receptor) expression in myocardium from patients with end-stage heart failure caused by ischemic versus dilated cardiomyopathy. Circ Res. 1992;71:18–26. doi: 10.1161/01.res.71.1.18. [DOI] [PubMed] [Google Scholar]
- 20.Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- 21.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 23.Piacentino V, 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 24.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–995. doi: 10.1016/S0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]
- 25.Clusin WT. Calcium and cardiac arrhythmias: DADs, EADs, and alternans. Crit Rev Clin Lab Sci. 2003;40:337–375. doi: 10.1080/713609356. [DOI] [PubMed] [Google Scholar]
- 26.Gwathmey JK, Slawsky MT, Briggs GM, Morgan JP. Role of intracellular sodium in the regulation of intracellular calcium and contractility. Effects of DPI 201-106 on excitation-contraction coupling in human ventricular myocardium. J Clin Invest. 1988;82:1592–1605. doi: 10.1172/JCI113771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraser H, Belardinelli L, Wang L, Light PE, McVeigh JJ, Clanachan AS. Ranolazine decreases diastolic calcium accumulation caused by ATX-II or ischemia in rat hearts. J Mol Cell Cardiol. 2006;41:1031–1038. doi: 10.1016/j.yjmcc.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;14:14. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Lindegger N, Hagen BM, Marks AR, Lederer WJ, Kass RS. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol. 2009;47:326–334. doi: 10.1016/j.yjmcc.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabbah HN, Goldberg AD, Schoels W, Kono T, Webb C, Brachmann J, et al. Spontaneous and inducible ventricular arrhythmias in a canine model of chronic heart failure: relation to haemodynamics and sympathoadrenergic activation. Eur Heart J. 1992;13:1562–1572. doi: 10.1093/oxfordjournals.eurheartj.a060102. [DOI] [PubMed] [Google Scholar]
- 31.Rao K, Fisher ML, Robinson S, Shorofsky S, Gottlieb SS. Effect of chronic changes in heart rate on congestive heart failure. J Card Fail. 2007;13:269–274. doi: 10.1016/j.cardfail.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Logeart D, Gueffet JP, Rouzet F, Pousset F, Chavelas C, Solal AC, et al. Heart rate per se impacts cardiac function in patients with systolic heart failure and pacing: a pilot study. Eur J Heart Fail. 2009;11:53–57. doi: 10.1093/eurjhf/hfn016. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- 35.Nagatomo T, January CT, Makielski JC. Preferential block of late sodium current in the LQT3 DeltaKPQ mutant by the class I(C) antiarrhythmic flecainide. Mol Pharmacol. 2000;57:101–107. [PubMed] [Google Scholar]
- 36.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, et al. Multiple mechanisms of Na+ channel–linked long-QT syndrome. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 37.Winslow RL, Rice J, Jafri S, Marban E, O’Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84:571–586. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- 38.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 39.Kaab S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, et al. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 40.Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, et al. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543–550. doi: 10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- 41.Wigle ED (1995) The failing heart. In: Diastolic dysfunction: pathology and treatment options. Lippincott-Raven, Philadelphia, pp 79–94
- 42.Pogwizd SM, Bers DM. Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci. 2002;976:454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- 43.Lakatta EG. Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc Res. 1992;26:193–214. doi: 10.1093/cvr/26.3.193. [DOI] [PubMed] [Google Scholar]
- 44.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 45.Wilson LD, Wan X, Rosenbaum DS. Cellular alternans: a mechanism linking calcium cycling proteins to cardiac arrhythmogenesis. Ann N Y Acad Sci. 2006;1080:216–234. doi: 10.1196/annals.1380.018. [DOI] [PubMed] [Google Scholar]
- 46.Johnson N, Danilo P, Jr, Wit AL, Rosen MR. Characteristics of initiation and termination of catecholamine-induced triggered activity in atrial fibers of the coronary sinus. Circulation. 1986;74:1168–1179. doi: 10.1161/01.cir.74.5.1168. [DOI] [PubMed] [Google Scholar]
- 47.Diaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weisser-Thomas J, Piacentino V, 3rd, Gaughan JP, Margulies K, Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–985. doi: 10.1016/S0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- 49.Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 50.Black SC, Gralinski MR, McCormack JG, Driscoll EM, Lucchesi BR. Effect of ranolazine on infarct size in a canine model of regional myocardial ischemia/reperfusion. J Cardiovasc Pharmacol. 1994;24:921–928. doi: 10.1097/00005344-199424060-00009. [DOI] [PubMed] [Google Scholar]
- 51.Gralinski MR, Black SC, Kilgore KS, Chou AY, McCormack JG, Lucchesi BR. Cardioprotective effects of ranolazine (RS-43285) in the isolated perfused rabbit heart. Cardiovasc Res. 1994;28:1231–1237. doi: 10.1093/cvr/28.8.1231. [DOI] [PubMed] [Google Scholar]
- 52.Chandler MP, Stanley WC, Morita H, Suzuki G, Roth BA, Blackburn B, et al. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ Res. 2002;91:278–280. doi: 10.1161/01.RES.0000031151.21145.59. [DOI] [PubMed] [Google Scholar]
- 53.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 54.Undrovinas AI, Maltsev VA, Higgins RSD, Silverman N, Goldstein S, Sabbah HN. Amiodarone blocks the late sodium current in isolated ventricular myocytes of explanted failed human hearts. J Am Coll Cardiol. 2000;35:97A. doi: 10.1016/S0735-1097(00)80002-9. [DOI] [Google Scholar]
- 55.Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–359. doi: 10.2174/187152508785909447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- 57.Beresewicz A, Horackova M. Alterations in electrical and contractile behavior of isolated cardiomyocytes by hydrogen peroxide: possible ionic mechanisms. J Mol Cell Cardiol. 1991;23:899–918. doi: 10.1016/0022-2828(91)90133-7. [DOI] [PubMed] [Google Scholar]
- 58.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 59.Haigney MC, Lakatta EG, Stern MD, Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. [DOI] [PubMed] [Google Scholar]
- 60.Ver Donck L, Borgers M. Myocardial protection by R 56865: a new principle based on prevention of ion channel pathology. Am J Physiol. 1991;261:H1828–H1835. doi: 10.1152/ajpheart.1991.261.6.H1828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.