

Abstract
We identified a family in Mali with two sisters affected by spastic paraplegia. In addition to spasticity and weakness of the lower limbs, the patients had marked atrophy of the distal upper extremities. Homozygosity mapping using single nucleotide polymorphism arrays showed that the sisters shared a region of extended homozygosity at chromosome 19p13.11-q12 that was not shared by controls. These findings indicate a clinically and genetically distinct form of hereditary spastic paraplegia with amyotrophy, designated SPG43.
Keywords: hereditary spastic paraplegia, amyotrophy, autosomal recessive, chromosome 19, SPG43
Introduction
The hereditary spastic paraplegias (HSPs) include over 40 disorders that are classified by phenotype (pure or complicated) and mode of transmission (autosomal dominant, autosomal recessive, or X-linked). Pure (uncomplicated) HSPs present with progressive lower limb spasticity and weakness, and the complicated forms also exhibit a variety of other findings such as dementia, thinning of the corpus callosum, ataxia, and distal amyotrophy,. Although the age of onset and disease severity vary, most patients share key features of spastic gait, lower extremity hypertonicity, pyramidal weakness, hyperreflexia, and Babinski's sign [1, 2].
We studied a family in an isolated region (Kemacina) of Mali, West Africa in which two affected sisters had complicated HSP with distal amyotrophy. The population in Mali is comprised of eight major ethnic groups, many of which intermarry for cultural reasons. The unaffected parents were from neighboring villages and are of the same ethnic origin (Bambara). We performed homozygosity mapping to test the hypothesis that this disease is caused by a recessive founder mutation and to localize the disease gene.
Materials and Methods
We obtained approval from the National Institute of Neurological Disorders and Stroke (NINDS) and Johns Hopkins University School of Medicine Institutional Review Boards and the University of Bamako Ethics Committee for this study.
Clinical Examination
The subjects who agreed to participate signed informed consent forms or provided a fingerprint to indicate consent. The patients were then evaluated by American and Malian neurologists and an American genetic counselor to obtain an extensive neurological exam and family history. (Figure 1).
Figure 1. Hereditary spastic paraparesis family pedigree.
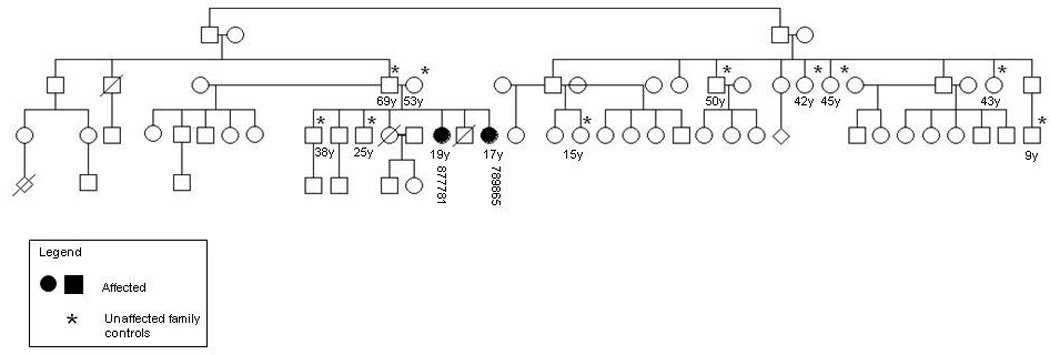
Pedigree showing the two affected sisters (877781, 789865) and 10 family controls (asterisk).
Diagnostic Testing
A blood specimen (4 ml) was obtained from the older affected sister. DNA was sent for genetic testing for Troyer syndrome (SPG20) to the Laboratory of Human Genetics at the University of Regensberg, and serum for serological testing for human T-cell lymphotropic virus (HTLV-1)-associated myelopathy was sent to the Neuroimmunology Branch at the NINDS. Genomic DNA was isolated from blood using PUREGENE DNA Isolation Kit (Gentra Systems) according to the manufacturer’s instructions. Serum was separated from whole blood within 1–3 hours of the blood draw by centrifugation (4000 rpm, 5°C, 10 min).
SNP Microarray Genotyping
SNP array genotyping was performed to identify shared homozygous regions greater than 500Kb in affected members based on apparent recessive transmission. Six family members, including the affected sisters, consented to provide saliva samples for DNA isolation. Later, five extended family members and 43 unrelated people of the same ethnic group consented to serve as control subjects. Saliva samples (4 ml) were collected from these participants directly into an OrageneTM (Genotek) container and extracted via the Oragene TM protocol. The two affected sisters and controls were genotyped using Illumina Infinium HumanHap550Kv3 Genotyping BeadChips (Illumina). The chips were scanned and data were loaded into the genotyping program BeadStudio (Illumina, CA), where calling rate and genotypes were calculated. Further sample error checking and quality assurance testing such as examining genotyping success rates, Log R ratio, B allele frequencies, and contamination, were performed by the Computational Biology Core at the National Institute of Aging (NIA).
Analysis of regions of homozygosity was performed using an in-house open source software program acting in concert with the GERON genotyping database (http://neurogenetics.nia.nih.gov/). Single nucleotide polymorphisms (SNPs) represented on the chips were analyzed by Log R ratio and B allele frequencies, and extended regions of homozygosity were identified. Common tracks of homozygosity among affected individuals, but not shared with controls, were identified as likely harboring the disease gene.
Candidate Gene Selection and Mutation Screening
Of the 140 known genes in the common region of homozygosity, candidates were assessed for functions related to genes implicated in other HSPs. Of these, RAB3A, εCOP, and FKBP8, were screened for mutations in both affected family members and unaffected family members with primers flanking the exons. Polymerase chain reaction (PCR) was performed under standard conditions (primer sequences and annealing temperatures available upon request). PCR products were then sequenced with BigDye Terminator (v3.1, Applied Biosystems). Reverse transcriptase (RT) PCR was performed from lymphoblastoid cell lines to rule out splicing abnormalities in the three candidate genes.
Results
Clinical Report
The family is of Bambara ethnicity with two affected sisters, examined at 17 and 19 years of age. The older sister (Patient 1) developed difficulty walking at age seven, with gradually progressive spastic paraparesis (Table 1). She remained ambulatory but complained of falling occasionally. Distal upper extremity muscle atrophy, particularly of the intrinsic hand muscles, and flexion contractures in the fingers were noted. She had decreased fine motor skills in her hands. The deltoids, biceps, and left triceps had normal strength bilaterally, however the right triceps, and both wrist extensors and flexors showed weakness against resistance (4/5). The intrinsic hand muscles were extremely weak bilaterally (0–1/5). There was weakness of hip flexion, slightly worse on the left than right, graded 4/5. Hamstrings and quadriceps muscles were also 4/5. Contractures of the knees and ankles were present, with limited ankle mobility. Reflexes were as follows (bilaterally, unless otherwise noted): biceps 3+, triceps 3+, knees 2–3+, ankles 2+ (left) and 0 (right). Plantar responses were extensor, and there was marked bilateral spasticity in the legs. The younger sister (Patient 2) was similarly, but more mildly, affected. Brain imaging was not feasible due to the family’s remote location.
Table 1.
Summary of neurological findings.
| Patient 1 | Patient 2 | |
|---|---|---|
| Age of onset | 7 years | 12 years |
| Age at evaluation | 19 years | 17 years |
| Cognition, vision | normal | normal |
| Bulbar function | mild dysarthria | normal |
| Muscle bulk | distal atrophy, especially of intrinsic hand muscles |
distal atrophy |
| Upper Extremities | decreased fine movement, flexion contractures of fingers |
difficulty with fine movement |
| Muscle tone | normal | normal |
| Muscle power | weak distally | weak intrinsic hand muscles |
| Reflexes | brisk | normal |
| Sensation | normal | normal |
| Lower extremities | pes cavus, contractures of knees and ankles |
increased |
| Muscle tone | severe spasticity | moderately decreased |
| Muscle power | moderately decreased | brisk |
| Reflexes | absent (ankle)-brisk (knees) | mild decrease in vibration and |
| Sensation | mild decrease in vibration | pinprick |
| Plantar responses | extensor | extensor |
| Cerebellar findings | normal | normal |
| Gait | spastic | mildly spastic |
Diagnostic Testing
The proband was negative on genetic testing for Troyer syndrome and serological testing for HTLV-1.
SNP Microarray Genotyping
Homozygosity mapping with genome-wide single nucleotide polymorphism arrays showed that the two affected siblings shared only one large (>500Kb) region of homozygosity bordering the centromere of chromosome 19 (19p13.11-q12), defined by SNP markers rs36692 and rs12460915. This 17.7 Mb region was not homozygous in 10 unaffected family members or 43 ethnically matched controls (Figure 2). Although other structural variations such as duplications and deletions were identified in the family, none were shared by the affected sisters alone. We excluded the other recessive SPG genes which can cause a similar phenotype (SPG5, 7, 11, 15, 20, 21, and 39) and ALS2 by identifying SNPs within and closely flanking these genes that do not segregate with the disease in the family we report. Lastly, SNPs across the SPG45 locus did not show blocks of homozygosity in the affecteds.
Figure 2. Homozygosity mapping results.

Large homozygous region shared in two affected family members only on chromosome 19. The three panels show results for patients 789865 (older affected sister) and 877781 (younger affected sister) and a 38 year old unaffected brother. The upper horizontal blue band in each panel represents heterozygous calls of two-allele SNPs across chromosome 19 (B allele frequency). Red arrows indicate the region of extended homozygosity (no heterozygous calls) present only in the affected sisters. The lower horizontal blue band in each panel represents DNA copy number (Log R ratio), which was normal.
Candidate Gene Selection and Mutation Screening
Based on their functions of exocytosis, intracellular trafficking and axonal growth, key candidate genes included RAB3A, εCOP, and FKBP8. RAB3A encodes a GTP-binding protein involved in exocytosis of synaptic vesicles [3] εCOP encodes the epsilon subunit of a heptameric coatomer complex, COPI, which mediates retrograde transport of certain transmembrane proteins (dilysine-tagged) from the Golgi apparatus to the endoplasmic reticulum [4]. The protein product of FKBP8 (FK506-binding protein 8) is an immunophilin highly expressed in neurons, particularly those associated with hippocampal formation as well as neocortex, striatum and cerebellum. FKBP8 may protect neurons from apoptosis and assists in the transport of antiapoptotic proteins to the outer mitochondrial membrane [5]. FKBP8 was shown to possibly interact with protrudin, a protein involved in axon growth and mutated in an autosomal dominant HSP, SPG33. Furthermore, protrudin interacts with spastin, another protein involved in axon elongation and branching, which is mutated in the most common autosomal dominant HSP, SGP4 [6]. However, Martignoni et al. recently challenged the claim of protrudin’s role in SGP33 and its interaction with spastin [7]. These genes were sequenced, but no variants were noted that change the encoded amino acid sequence. RT PCR showed normal, previously reported splice variants in εCOP in lymphoblastoid cDNA, though this analysis does not exclude abnormal tissue-specific splicing.
Discussion
We identified a putative recessive locus associated with a complicated form of HSP on chromosome 19, adding a novel locus to this family of disorders. The study included only two affected patients; however, the power of homozygosity mapping by genome-wide SNP array serves to confirm this locus, as in other studies of neurological disorders [8–13].
Other forms of HSP with distal amyotrophy have been identified, including Silver syndrome (SPG17), Troyer syndrome (SPG20), and neuropathy target esterase-related motor neuron disorder (NTE-MND; SPG39). However, the proband was negative upon genetic testing for Troyer syndrome, which additionally presents with short stature, hyperextensible joints, cognitive difficulties, and spastic dysarthria [14]. Furthermore, Troyer syndrome and autosomal dominant Silver syndrome map to other genomic loci: 13q12.3 and 11q13, respectively [15, 16] NTE-MND is another progressive, autosomal recessive HSP with distal upper and lower extremity wasting and maps to chromosome 19p13, but the NTE gene falls 10 Mb outside the region of homozygosity in the family we studied [17]. Juvenile amyotrophic lateral sclerosis 2 (ALSJ; ALS2), which presents with hyperreflexia, fasciculations, spasticity, and amyotrophy in some cases, maps to 2q33 [18]. Although we excluded other recessive genes for SPG with similar presentation (SPG5, 7, 11, 15, 20, 21, and 39) as well as the locus for SPG45 and the gene for ALS2, it remains possible, although less likely, that the disease was caused by a compound heterozygous or dominant mutation or has a non-genetic etiology. Hopefully this report will serve to identify other cases with a similar clinical presentation in order to investigate this locus further with additional families. The clinical and genomic findings in this family differ from related forms of HSP with amyotrophy and juvenile ALS, indicating a distinct addition to the family of HSPs.
Our study of individuals with HSP in Mali revealed an extended region of homozygosity shared between two affected sisters. By sequence analysis, we evaluated the coding regions of three candidate genes within this interval involved in cell trafficking and axon growth, RAB3A, εCOP, and FKBP8. No causative mutation was found, and further research is needed to identify the genetic mutation responsible. Such future studies will ultimately promote earlier diagnosis and intervention, improve disease management, and perhaps inspire new ideas for therapy based on the disease gene's function.
Acknowledgements
We thank the patients and staff of the Neurology Department of Point G Hospital for their warmth, participation, and flexibility. The Intramural Research Programs of the NINDS, NINR, and NIA provided funding for this study.
References
- 1.Hanein S, Martin E, Boukhris A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dürr A. Genetic testing for the spastic paraplegias: Drowning by numbers. Neurology. 2008;71:236–238. doi: 10.1212/01.wnl.0000320131.36091.a5. [DOI] [PubMed] [Google Scholar]
- 3.Geppert M, Goda Y, Stevens CF, Sudhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- 4.Harter C, Wieland FT. A single binding site for dilysine retrieval motifs and p23 within the gamma subunit of coatomer. Proc Natl Acad Sci U S A. 1998;95:11649–11654. doi: 10.1073/pnas.95.20.11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen JV, Mitchelmore C, Pedersen KM, et al. Fkbp8: Novel isoforms, genomic organization, and characterization of a forebrain promoter in transgenic mice. Genomics. 2004;83:181–192. doi: 10.1016/j.ygeno.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Shirane M, Nakayama KI. Protrudin induces neurite formation by directional membrane trafficking. Science. 2006;314:818–821. doi: 10.1126/science.1134027. [DOI] [PubMed] [Google Scholar]
- 7.Martignoni M, Riano E, Rugarli EI. The role of ZFYVE27/protrudin in hereditary spastic paraplegia. Am J Hum Genet. 2008;83:127–128. doi: 10.1016/j.ajhg.2008.05.014. author reply 128-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lander ES, Botstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 9.Gibbs JR, Singleton A. Application of genome-wide single nucleotide polymorphism typing: Simple association and beyond. PLoS Genet. 2006;2:e150. doi: 10.1371/journal.pgen.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garshasbi M, Motazacker MM, Kahrizi K, et al. SNP array-based homozygosity mapping reveals MCPH1 deletion in family with autosomal recessive mental retardation and mild microcephaly. Hum Genet. 2006;118:708–715. doi: 10.1007/s00439-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 11.Morrow EM, Yoo SY, Flavell SW, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camargos S, Scholz S, Simon-Sanchez J, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol 2008. 2008;7:207–215. doi: 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- 13.Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakowska JC, Wang H, Xin B, et al. Lack of spartin protein in Troyer syndrome: A loss-of-function disease mechanism? Arch Neurol. 2008;65:520–524. doi: 10.1001/archneur.65.4.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel H, Cross H, Proukakis C, et al. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 16.Windpassinger C, Wagner K, Petek E, Fischer R, Auer-Grumbach M. Refinement of the 'Silver syndrome locus' on chromosome 11q12–q14 in four families and exclusion of eight candidate genes. Hum Genet. 2003;114:99–109. doi: 10.1007/s00439-003-1021-6. [DOI] [PubMed] [Google Scholar]
- 17.Rainier S, Bui M, Mark E, et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hadano S, Nichol K, Brinkman RR, et al. A yeast artificial chromosome-based physical map of the juvenile amyotrophic lateral sclerosis (ALS2) critical region on human chromosome 2q33–q34. Genomics. 1999;55:106–112. doi: 10.1006/geno.1998.5637. [DOI] [PubMed] [Google Scholar]