

Abstract
Transforming growth factor β (TGF-β) plays an important role in tumor initiation and progression, functioning as both a suppressor and a promoter. The mechanisms underlying this dual role of TGF-β remain unclear. TGF-β exerts systemic immune suppression and inhibits host immunosurveillance. Neutralizing TGF-β enhances CD8+ T-cell- and NK-cell-mediated anti-tumor immune responses. It also increases neutrophil-attracting chemokines resulting in recruitment and activation of neutrophils with an antitumor phenotype. In addition to its systemic effects, TGF-β regulates infiltration of inflammatory/immune cells and cancer-associated fibroblasts in the tumor microenvironment causing direct changes in tumor cells. Understanding TGF-β regulation at the interface of tumor and host immunity should provide insights into developing effective TGF-β antagonists and biomarkers for patient selection and efficacy of TGF-β antagonist treatment.
TGF-β signaling, a tumor suppressor and a tumor promoter
Transforming growth factor β (TGF-β) signaling plays a very important role in tumor initiation and progression [1,2]. There are three TGF-β ligands, TGF-β1, TGF-β2 and TGF-β3. TGF-β1 is the most commonly upregulated in tumor cells [3]. The TGF-β ligands signal through type I and type II TGF-β receptors (TβRI and TβRII, respectively). Upon binding of TGF-β to TβRII, TβRI is recruited, transphosphorylated and activated to phosphorylate the downstream mediators, SMAD2 and SMAD3. Phosphorylated SMAD2 and SMAD3 combine with SMAD4 to enter the nucleus to modulate gene transcription. SMAD7 is a negative regulator of the SMAD signaling pathway [4,5] Related to TGFβ–SMAD signaling, bone morphology protein (BMP) signals through SMAD1, SMAD5 or SMAD8 (termed BR-SMADs). Once phosphorylated, BR-SMADs form complexes with SMAD4 and activate or repress targeted gene transcription important for tissue and organ development. In addition to SMAD-mediated signaling, TGF-β activates many non-canonical signaling pathways such as PI3-kinase, p38 kinase and small GTPase pathways (RhoA, PKN, and Rock) (Figure 1).
Figure 1.
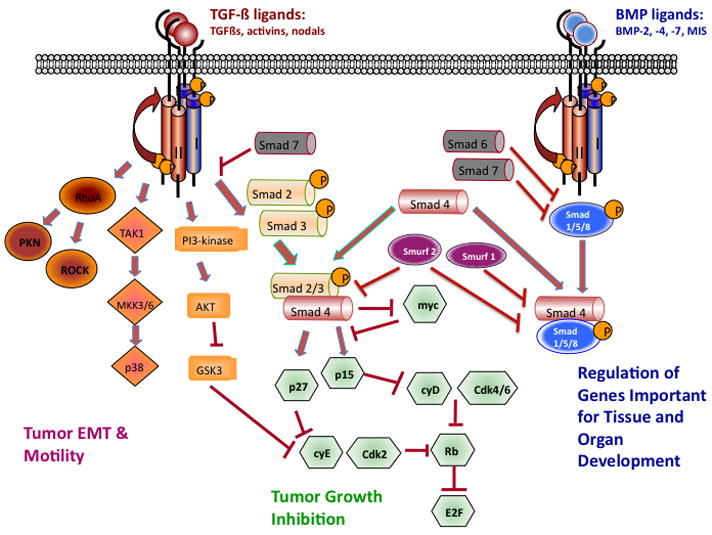
The TGF-β ligands signal through the type I and type II TGF-β receptors. Canonical signaling proceeds with phosphorylation of SMAD2 and SMAD3, which then combine with SMAD4 to enter the nucleus and mediate growth inhibition. TGF-β binding to its receptors activates many non-canonical signaling pathways, including small GTPases (RhoA, PKN, and Rock), p38 kinase and PI3 kinase pathways. These pathways are important in regulating tumor-cell migration and metastasis. In addition, bone morphogenetic proteins (BMPs) signal through SMAD1, SMAD5 or SMAD8, which form complexes with SMAD4, which activate or repress targeted gene transcription that is important for tissue and organ development. Smad 6 and 7 are negative mediators in the TGF-β signaling pathway.
TGF-β is an important tumor suppressor. Mutations of the genes encoding TβRI and TβRII or decreased expression and phosphorylation of other components of this pathway have been reported in human cancers [6]. Mutations in the type II TGF-β receptor gene, TGFBR2, are particularly frequent in tumors with microsatellite instability [7]. Lack of or downregulation of TGF-β receptors or SMADs is often associated with a worse prognosis [8]. In a number of mouse models, genetic deletion or downregulation of TGF-β signaling often results in a more malignant tumor phenotype, including breast, intestine, pancreatic, colon, head-and-neck squamous cell carcinoma [9]. Deletion of BR-SMADs 1, 5 and 8 from somatic cells of ovaries and testes leads to malignant transformation [10]. Abrogation of TGF-β signaling specifically in stratified epithelia causes spontaneous squamous cell carcinomas in the anogenital region through destabilization of epithelium homeostasis [11]. TGF-β suppresses tumor initiation and early development through the inhibition of cell cycle progression, induction of apoptosis, and suppression of growth factor, cytokine and chemokine expression.
However, TGF-β is a well-known tumor promoter too. TGF-β is often produced in large quantities by many tumor types and is known to be pro-oncogenic. Mutation of TGFBR2 points to a favorable outcome after adjuvant chemotherapy in colon cancer with high microsatellite instability [12]. High TGF-β–SMAD activity is present in aggressive, highly proliferative gliomas and confers poor prognosis in patients [13]. In mouse models, enhancement of TGF-β signaling by expression of a constitutively active TGFβ1 or TβRI in mammary epithelial cells increases pulmonary metastases whereas systemic inhibition of TGF-β signaling suppresses pulmonary metastases [14]. TGF-β signaling is also important in the ‘vicious cycle’ of osteolytic bone metastases [15,16]. The mechanisms of TGF-β tumor promotion include dysregulation of cyclin-dependent kinase inhibitors, alteration in cytoskeletal architecture, increases in proteases and extracellular matrix formation, decreased immune surveillance and increased angiogenesis [2].
Challenges in understanding the dual role of TGF-β
The mechanisms underlying the dual role of TGF-β described above are very complex and intricate [2,17,18]. Tumor progression from premalignant to metastatic disease is generally accompanied by decreased or altered TGF-β responsiveness and increased expression or activation of the TGF-β ligand. When combined with genetic or epigenetic perturbations in tumor cells, along with alteration in the tumor microenvironment, the spectrum of biological responses to TGF-β is altered (Figure 2). In the past, most work to dissect the underlying mechanisms was largely focused on differential regulation of signaling pathways by tumor autonomous TGF-β signaling. It was thought that changes in signal intensity and connectivity of SMAD-dependent and SMAD-independent pathways might explain the complex transition of TGF-β from a tumor suppressor to a tumor promoter. In early tumor development, SMAD-dependent pathways mediate the growth inhibition; in later tumor progression, the SMAD-independent pathways probably contribute to the tumor-promoting effect. However, recent data also demonstrate that SMAD-dependent pathways are involved in the tumor-promoting activities of TGF-β. For example, SMAD signaling is responsible for lung metastasis through induction of angiopoietin-like 4 (ANGPTL4) by TGF-β [19]. High TGF-β–SMAD activity is present in aggressive, highly proliferative gliomas and confers poor prognosis in patients with gliomas [13]. Interestingly, in concert with oncogenic Ras and mutant p53, SMADs are associated with a mutant-p53/p63 protein complex, which inhibits p63 suppressor functions. Thus mutant p53 promotes SMAD-mediated tumor metastasis [20]. In addition, SMAD4 activity is tightly controlled through Ecto (Ectodermin/Tif1γ), a monoubiquitinating factor and FAM (Fat Facets in Mouse), a deubiquitinase. Ecto induces monoubiquitination in Lys519 in SMAD4. This modification inhibits SMAD4 by impeding association with phospho-SMAD2. FAM opposes the activity of Ecto through deubiquitination of SMAD4 [21]. Another non-canonical mechanism involves the phosphorylation of SMAD1 and SMAD5 through kinase activity of ALK5 (activin receptor-like kinase 5) that mediates the pro-tumor migratory activity switch in breast cancer [22]. The literature in the field is very diverse, but is primarily based on autonomous tumor cell signaling. Recent progress in elucidating the effects of TGF-β on host immune cells and inflammatory cell infiltration in the tumor microenvironment might bring a new perspective to the field, which is discussed below.
Figure 2.
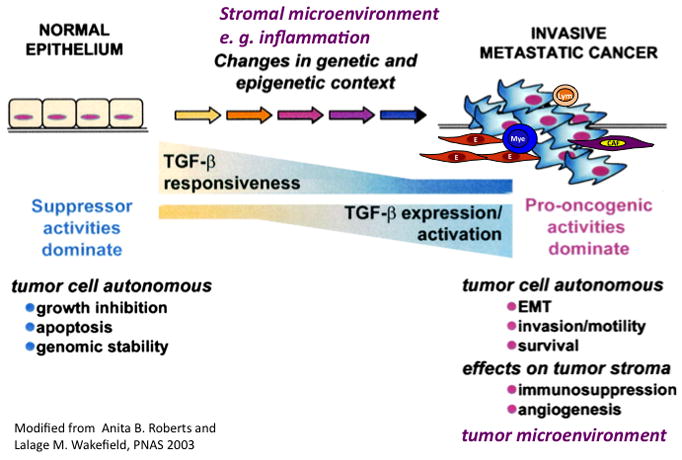
TGF-β switches from tumor suppressor in the premalignant stages of tumorigenesis to tumor promotion in later stages of the disease leading to metastasis. Progression to metastatic disease is generally accompanied by decreased or altered TGF-β responsiveness and increased expression or activation of the TGF-β ligand. When combined with genetic or epigenetic perturbations in tumor cells, along with alteration in the tumor microenvironment, the spectrum of biological responses to TGF-β are altered. EMT: epithelial mesenchymal transition (Figure modified from Ref. 32)
Effect of TGF-β on systemic immune surveillance of tumor host
TGF-β controls immune responses and maintains immune homeostasis through its impact on proliferation, differentiation and survival of multiple immune cell lineages [23–25]. TGF-β has an adverse effect on anti-tumor immunity and significantly inhibits host tumor immune surveillance [23,26]. The effect of TGF-β on T cells, was clearly demonstrated by genetic deletion or attenuation of TGF-β signaling. Transgenic mice with a dominant-negative Tgfbr2 under a T-cell-specific promoter showed spontaneous T-cell differentiation and autoimmune disease [24]. Interestingly, this T-cell-specific blockade of TGF-β signaling allows the generation of tumor-specific cytotoxic T lymphocytes (CTLs) that are capable of eradicating tumors in mice challenged with EL-4 thymoma or B16-F10 melanoma tumor cells [27]. TGF-β markedly and directly suppresses the ‘cytotoxic program’ of CTLs, through transcriptional repression of genes encoding multiple key proteins, such as perforin, granzymes and cytotoxins [28,29]. TGF-β regulates both the clonal expansion of CD8+ T cells and CD8+ T-cell cytotoxicity in vivo. Inhibition of T-cell proliferation and repression of the cytotoxic gene program in T cells are two distinct effects, which, together, would ultimately favor tumor progression [28].
TGF-β also has a significant impact on CD4+ T-cell differentiation and function. TGF-β induces Foxp3 and generates induced regulatory T cells (Tregs) [29–32]. In addition, TGF-β, together with IL-6, induces Th-17 cells [25,32,33]. Th17 is a newly defined Th-cell population that expresses IL-17 and regulates leukocyte recruitment and activation [34]. TGF-β was found to mediate the lineage-specific differentiation of these Th17 cells [35,36]. TGF-β, coordinating with IL-21, induces CD4+CD25+ Tregs that counterbalance the effect of IL-6 that promotes proinflammatory IL-17-producing T cells [37]. Studies suggest that TGF-β signaling in T cells is pro-tumorigenic; when deleted or inhibited by neutralizing antibodies, TGF-β enhances T-cell differentiation and function [30,38]. However, contradictory results were observed with selective deletion of SMAD4-dependent TGF-β signaling in T cells, which leads to spontaneous epithelial cancers throughout the gastrointestinal tract in mice (whereas epithelial-specific deletion of the SMAD4 gene does not). These SMAD4−/− T cells produce abundant Th2-type cytokines, which promote plasma cell and stromal expansion [39]. These results show that SMAD4 signaling in T cells is required for suppression of gastrointestinal cancer. It is unclear what underlies the differences between these studies. Mechanisms under the former observations include TGF-β inhibition of IL-2 production thus suppressing T-cell proliferation. This involves the anti-mitogenic effect of TGF-β. Mechanisms responsible for the latter observation include the significant role of TGF-β in maintaining intestinal homeostasis, preventing cancer by control over the intestinal lymphocytes and their response to signals within the mucosal environment.
TGF-β also inhibits NK-cell proliferation and function, which is in part modulated by CD4+CD25+ regulatory T cells that are known to produce high levels of TGF-β [31]. TGF-β inhibits NKp30 and NKG2D receptor expression, which suppresses NK-cell cytolytic activity [40,41]. Blockade of TGF-β signaling in NK cells caused the accumulation of NK cells that produce IFN-γ [42]. Neutralization of TGF-β not only prevented NKG2D downregulation but also restored NK cell anti-tumor reactivity [43]. In human glioma patients, TGF-β decreases the expression of the activating immunoreceptor NKG2D in CD8+ T cells and NK cells and represses the expression of the NKG2D ligand MICA [44].
B cells were shown to have high-affinity TGF-β receptors and to secrete TGF-β. Exogenous administration of TGF-β suppressed B-cell proliferation and Ig secretion [45,46]. In mice with B-cell-specific deletion of TβRII, there was significant B-cell expansion [46], which indicates that TGF-β suppresses B-cell proliferation. In addition, these mice showed complete absence of IgA in serum, suggesting an important role of TGF-β in IgA class switching in vivo [46]. In EL-4 leukemia and D5 melanoma mouse models, when spleen cells from WT C57BL/6 and B-cell knockout mice were cultured with irradiated tumor cells in vitro, IFN-gamma production from CD8 T cells and natural killer cells was markedly decreased in WT compared with B-cell KO (BKO) cultures. This is due to IL-10 produced by B cells that diminishes anti tumor CTLs [47]. These results and others suggest the possible existence of regulatory B cells (Breg) [48,49]. The immune suppressive function of these B cells appears to be directly mediated by TGF-β (and/or IL-10) [48]. Recently, it was found that B-lymphocytes are required for establishing chronic inflammatory states that promote de novo carcinogenesis in a skin carcinogenesis model of K14-HPV16 [50]. B-cell functions in anti-tumor immunity are not well understood; studies investigating how TGF-β regulates B-cell function are needed.
In addition to lymphoid cells, TGF-β also has a profound impact on the myeloid lineage. There are three major myeloid cell types, Gr-1+CD11b+ immature myeloid cells, tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs). While TAMs and TANs are well differentiated and mature, the Gr-1+CD11b+ cells are a heterogeneous set of immature myeloid cells. It remains to be elucidated how these three populations of cells are interrelated in terms of phenotype and function under tumor conditions and in the tumor microenvironment.
Gr-1+CD11b+ immature myeloid cells are also called myeloid derived suppressor cells (MDSCs). They are overproduced in tumor hosts including cancer patients with a variety of tumors. These cells have a profound immune suppressive effect. Gr-1+CD11b+ cells inhibit the function of NK, B and T cells through the production of arginase and reactive oxygen species. They also inhibit functional maturation of dendritic cells, and promote type II macrophage development. Gr-1+CD11b+ cells represent one of the mechanisms by which tumors escape from immune system control and compromise the efficacy of cancer immunotherapy [51]. There are two major subpopulations of Gr-1+CD11b+ cells: mononuclear cells (precursor for macrophages), and low-density polymorphonuclear cells (immature neutrophils). Both populations suppress antigen-specific T-cell responses, but by distinct effector molecules and signaling pathways [52]. Tumor-infiltrating Gr-1+CD11b+ myeloid cells also have non-immune suppressive effects that profoundly affect tumor progression and metastasis. For example, these cells are recruited into the tumor microenvironment in response to chemokine cues and produce high levels of MMPs and TGF-β [53]. They contribute to tumor angiogenesis and vasculogenesis [54]. TGF-β produced by these Gr-1+CD11b+ cells is a significant component of the tumor-promoting effect of TGF-β signaling. It affects both the tumor microenvironment and the host immune system. However, it is unclear whether systemic immune suppression and direct participation in tumor microenvironment and progression are two different properties or different manifestations of the same process.
TAMs are identified as Mac-1 (CD11b+) and/or F4/80+. They have been shown to promote tumor progression and metastasis [55–59]. Mechanisms include increased CSF-1 production and enhanced EGF signaling in cancer cells [55]. Similarities between TAM and Gr-1+CD11b+ immature myeloid cells were noticed from profiling work [60]; however, differences between the two populations were also evident. For example, myeloid suppressor cells produce high levels of TGF-β1, whereas TGF-β1 expression in TAMs was restricted to unstimulated TAMs and was not further increased by M2-biasing cytokines [60].
Information related to TANs is very incomplete. One recent study showed that TGF-β within the tumor microenvironment induces TAN with a pro-tumor phenotype [61]. TGF-β blockade increases neutrophil-attracting chemokines and results in the recruitment and activation of TANs with an anti-tumor phenotype. These TANs were identified as CD11b+Ly6G+ that have hypersegmented nuclei, are more cytotoxic to tumor cells, and express higher levels of proinflammatory cytokines. Depletion of these neutrophils following TGF-β blockade significantly blunts anti-tumor effects of treatment and reduces CD8 T-cell activation [61]. These findings suggest that TGF-β regulate N1–N2 polarization of neutrophils. This N1–N2 polarization of neutrophils might mirror the M1–M2 polarization of macrophages that are defined by interferon-γ and IL-4 production as Th1 and Th2 cells, respectively [62].
The effects of TGF-β on immune cells are summarized in Figure 3. Although the effects are distinct for each immune cell population, they are also interrelated. For example, TGF-β inhibition of CD8 CTLs is largely due to TGF-β produced by Gr-1+CD11b+ myeloid cells, which is further regulated by IL-13 produced in natural killer T (NKT) cells, a new natural killer cell type discovered in recent years [63].
Figure 3.
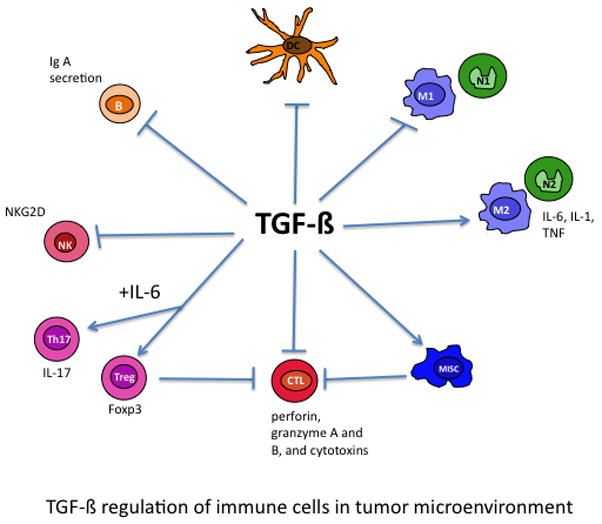
TGF-β affects multiple components of the immune system. TGF-β inhibits the function of natural killer (NK) and CD8+ CTL (cytotoxic T lymphocytes), blocking the ‘cytotoxic program’ key proteins such as perforin, granzymes and cytotoxins. TGF-β induces Treg and Th17 cell differentiation and inhibits B-cell proliferation and IgA secretion. In addition, TGF-β inhibits dendritic function, block type I macrophage and neutrophil development but promotes type II macrophages and neutrophils, and mediates the immune suppression function of MISCs.
TGF-β regulation of the tumor microenvironment
The tumor microenvironment is filled with various inflammatory cells, including myeloid cell subpopulations, innate and adaptive immune cells NK(T) cells, T cells and B cells (Figure 4a). In addition, there is an abundance of cancer-associated fibroblasts (CAFs) and endothelial progenitor cells (EPCs) [64] (Figure 4a). This very dynamic tumor microenvironment probably serves as a selective pressure for tumor cell variants through genomic instability, genomic heterogeneity and epigenetic alteration [65].
Figure 4.
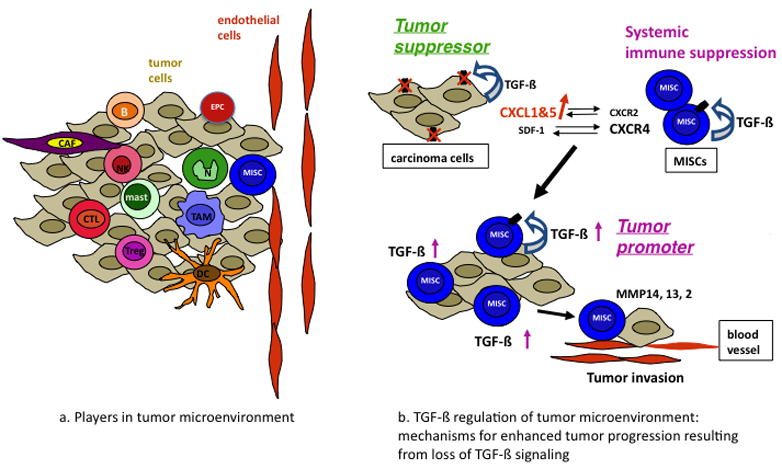
TGF-β regulation of tumor microenvironment. (a) Cellular players in tumor microenvironment. (b) The mechanisms by which TGFβ signaling switches from a tumor suppressor to a tumor promoter are shown. TGFβ signals through the type II receptor mediate growth inhibition of carcinoma cells. When TβRII is deleted or downregulated, the result is increased chemokine/chemokine-receptor signaling, such as CXCL1–CXCL5/CXCR2 and SDF-1–CXCR4. Host-derived immature myeloid Gr-1+CD11b+ cells are recruited into the tumor microenvironment through these chemokine mechanisms. These Gr-1+CD11b+ cells express high level of MMPs and TGFβ1, which promote tumor invasion and immune suppression. The effect of Gr-1+CD11b+ cells on the tumor microenvironment and host immune surveillance constitute a tumor-promoting mechanism of TGFβ signaling.
The evidence supporting TGF-β as a potent regulator of the tumor microenvironment comes from studies with specific deletion of Tgfbr2 in a variety of epithelial cells, including mammary, pancreatic, intestinal, colon, and head-and-neck squamous cell carcinoma [9]. Deletion of Tgfbr2 in these epithelial cells results in increased tumor progression and metastasis. Deletion of Tgfbr2 in mammary epithelial cells results in increased chemokine/chemokine receptor signaling CXCL1/CXCL5–CXCR2 and SDF-1–CXCR4 [53,66], which leads to increased Gr-1+CD11b+ cell recruitment into the tumor microenvironment. These Gr-1+CD11b+ cells produce high levels of MMPs and TGF-β (Figure 4b) [53]. The increased infiltration of myeloid cells into the invasive front of tumors, and their production of MMPs and TGFβ1 are found to be one of the major contributors to enhanced tumor invasion and metastasis due to deletion of TGF-β signaling in epithelial cells [9,53,56]. This complete abrogation of TGF-β signaling correlated with reduced relapse-free survival in four human breast cancer data sets particularly in patients with estrogen-receptor-positive breast cancer [67]. Deletion of SMAD4 in a colon cancer model resulted in an increased recruitment of CCR1+ myeloid cells (CD34+) that promote tumor invasion [68]. Indeed, inflammatory cells (CD45 and BM8 positive cells) have been observed in head-and-neck tumors that lack TGF-β signaling [69]. In TGFβ1-deficient mice, inflammation causes precancerous lesions to progress to colon cancer [70]. However, in contrast to the above observations, over-expression of TGFβ1 in head and neck epithelia results in inflammation, angiogenesis and epithelial hyperproliferation [71]. It is unclear what underlies these different observations and whether different chemokine/chemokine-receptor mechanisms are involved for deletion of TGF-β signaling versus increased TGF-β signaling.
In addition to epithelial cells, TGF-β signaling in stromal cells also exerts significant effects on tumor development and growth. This is one of the most recent developments in the field. These stromal cells include fibroblasts and T cells but perhaps many more types discovered in the future. Loss of Tgfbr2 in a subset of fibroblasts (FSP promoter driving cre expression) leads to intraepithelial neoplasia in the prostate and invasive squamous cell carcinoma in the forestomach involving upregulation of TGFα, MSP- and HGF-mediated signaling networks [72]. Which is often accompanied by an increased abundance of stromal cells and inflammatory cell infiltration. Deletion of TGF-β signaling in T lymphocytes results in the development of carcinomas throughout the gastrointestinal tract, which is also accompanied by increased expression of inflammatory gene (such as IL5, IL6 and IL13) and inflammatory cell infiltration [39]. These results demonstrate that TGF-β, the tumor suppressor, also functions as an inflammation suppressor. When it is deleted in the epithelial or stromal compartment it causes an increased inflammatory reaction that promotes tumor development and progression.
One of the most important mechanisms underlying TGF-β regulation of inflammation that orchestrates the tumor microenvironment is through NF-κB signaling, the master regulator of the inflammation. For example, in the gut, TGFβ1 negatively regulates NF-κB activation through SMAD7 [73]. Mice deficient in SMAD3 develop colon cancer because of increased inflammation caused by Helicobacter infection [74]. In TGFβ1-deficient mice, inflammation causes precancerous lesions to progress to colon cancer [70]. Furthermore, TGF-β crosstalks with inflammatory pathways through the modulation of IL-1 [75].
TGF-β regulates the production of chemokines–chemokine receptors that are important in inflammatory cell recruitment. These include stromal-derived factor 1 (SDF-1 or CXCL12), which signals through CXCR4 that is highly expressed on putative stem and progenitor cells [64,76]. TGF-β also suppresses expression of CXCL1 and CXCL5. Deleted or diminished TGF-β signaling in tumor epithelial cells significantly increases the expression of CXCL1 and CXCL5 [53,66]. In the distant premetastatic lung of tumor-bearing mice, TGF-β regulates the production of the chemoattractants S100A8 and S100A9. These chemoattractants attract Mac1+ myeloid cells that activate mitogen-activated protein kinase p38 in tumor cells [77]. TGF-β also induces tumor cells to produce angiopoietin-like 4 that disrupts vascular endothelial cell–cell junctions, increases the permeability of lung capillaries and facilitates the transendothelial passage of tumor cells [19].
Antitumor activity of TGF-β inhibition is dependent on the host immune and inflammatory response: implications for therapy
TGF-β has been identified as a therapeutic target because of its significant tumor- promoting roles. Approaches include a variety of neutralizing antibodies and small molecular inhibitors. However, the goal of these therapies is to abolish the tumor- promoting effect of TGF-β, while maintaining its tumor suppressive properties. Recent developments in the field point out that the efficacy of TGF-β antagonist therapy might not derive from a direct effect on tumor cells as was originally thought. Rather, several mechanisms are probably acting on the host compartment. First, blocking TGF-β might decrease the suppression of host immune surveillance. This effect of TGF-β antagonism is particularly important as it could be combined with anti-tumor immunotherapy. For example, the efficacy of immunotherapy is potentiated by systemic blockade of TGF-β signaling in mice bearing lung cancer (TC1) [78,79]. Second, TGF-β antagonism could abrogate the tumor-promoting effect of Gr-1+CD11b+ cells in the tumor microenvironment by neutralizing the high level of TGF-β1 that these cells produce [53]. Third, TGF-β antagonism would be very effective for blocking the ‘vicious cycle’ of bone metastasis, because TGF-β signaling is a key player in the differentiation of osteoclasts that destroy the bone [15]. Lastly, TGF-β antagonism might be effective for patients immediately after radiation or chemotherapy because there is a surge in TGF-β level after these treatments [80].
Indeed, TGF-β blockade significantly slows tumor progression through a synergistic combination of effects on both the tumor parenchyma and microenvironment. These effects include enhancement of the CD8+ T-cell-mediated anti-tumor immune response, increased infiltration of natural killer cells and T cells at the metastatic sites, and increased expression of an NKG2D ligand (Rae1gamma) and of a death receptor (TNFRSF1A) on tumor cells [81]. Treatment of mice with anti-TGF-β antibodies in vivo reduced the expression of IL-17, which suppresses tumor apoptosis [82]. Anti-TGF-β antibody treatment also reverses the suppressive function of Tregs [30]. In the E.G7 tumor model, the effect of TGF-β inhibition was clearly shown in draining lymph nodes, in which suppression of Treg proliferation and an increase in the number of tumor antigen-specific CD4+ or CD8+ cells producing IFN-gamma was found [83]. However, one should note that while TGF-β is known to play a crucial role in CD4+Foxp3+ Treg cell induction, there are few reports to show that the improved immune responses from TGF-β antagonism is mediated by CD4 Treg cells ([79] and personal communication with Dr Lalage Wakefield).
Gr-1+CD11b+ cells infiltrate into the tumor microenvironment in response to loss of TGF-β signaling in mammary epithelial cells, which indicates that autologous TGF-β signaling in epithelial cells acts as a tumor suppressor; deletion of TGF-β results in recruitment of Gr-1+CD11b+ cells. This recruitment leads to increased MMP and TGF-β production, which enhances tumor invasion and metastasis. The switch of TGF-β signaling from tumor suppressor to tumor promoter thus involves an additional component, which is the recruitment of Gr-1+CD11b+ cells in the tumor microenvironment. In addition, Gr-1+CD11b+ cells produce a high level of TGF-β and correlates with stage of tumor progression [84]. These data suggest that Gr-1+CD11b+ cells can be used as a biomarker for patient selection in ongoing phase I/II clinical trials of TGF-β therapy. This concept is supported by recent findings that the immune/inflammatory responses are reliable markers for predicting the clinical outcome in human hepatocellular carcinoma [85] and colorectal tumors [86]. These studies also point out a novel therapeutic strategy for advanced cancer – the prevention of the recruitment of MMP-expressing cells by chemokine receptor antagonists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6 (7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGFbeta in Cancer. Cell. 2008;134 (2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425 (6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 4.Nakao A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389 (6651):631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi H, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89 (7):1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 6.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11 (11):S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 7.Markowitz S, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268 (5215):1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 8.Xie W, et al. Alterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray study. Cancer Res. 2002;62 (2):497–505. [PubMed] [Google Scholar]
- 9.Yang L, Moses HL. Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008;68 (22):9107–9111. doi: 10.1158/0008-5472.CAN-08-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pangas SA, et al. Conditional deletion of Smad1 and Smad5 in somatic cells of male and female gonads leads to metastatic tumor development in mice. Mol Cell Biol. 2008;28 (1):248–257. doi: 10.1128/MCB.01404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guasch G, et al. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12 (4):313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344 (16):1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruna A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11 (2):147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 14.Arteaga CL. Inhibition of TGFbeta signaling in cancer therapy. Curr Opin Genet Dev. 2006;16 (1):30–37. doi: 10.1016/j.gde.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 15.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2 (8):584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 16.Guise TA, Chirgwin JM. Transforming growth factor-beta in osteolytic breast cancer bone metastases. Clin Orthop Relat Res. 2003;(415 Suppl):S32–38. doi: 10.1097/01.blo.0000093055.96273.69. [DOI] [PubMed] [Google Scholar]
- 17.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100 (15):8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bierie B, Moses HL. Gain or loss of TGFbeta signaling in mammary carcinoma cells can promote metastasis. Cell Cycle. 2009;8 (20):3319–3327. doi: 10.4161/cc.8.20.9727. [DOI] [PubMed] [Google Scholar]
- 19.Padua D, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133 (1):66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adorno M, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137 (1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 21.Dupont S, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136 (1):123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 22.Liu IM, et al. TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J. 2009;28 (2):88–98. doi: 10.1038/emboj.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li MO, et al. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 24.Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2 (1):46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 25.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134 (3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wahl SM, et al. TGF-beta: a mobile purveyor of immune privilege. Immunol Rev. 2006;213:213–227. doi: 10.1111/j.1600-065X.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- 27.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7 (10):1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 28.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8 (5):369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Trapani JA. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell. 2005;8 (5):349–350. doi: 10.1016/j.ccr.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura K, et al. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194 (5):629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghiringhelli F, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202 (8):1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30 (5):636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Bettelli E, et al. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8 (4):345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 34.Korn T, et al. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 35.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441 (7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 36.Veldhoen M, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24 (2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448 (7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen ML, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102 (2):419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim BG, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441 (7096):1015–1019. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 40.Castriconi R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A. 2003;100 (7):4120–4125. doi: 10.1073/pnas.0730640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JC, et al. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172 (12):7335–7340. doi: 10.4049/jimmunol.172.12.7335. [DOI] [PubMed] [Google Scholar]
- 42.Laouar Y, et al. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6 (6):600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 43.Kopp HG, et al. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009;69 (19):7775–7783. doi: 10.1158/0008-5472.CAN-09-2123. [DOI] [PubMed] [Google Scholar]
- 44.Friese MA, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64 (20):7596–7603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 45.Kehrl JH, et al. Transforming growth factor-beta suppresses human B lymphocyte Ig production by inhibiting synthesis and the switch from the membrane form to the secreted form of Ig mRNA. J Immunol. 1991;146 (11):4016–4023. [PubMed] [Google Scholar]
- 46.Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13 (4):443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 47.Inoue S, et al. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006;66 (15):7741–7747. doi: 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- 48.Mauri C, Ehrenstein MR. The ‘short’ history of regulatory B cells. Trends Immunol. 2008;29 (1):34–40. doi: 10.1016/j.it.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Yanaba K, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28 (5):639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 50.de Visser KE, et al. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7 (5):411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 51.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9 (3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T-cell suppressive activity. Blood. 2008 doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 53.Yang L, et al. Abrogation of TGFbeta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13 (1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6 (4):409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 55.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124 (2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 56.Mantovani A, et al. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25 (3):315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 57.Mantovani A, et al. Cancer-related inflammation. Nature. 2008;454 (7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 58.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9 (4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9 (4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biswas SK, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107 (5):2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 61.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16 (3):183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mantovani A. The yin-yang of tumor-associated neutrophils. Cancer Cell. 2009;16 (3):173–174. doi: 10.1016/j.ccr.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 63.Terabe M, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198 (11):1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13 (3):206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8 (3):180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 66.Bierie B, et al. Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res. 2008;68 (6):1809–1819. doi: 10.1158/0008-5472.CAN-07-5597. [DOI] [PubMed] [Google Scholar]
- 67.Bierie B, et al. Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest. 2009;119 (6):1571–1582. doi: 10.1172/JCI37480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kitamura T, et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat Genet. 2007;39 (4):467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 69.Lu SL, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20 (10):1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Engle SJ, et al. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002;62 (22):6362–6366. [PubMed] [Google Scholar]
- 71.Lu SL, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64 (13):4405–4410. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- 72.Bhowmick NA, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303 (5659):848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 73.Monteleone G, et al. A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem. 2004;279 (6):3925–3932. doi: 10.1074/jbc.M303654200. [DOI] [PubMed] [Google Scholar]
- 74.Maggio-Price L, et al. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66 (2):828–838. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu T, et al. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104 (11):4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431 (7007):405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 77.Hiratsuka S, et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8 (12):1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 78.Kim S, et al. Systemic blockade of transforming growth factor-beta signaling augments the efficacy of immunogene therapy. Cancer Res. 2008;68 (24):10247–10256. doi: 10.1158/0008-5472.CAN-08-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Terabe M, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res. 2009;15 (21):6560–6569. doi: 10.1158/1078-0432.CCR-09-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biswas S, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117 (5):1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nam JS, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68 (10):3835–3843. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nam JS, et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68 (10):3915–3923. doi: 10.1158/0008-5472.CAN-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fujita T, et al. Inhibition of transforming growth factor-beta-mediated immunosuppression in tumor-draining lymph nodes augments antitumor responses by various immunologic cell types. Cancer Res. 2009;69 (12):5142–5150. doi: 10.1158/0008-5472.CAN-08-2499. [DOI] [PubMed] [Google Scholar]
- 84.Almand B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6 (5):1755–1766. [PubMed] [Google Scholar]
- 85.Budhu A, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10 (2):99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 86.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313 (5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]