

Abstract
Neonatal intravenous injection of gammaretroviral vectors (γ-RVs) with an intact long terminal repeat (LTR) and an internal liver promoter can result in long-term expression in liver cells and correction of mucopolysaccharidosis. Some expression also occurs in blood cells and brain, which likely derives from the LTR, and may contribute to clinical efficacy. The goal of this project was to determine whether neonatal gene therapy with an LTR-intact γ-RV would induce tumors in mice. Fifty-one normal newborn C57BL/6 mice were injected intravenously at 1010 transducing units/kg with a γ-RV expressing canine β-glucuronidase (GUSB) cDNA. This resulted in transduction of 23 ± 9% of hepatocytes as determined by histochemical staining, and 0.24 ± 0.20 copy of γ-RV DNA per cell in liver as determined by real-time polymerase chain reaction. Serum GUSB activity was stable for 1.75 years after transduction at 705 ± 119 units/ml. Ninety-six percent of mice survived for the duration of evaluation, which was similar to the survival rate for 65 control mice that were not injected with γ-RV. One γ-RV-treated mouse (2%) developed a small (diameter, 2 mm) liver adenoma, which was similar to the frequency of liver adenomas (2%) or hepatocellular carcinoma (2%) in untreated mice. Although 22% of γ-RV-treated mice developed hematopoietic tumors, none contained high γ-RV DNA copy numbers, and the frequency was similar to that in the control group (22%). We conclude that neonatal intravenous injection of an LTR-intact γ-RV does not have a high risk of inducing cancer in mice.
Introduction
Intravenous injection of gammaretroviral vectors (γ-RVs) with an intact long terminal repeat (LTR) into newborn mice and dogs has successfully treated mucopolysaccharidosis I (MPS I) and MPS VII in mice and dogs (Ponder et al., 2002; Xu et al., 2002a, b; Liu et al., 2005; Sands and Davidson, 2006; Ponder and Haskins, 2007; Traas et al., 2007). This approach resulted in transduction of ~20% (Xu et al., 2002b) and ~2% (Xu et al., 2002a) of hepatocytes in mice and dogs, respectively, which expanded clonally as the animals underwent normal growth (Ponder, 1996). In addition, ~1% of blood cells appeared to express the transgene in both mice (Xu et al., 2002b; and L.X. and K.P.P., data not shown) and dogs (Wang et al., 2006), and there was substantial expression from the γ-RV in brain in dogs (Traas et al., 2007). Expression in brain could derive from migration of transduced hematopoietic cells into brain, or from transduction of brain cells at the time of intravenous injection of vector, and may be important for reducing lysosomal storage in neurons (Traas et al., 2007).
Although this gene therapy approach has been effective in treating MPS long-term, there are concerns about the risk of cancer or leukemia. Indeed, leukemia developed in 5 of 21 children with X-linked severe combined immunodeficiency (SCID; due to deficiency of the common γ chain of cytokine receptors) and who received hematopoietic stem cell (HSC)-directed gene therapy with an LTR-intact γ-RV (Nienhuis et al., 2006; Baum, 2007a,b; Cole, 2008). Insertional mutagenesis into the LMO2 locus was clearly a factor in some patients (Hacein-Bey-Abina et al., 2003), although growth-promoting activity of the therapeutic gene, selection for immortalizing mutations during in vitro culture, and extensive in vivo amplification of genetically modified cells may have contributed (Calmels et al., 2005; Du et al., 2005; Thrasher et al., 2006; Woods et al., 2006; Métais and Dunbar, 2008). In addition, a patient who received gene therapy for chronic granulomatous disease had clonal expansion of cells with insertions near immortalization genes without leukemic transformation (Ott et al., 2006), and a myeloid sarcoma developed in a rhesus macaque that received HSC-directed transfer of a γ-RV (Seggewiss et al., 2006). In contrast, leukemias and/or clonal expansion have not occurred after gene therapy for adenosine deaminase (ADA) deficiency (Aiuti et al., 2007; Bushman, 2007; Deichmann et al., 2007).
The risk of cancer after gene therapy to other cell types with genes that do not confer a selective advantage has not been as well defined. Liver tumors were reported in one study in 58% of mice that received fetal or neonatal transfer with an equine infectious anemia virus (EIAV)-based lentiviral vector, although tumors did not develop in animals that received other lentiviral vectors or γ-RV, suggesting that a specific element in the EIAV lentiviral vector functioned as an oncogene (Themis et al., 2005). The goal of this project was to assess the risk of cancer in mice after neonatal intravenous injection of an LTR-intact γ-RV that has been highly effective at treating models of MPS.
Materials and Methods
Reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Animals
Animal studies were approved by the Institutional Review Board of the Washington University School of Medicine (St. Louis, MO). Normal C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were bred in a barrier facility. Newborn mice were injected intravenously with 1010 transducing units (TU)/kg of the retroviral vector hAAT-cGUSB-WPRE (Xu et al., 2002a) 2 to 3 days after birth as described. Serum was tested periodically for β-glucuronidase (GUSB) activity as noted below. Liver biopsies were obtained from some animals at 4 months of age, and frozen sections were stained for GUSB activity. At the end point of ~1.75 years, serum was collected from the inferior vena cava. Liver and spleen were sliced into ~0.5-cm-thick pieces and evaluated for nodules, and enlarged lymph nodes or lung masses were collected. Normal pieces of liver, spleen, and lung were fixed in formalin. For abnormal-appearing regions that were ≤5 mm in diameter, normal tissue was removed and half was fixed in formalin; the other half was frozen for DNA analysis. Six-micrometer-thick sections of paraffin-embedded specimens were stained with hematoxylin and eosin and evaluated by a murine pathologist.
Serum GUSB activity
Serum was tested for GUSB activity by mixing 5 μl of serum with 95 μl of 2.5 mM 4-methylumbelliferyl-β-d-glucuronide in 0.1 M sodium acetate (pH 4.6) in a 96-well plate. The fluorescence was determined every 10 min for 2 hr with a Fluoroskan Ascent microplate fluorometer (Thermo Scientific, Waltham, MA) with excitation at 355 nm and emission at 460 nm. Standards of 4-methylumbelliferone in water were protected from light and stored at 4°C, and 5 μl of each was mixed with 95 μl of reaction buffer for the standard curve. One unit of enzyme produces 1 nmol of product in 1 hr at 37°C. The activity obtained with this assay was ~40% of the activity obtained in our previous protocol, which was performed in a test tube and involved termination of the reaction with a high-pH buffer (Wolfe and Sands, 1996).
GUSB staining of liver
Organs were embedded in optimal cutting temperature (O.C.T.) compound (Sakura Finetek USA, Torrance, CA) and 8-μm-thick sections were stained for GUSB activity with 0.25 mM naphthol AS-BI-glucuronide as described (Wolfe and Sands, 1996), except that the reactions were terminated after 1 hr.
Analysis of DNA copies
For real-time polymerase chain reaction (PCR) of DNA, a portion of organ that weighed ~300 mg was homogenized in guanidinium, and DNA was extracted. To remove low molecular weight nucleic acids, ~20 μg of DNA was bound to beads from a QIAEX II gel extraction kit (Qiagen, Valencia, CA) and eluted according to the manufacturer's instructions. DNA was used for real-time PCR with TaqMan technology (Applied Biosystems, Foster City, CA) and primers specific for the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) of the γ-RV, with normalization to the mouse β-actin sequence (Xu et al., 2002b). Standards were DNA from the murine MPS VII fibroblast cell line 3521, which was transduced with a single copy of hAAT-cGUSB-WPRE (Xu et al., 2002b); the DNA was diluted with DNA from liver of a nontransduced mouse.
Statistical evaluations
Averages ± the standard deviation (SD) were calculated for all values. The program SigmaStat 3.1 (Systat Software, San Jose, CA) was used to determine the statistical significance of differences between groups.
Results
Transduction of newborn mice with an LTR-intact γ-RV
We have previously demonstrated that neonatal injection of a γ-RV with an internal liver-specific human α1-antitrypsin (hAAT) promoter and an intact LTR resulted in stable expression from liver for up to 1.5 years in mice and up to 7 years in dogs. However, it is possible that this vector could induce cancer by activating an oncogene close to an integration site. We therefore initiated this study to assess the carcinogenicity of an LTR-intact γ-RV in mice after neonatal administration. Fifty-one normal C57BL/6 mice were injected intravenously with 1010 TU/kg of the γ-RV hAAT-cGUSB-WPRE 2–3 days after birth, and 65 uninjected mice of the same breeding colony were set aside as controls. C57BL/6 mice were chosen for analysis, as they have a detectable but relatively low risk of liver adenoma or carcinoma, 2.3 to 13.9% after ~2 years of observation (Frith et al., 1993; Volk et al., 1994; Ward et al., 2000), which might allow a single mutation that results in increased proliferative potential to have a carcinogenic effect. Normal mice were chosen for analysis, as the short life span (less than 1 year) of untreated MPS VII mice would make it impossible to compare the frequency of tumor development at late times in untreated mice with that in treated mice.
GUSB activity in serum can be used to assess the stability and the level of expression, as some of the enzyme that is produced by hepatocytes or other cells is secreted. All γ-RV-treated mice had high levels of serum GUSB activity at 1.5 months after transduction, as shown in Fig. 1A. Expression was stable for the duration of evaluation, and averaged 705 ± 119 [standard deviation (SD)] U/ml at the end point of 1.75 years, which was 28-fold the value of 26 ± 2 U/ml in normal mice (p < 0.001) (Table 1). Liver biopsy was performed on 5 mice whose serum GUSB activity was near the mean for the group at 4 months after transduction, and histochemical staining was used to determine the percentage of cells that expressed GUSB, as shown in Fig. 1 and summarized in Table 1. A short period (1 hr) of staining was used, which allowed the transduced hepatocytes with high GUSB activity to be identified over the endogenous GUSB activity in the nontransduced normal hepatocytes. This demonstrated that 23 ± 9% of hepatocytes expressed the transgene. No large clusters of GUSB-positive or GUSB-negative cells were observed in any of the livers that were evaluated, making it unlikely that there was extensive expansion of an individual transduced cell that did or did not express the transgene, respectively. In addition, DNA analysis was performed on livers collected from transduced mice at ~1.75 years after transduction. Real-time PCR demonstrated that the γ-RV DNA copy number was 0.24 ± 0.20 copy per cell in liver (n = 8; Table 1), while γ-RV DNA was undetectable in nontransduced controls (n = 2; p < 0.001). These data demonstrate that a relatively high transduction efficiency was achieved.
FIG. 1.
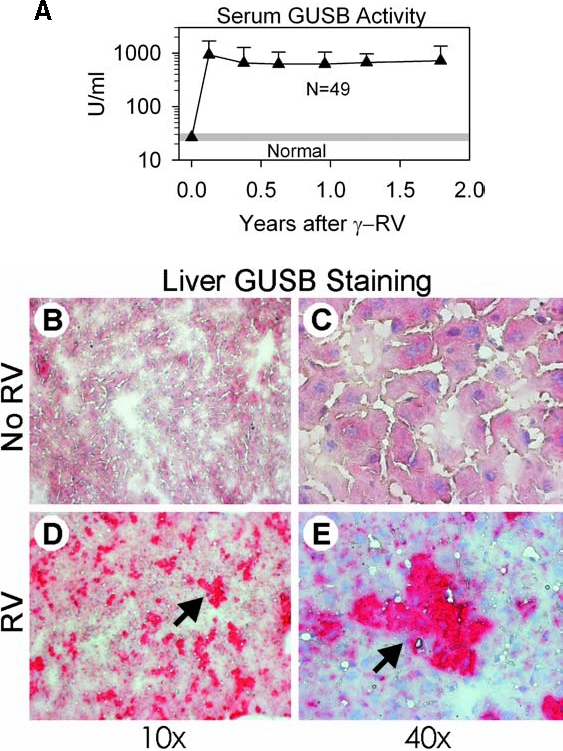
Serum GUSB activity and histochemical stain of liver for GUSB activity. Some normal C57BL/6 mice received neonatal intravenous injection of LTR-intact γ-RV, designated hAAT-cGUSB-WPRE, at 1010 TU/kg 2 or 3 days after birth. Other normal C57BL/6 mice were not treated. (A) Serum GUSB activity. Average serum GUSB activity ± SD was determined for all surviving mice at the indicated age after transduction. The average serum GUSB activity ± 2 SD in nontransduced normal mice (22 to 31 U/ml) is indicated by the shaded region. (B–E) Histochemical assay for liver GUSB activity. Liver biopsies were obtained 4 months after birth and stained for 1 hr for GUSB activity, which results in a red color in the cytoplasm. Nuclei were counterstained with hematoxylin, which results in a blue color. Samples from nontransduced normal C57BL/6 mice (B and C) have a low level of endogenous GUSB activity. Neonatal γ-RV-transduced normal mice (D and E) have several clusters of hepatocytes with high enzyme activity, as indicated by the arrows.
Table 1.
Neonatal Gene Therapy in Mice: Resultsa
| No gene transfer | Neonatal γ-RV | |
|---|---|---|
| Dose of γ-RV | None | 1010 TU/kg |
| Number of mice at start | 65 | 51 |
| Number of mice that survived 1.75 years (%) | 63 (97%) | 49 (96%) (NS) |
| Average age (months) at sacrifice | 21.0 ± 2.6 | 21.9 ± 1.6 (NS) |
| Average serum GUSB activityb (U/ml) | 26 ± 2 (n = 4) | 705 ± 119 (n = 49; p < 0.001) |
| Percent liver cells strongly positive for GUSBc | 0 ± 0 (n = 2) | 23 ± 9% (n = 5; p < 0.001) |
| γ-RV DNA copies/cell in liverd | 0 ± 0 (n = 2) | 0.24 ± 0.20 (n = 8; p < 0.001) |
| Frequency of all neoplasms | 26% | 24% (NS) |
| γ-RV DNA copies/cell in hematopoietic tumors | Not tested | 0.04 ± 0.05 (n = 8) |
Abbreviations: γ-RV, gammaretroviral vector; GUSB, β-glucuronidase; NS, not significant; TU, transducing units.
The dose of the LTR-intact γ-RV designated hAAT-cGUSB-WPRE that was given intravenously to newborn normal C57BL/6 mice in transducing units (TU) per kilogram is shown. Control mice did not receive γ-RV.
Average serum GUSB activity was determined at 1.75 years of age.
The percentage of hepatocytes with high GUSB activity was determined in liver biopsy samples obtained 4 months after transduction.
The number of γ-RV DNA copies per cell was determined in liver, spleen, or lymph node samples obtained 1.75 years after transduction. Statistical comparisons used the Student t test (age at sacrifice, serum GUSB activity, percent liver cells strongly positive for GUSB, and γ-RV DNA copies in liver) or the Fisher exact test (the percentage of mice that survived to 1.75 years and the frequency of tumor development).
Long-term survival of mice
Mice were evaluated approximately twice per month for their general health and for the presence of abdominal or other masses. Two animals in each group died before the end point of 1.75 years, for a mortality of 4 and 3% for the γ-RV-treated and the control group, respectively, as shown in Fig. 2A. Although the cause of death was unclear and pathology was not performed, as the bodies were in a poor condition when found in the cage, none had gross evidence of tumors.
FIG. 2.

Survival and tumor incidence in mice. (A) Survival curve. The survival of all mice from birth until the age of sacrifice at 1.75 years is shown for γ-RV-transduced mice and for nontransduced controls. There was no significant difference in the survival rate between the two groups. (B) Incidence of tumors. Autopsy was performed, and slides of organs were evaluated by a murine pathologist. The percentage of animals with hepatocellular carcinomas, liver adenomas, hematopoietic neoplasms (lymphoma or histiocytic sarcoma), or lung adenomas relative to the total number of mice in the group that were evaluated histochemically was determined. There were no significant differences in the incidence of tumors of specific types, using the Fisher exact test.
Frequency and type of tumors
The remaining animals were killed at ~1.75 years of age as detailed in Table 1, and the abdomens and chests were evaluated for overt abnormalities. Histopathological analysis was performed on liver, spleen, and lung from all animals, and on any abnormal-appearing regions. As shown in Fig. 2B, 1 of 49 (2%) of the γ-RV-treated mice that survived to the end point of 1.75 years had a small (2 mm) liver nodule, 1 untreated control of 63 that were evaluated had a liver nodule (2%), and 1 untreated control had a hepatocellular carcinoma (2%). Eleven γ-RV-treated mice (22%) had a hematopoietic tumor in the spleen and/or lymph nodes, the phenotype of which was classified as lymphoma (9 total or 18% of all mice) or histiocytic sarcoma (2 total or 4% of all mice). For the control group that did not receive γ-RV, 14 of 63 that were evaluated had hematopoietic tumors (22%), of which 10 were lymphomas (16%) and 4 were histiocytic sarcomas (6%). Two γ-RV-treated mice (4%) had small (<2 mm) lung nodules, and one normal control had a lung adenoma (2%). The incidence of tumors, according to the Fisher exact test, was not different between the groups for any tumor type.
Hematopoietic tumors from γ-RV-treated mice were evaluated to determine the γ-RV DNA copy number. Although livers of γ-RV-treated mice had an average of 0.24 ± 0.20 copy of γ-RV per cell (n = 8), the tumors had only 0.04 ± 0.05 copy of γ-RV per cell (n = 8). The presence of low numbers of γ-RV sequences in the tumors is likely due to contamination with normal transduced blood cells, as discussed in the next section.
Discussion
The goal of this study was to determine the risk of development of malignancy in mice after neonatal intravenous injection of an LTR-intact γ-RV with an internal liver-specific hAAT promoter. This vector has been extremely successful at preventing the clinical manifestations of MPS I and MPS VII in mice and dogs. The LTR may contribute to the therapeutic effect for MPS, as 1% of blood cells in mice (Xu et al., 2002b; and L.X. and K.P.P., unpublished data) and dogs (Wang et al., 2006) were strongly positive for GUSB activity on the basis of histochemical staining, and blood cells could migrate into other tissues and secrete enzyme locally. In addition, an undetermined cell type in brain expressed α-l-iduronidase RNA from an LTR-intact γ-RV in MPS I dogs (Traas et al., 2007). Expression in nonhepatic cells likely derives from the LTR and not the hAAT promoter, and thus elimination of the enhancer region of the LTR may reduce the clinical efficacy of this gene therapy approach if expression in these sites is important. In addition, other organs such as spleen, kidney, and thymus had detectable copies of γ-RV after neonatal administration of this vector in a previous study and could possibly develop tumors, although the levels of γ-RV were low in these other organs, at <14% of that found in liver (Xu et al., 2002b).
This study demonstrates that neonatal intravenous injection of 1010 TU/kg of this γ-RV, which was sufficient to result in expression in 23 ± 9% of hepatocytes and 0.24 ± 0.20 copy of γ-RV DNA per cell in liver, did not increase the risk of malignancy at 1.75 years over that observed in control mice of the same strain that did not receive the γ-RV. Although the hematopoietic tumors that developed in γ-RV-treated mice had 0.04 copy of γ-RV per cell, this was likely due to contamination of the tumor with normal hematopoietic cells, as this value was lower than the value of 0.10 copy per cell that we previously observed in peripheral blood and spleen after neonatal intravenous injection of a similar γ-RV into mice (Xu et al., 2004). If integration adjacent to an oncogene were driving the development of the tumor, the γ-RV DNA copy number should be >0.5 copy per cell, as histological evaluation of these tumors suggests that >50% of the cells were abnormal (data not shown). In addition, we have analyzed ~50 additional mice that received neonatal injection of various γ-RVs expressing other transgenes 0.5 to 2 years after transduction, and none developed liver tumors. We therefore conclude that the risk of insertional mutagenesis resulting in malignancy with this gene therapy approach is low in mice. In addition, we have not seen tumors or other adverse effects in any of four MPS VII dogs (Ponder et al., 2002) that have now been monitored for 7.5 years after neonatal gene therapy (M. Haskins and K.P.P., unpublished data), or in any of ~30 MPS I, hemophilia A, or hemophilia B dogs (Ponder, 2006; Traas et al., 2007) that were monitored for 0.5 to 3 years after transduction. Although the number of animals and the time of evaluation are not sufficient to rule out a small carcinogenic effect, these data suggest that this risk is low. The low incidence of tumors in mice that received a replication-incompetent γ-RV in this study is in contrast to the high frequency of leukemia or lymphoma in newborn mice that received a replication-competent gammaretrovirus (Ott et al., 1992), and in rhesus macaques that received a replication-competent gammaretrovirus during hematopoietic stem cell-directed gene therapy (Vanin et al., 1994). This study does not examine γ-RV integration sites in the liver, which are of interest and will be evaluated in future.
Despite these data suggesting that the risk of cancer induction is low with an LTR-intact γ-RV after neonatal intravenous injection, it has been shown that an LTR-intact γ-RV has increased oncogenic potential in HSCs as compared with a self-inactivating (SIN) vector that has a deletion in the 3′ LTR that is copied to the 5′ end after transduction (Modlich et al., 2006; Schambach et al., 2006; Zychlinski et al., 2008). It therefore remains possible that the LTR of the γ-RV could enhance expression of a nearby oncogene, and that this could serve as one step in the multistep process (Vogelstein and Kinzler, 1993) toward the development of malignancy. We are currently testing the efficacy of an SIN γ-RV for treating MPS I and MPS VII. If an SIN vector is as effective as an LTR-intact γ-RV, it would be preferred for translation into humans, as it should be safer.
Another issue concerns whether or not lentiviral vectors are safer than γ-RV because of reductions in their propensity to integrate close to promoters (Nienhuis et al., 2006; Beard et al., 2007a,b; Bushman, 2007; Deichmann et al., 2007; Schwarzwaelder et al., 2007). However, these differences are modest, and the biological significance is unclear, as enhancers by definition can activate promoters from a distance. Although lentiviral vectors had a reduced incidence of leukemia in one study as compared with γ-RV (Montini et al., 2006), differences in the strength of the promoter and the use of an intact LTR in the γ-RV complicate comparisons. There are potential advantages to using a γ-RV for in vivo delivery, as the requirement for division for transduction by γ-RV (Miller et al., 1990) but not lentiviral vectors (Naldini et al., 1996) should reduce promiscuous transduction of cells that will not contribute to a therapeutic effect, and eliminate the risk of transduction of the nonreplicating germ cells in newborns.
Implications for gene therapy
These data suggest that the risk of cancer induction with neonatal intravenous injection of an LTR-intact γ-RV expressing a transgene that does not confer a selective advantage is low in mice. Nevertheless, if an SIN vector were to be as effective as an LTR-intact vector, the SIN vector would be preferred because of its reduced risk of carcinogenesis in HSCs. However, if an LTR-intact vector proves to be more effective than an SIN vector in some sites, such as brain, the decision as to whether or not to go forward with an LTR-intact vector would be complicated, and would need to consider these data showing a low risk of cancer with this LTR-intact γ-RV, and the fact that alternative treatments for MPS have substantial toxicity. For example, HSC transplantation, an established treatment for MPS, has a 15% short-term mortality rate (Boelens et al., 2007). In addition, HSC transplantation has a 7% incidence of malignancy within 10 years that may be due to the mutagenic effect of radiation and DNA-damaging agents (Ferry et al., 2007). We will continue to evaluate large animals long-term for the development of malignancy after neonatal gene therapy with an LTR-intact vector and with the SIN vectors that we are currently developing. In addition, others are testing SIN lentiviral vectors for efficacy in MPS (Kobayashi et al., 2005; Di Domenico et al., 2006). The results of these studies should further define the efficacy and the risks of various vectors, and may lead to using the vector with the greatest benefit-to-risk ratio to treat patients with MPS.
Acknowledgments
The authors thank Suellen Greco (Comparative Medicine, Washington University School of Medicine) for assistance with analysis of pathology. This work was supported by the Ryan Foundation, the National MPS Society, and DK66448 from the National Institutes of Health. Histology was supported by P30 DK52574 and real-time PCR was supported by DK20579 awarded to Clay Semenkovich.
Author Disclosure Statement
No competing financial interests exist.
References
- Aiuti A. Cassani B. Andolfi G. Mirolo M. Biasco L. Recchia A. Urbinati F. Valacca C. Scaramuzza S. Aker M. Slavin S. Cazzola M. Sartori D. Ambrosi A. Di Serio C. Roncarolo M.G. Mavilio F. Bordignon C. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest. 2007;117:2233–2240. doi: 10.1172/JCI31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum C. What are the consequences of the fourth case? Mol. Ther. 2007a;15:1401–1402. doi: 10.1038/sj.mt.6300263. [DOI] [PubMed] [Google Scholar]
- Baum C. Insertional mutagenesis in gene therapy and stem cell biology. Curr. Opin. Hematol. 2007b;14:337–342. doi: 10.1097/MOH.0b013e3281900f01. [DOI] [PubMed] [Google Scholar]
- Beard B.C. Dickerson D. Beebe K. Gooch C. Fletcher J. Okbinoglu T. Miller D.G. Jacobs M.A. Kaul R. Kiem H.P. Trobridge G.D. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol. Ther. 2007a;15:1356–1365. doi: 10.1038/sj.mt.6300159. [DOI] [PubMed] [Google Scholar]
- Beard B.C. Keyser K.A. Trobridge G.D. Peterson L.J. Miller D.G. Jacobs M. Kaul R. Kiem H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007b;18:423–434. doi: 10.1089/hum.2007.011. [DOI] [PubMed] [Google Scholar]
- Boelens J.J. Wynn R.F. O'Meara A. Veys P. Bertrand Y. Souillet G. Wraith J.E. Fischer A. Cavazzana-Calvo M. Sykora K.W. Sedlacek P. Rovelli A. Uiterwaal C.S. Wulffraat N. Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: A risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40:225–233. doi: 10.1038/sj.bmt.1705718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman F.D. Retroviral integration and human gene therapy. J. Clin. Invest. 2007;117:2083–2086. doi: 10.1172/JCI32949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmels B. Ferguson C. Laukkanen M.O. Adler R. Faulhaber M. Kim H.J. Sellers S. Hematti P. Schmidt M. von Kalle C. Akagi K. Donahue R.E. Dunbar C.E. Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood. 2005;106:2530–2533. doi: 10.1182/blood-2005-03-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A. Child in gene therapy programme develops leukaemia. BMJ. 2008;336:13. doi: 10.1136/bmj.39436.582292.DB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deichmann A. Hacein-Bey-Abina S. Schmidt M. Garrigue A. Brugman M.H. Hu J. Glimm H. Gyapay G. Prum B. Fraser C.C. Fischer N. Schwarzwaelder K. Siegler M.L. de Ridder D. Pike-Overzet K. Howe S.J. Thrasher A.J. Wagemaker G. Abel U. Staal F.J. Delabesse E. Villeval J.L. Aronow B. Hue C. Prinz C. Wissler M. Klanke C. Weissenbach J. Alexander I. Fischer A. von Kalle C. Cavazzana-Calvo M. Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J. Clin. Invest. 2007;117:2225–2232. doi: 10.1172/JCI31659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico C. Di Napoli D. Gonzalez Y. Reyero E. Lombardo A. Naldini L. Di Natale P. Limited transgene immune response and long-term expression of human α-l-iduronidase in young adult mice with mucopolysaccharidosis type I by liver-directed gene therapy. Hum. Gene Ther. 2006;17:1112–1121. doi: 10.1089/hum.2006.17.1112. [DOI] [PubMed] [Google Scholar]
- Du Y. Jenkins N.A. Copeland N.G. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005;106:3932–3939. doi: 10.1182/blood-2005-03-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry C. Gemayel G. Rocha V. Labopin M. Esperou H. Robin M. de Latour R.P. Ribaud P. Devergie A. Leblanc T. Gluckman E. Baruchel A. Socié G. Long-term outcomes after allogeneic stem cell transplantation for children with hematological malignancies. Bone Marrow Transplant. 2007;40:219–224. doi: 10.1038/sj.bmt.1705710. [DOI] [PubMed] [Google Scholar]
- Frith C.H. Ward J.M. Chandra M. The morphology, immunohistochemistry, and incidence of hematopoietic neoplasms in mice and rats. Toxicol. Pathol. 1993;21:206–218. doi: 10.1177/019262339302100213. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. Von Kalle C. Schmidt M. McCormack M.P. Wulffraat N. Leboulch P. Lim A. Osborne C.S. Pawliuk R. Morillon E. Sorensen R. Forster A. Fraser P. Cohen J.I. de Saint Basile G. Alexander I. Wintergerst U. Frebourg T. Aurias A. Stoppa-Lyonnet D. Romana S. Radford-Weiss I. Gross F. Valensi F. Delabesse E. Macintyre E. Sigaux F. Soulier J. Leiva L.E. Wissler M. Prinz C. Rabbitts T.H. Le Deist F. Fischer A. Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Kobayashi H. Carbonaro D. Pepper K. Petersen D. Ge S. Jackson H. Shimada H. Moats R. Kohn D.B. Neonatal gene therapy of MPS I mice by intravenous injection of a lentiviral vector. Mol. Ther. 2005;11:776–789. doi: 10.1016/j.ymthe.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Liu Y. Xu L. Hennig A.K. Kovacs A. Fu A. Chung S. Lee D. Wang B. Herati R.S. Mosinger Ogilvie J. Cai S.R. Parker Ponder K. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol. Ther. 2005;11:35–47. doi: 10.1016/j.ymthe.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Métais J.Y. Dunbar C.E. The MDS1-EVI1 gene complex as a retrovirus integration site: Impact on behavior of hematopoietic cells and implications for gene therapy. Mol. Ther. 2008;16:439–449. doi: 10.1038/sj.mt.6300372. [DOI] [PubMed] [Google Scholar]
- Miller D.G. Adam M.A. Miller A.D. Gene transfer by retroviral vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U. Bohne J. Schmidt M. von Kalle C. Knoss S. Schambach A. Baum C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E. Cesana D. Schmidt M. Sanvito F. Ponzoni M. Bartholomae C. Sergi Sergi L. Benedicenti F. Ambrosi A. Di Serio C. Doglioni C. von Kalle C. Naldini L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- Naldini L. Blomer U. Galley P. Ory D. Mulligan R. Gage F.H. Verma I.M. Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nienhuis A.W. Dunbar C.E. Sorrentino B.P. Genotoxicity of retroviral integration in hematopoietic cells. Mol. Ther. 2006;13:1031–1049. doi: 10.1016/j.ymthe.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Ott D.E. Keller J. Sill K. Rein A. Phenotypes of murine leukemia virus-induced tumors: Influence of 3′ viral coding sequences. J. Virol. 1992;66:6107–6116. doi: 10.1128/jvi.66.10.6107-6116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M.G. Schmidt M. Schwarzwaelder K. Stein S. Siler U. Koehl U. Glimm H. Kuhlcke K. Schilz A. Kunkel H. Naundorf S. Brinkmann A. Deichmann A. Fischer M. Ball C. Pilz I. Dunbar C. Du Y. Jenkins N.A. Copeland N.G. Luthi U. Hassan M. Thrasher A.J. Hoelzer D. von Kalle C. Seger R. Grez M. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Ponder K.P. Analysis of liver development, regeneration, and carcinogenesis by genetic marking studies. FASEB J. 1996;10:673–682. doi: 10.1096/fasebj.10.7.8635684. [DOI] [PubMed] [Google Scholar]
- Ponder K.P. Gene therapy for hemophilia. Curr. Opin. Hematol. 2006;13:301–307. doi: 10.1097/01.moh.0000239700.94555.b1. [DOI] [PubMed] [Google Scholar]
- Ponder K.P. Haskins M.E. Gene therapy for mucopolysaccharidosis. Expert Opin. Biol. Ther. 2007;7:1333–1345. doi: 10.1517/14712598.7.9.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder K.P. Melniczek J.R. Xu L. Weil M.A. O'Malley T.M. O'Donnell P.A. Knox V.W. Aguirre G.D. Mazrier H. Ellinwood N.M. Sleeper M. Maguire A.M. Volk S.W. Mango R.L. Zweigle J. Wolfe J.H. Haskins M.E. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13102–13107. doi: 10.1073/pnas.192353499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands M.S. Davidson B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Schambach A. Mueller D. Galla M. Verstegen M.M. Wagemaker G. Loew R. Baum C. Bohne J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006;13:1524–1533. doi: 10.1038/sj.gt.3302807. [DOI] [PubMed] [Google Scholar]
- Schwarzwaelder K. Howe S.J. Schmidt M. Brugman M.H. Deichmann A. Glimm H. Schmidt S. Prinz C. Wissler M. King D.J. Zhang F. Parsley K.L. Gilmour K.C. Sinclair J. Bayford J. Peraj R. Pike-Overzet K. Staal F.J. de Ridder D. Kinnon C. Abel U. Wagemaker G. Gaspar H.B. Thrasher A.J. von Kalle C. Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J. Clin. Invest. 2007;117:2241–2249. doi: 10.1172/JCI31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seggewiss R. Pittaluga S. Adler R.L. Guenaga F.J. Ferguson C. Pilz I.H. Ryu B. Sorrentino B.P. Young W.S., 3rd Donahue R.E. von Kalle C. Nienhuis A.W. Dunbar C.E. Acute myeloid leukemia is associated with retroviral gene transfer to hematopoietic progenitor cells in a rhesus macaque. Blood. 2006;107:3865–3867. doi: 10.1182/blood-2005-10-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themis M. Waddington S.N. Schmidt M. von Kalle C. Wang Y. Al-Allaf F. Gregory L.G. Nivsarkar M. Themis M. Holder M.V. Buckley S.M. Dighe N. Ruthe A.T. Mistry A. Bigger B. Rahim A. Nguyen T.H. Trono D. Thrasher A.J. Coutelle C. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005;12:763–771. doi: 10.1016/j.ymthe.2005.07.358. [DOI] [PubMed] [Google Scholar]
- Thrasher A.J. Gaspar H.B. Baum C. Modlich U. Schambach A. Candotti F. Otsu M. Sorrentino B. Scobie L. Cameron E. Blyth K. Neil J. Abina S.H. Cavazzana-Calvo M. Fischer A. Gene therapy: X-SCID transgene leukaemogenicity. Nature. 2006;443:E5–E6. doi: 10.1038/nature05219. [DOI] [PubMed] [Google Scholar]
- Traas A.M. Wang P. Ma X. Tittiger M. Schaller L. O'Donnell P. Sleeper M.M. Vite C. Herati R. Aguirre G.D. Haskins M. Ponder K.P. Correction of clinical manifestations of canine mucopolysaccharidosis I with neonatal retroviral vector gene therapy. Mol. Ther. 2007;15:1423–1431. doi: 10.1038/sj.mt.6300201. [DOI] [PubMed] [Google Scholar]
- Vanin E.F. Kaloss M. Broscius C. Nienhuis A.W. Characterization of replication-competent retroviruses from nonhuman primates with virus-induced T-cell lymphomas and observations regarding the mechanism of oncogenesis. J. Virol. 1994;68:4241–4250. doi: 10.1128/jvi.68.7.4241-4250.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B. Kinzler K.W. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- Volk M.J. Pugh T.D. Kim M. Frith C.H. Daynes R.A. Ershler W.B. Weindruch R. Dietary restriction from middle age attenuates age-associated lymphoma development and interleukin 6 dysregulation in C57BL/6 mice. Cancer Res. 1994;54:3054–3061. [PubMed] [Google Scholar]
- Wang B. O'Malley T.M. Xu L. Vite C. Wang P. O'Donnell P.A. Ellinwood N.M. Haskins M.E. Ponder K.P. Expression in blood cells may contribute to biochemical and pathological improvements after neonatal intravenous gene therapy for mucopolysaccharidosis VII in dogs. Mol. Genet. Metab. 2006;87:8–21. doi: 10.1016/j.ymgme.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Ward J.M. Anver M.R. Mahler J.F. Devor-Henneman D.E. Pathology of mice commonly used in genetic engineering (C57BL/6; 129; B6,129; and FVB/N) In: Ward J.M., editor; Mahler J.F., editor; Maronpot R.R., editor; Sundberg J.P., editor; Frederickson R.M., editor. Pathology of Genetically Engineered Mice. Iowa State University Press; Ames, IA: 2000. pp. 161–179. [Google Scholar]
- Wolfe J. Sands M.S. Murine mucopolysaccharidosis type VII: A model system for somatic gene therapy of the central nervous system. In: Lowenstein P., editor; Enquist L., editor. Gene Transfer into Neurons: Towards Gene Therapy of Neurological Disorders. John Wiley & Sons; Essex, UK: 1996. pp. 263–274. [Google Scholar]
- Woods N.B. Bottero V. Schmidt M. von Kalle C. Verma I.M. Gene therapy: Therapeutic gene causing lymphoma. Nature. 2006;440:1123. doi: 10.1038/4401123a. [DOI] [PubMed] [Google Scholar]
- Xu L. Haskins M.E. Gao C. Weil M.A. O'Malley T.M. Melniczek J.R. O'Donnell P.A. Mazrier H. Ellinwood N.M. Ponder K.P. Transduction of hepatocytes after neonatal delivery of a Moloney murine leukemia virus-based retroviral vector results in long-term expression of β-glucuronidase in mucopolysaccharidosis VII dogs. Mol. Ther. 2002a;5:141–153. doi: 10.1006/mthe.2002.0527. [DOI] [PubMed] [Google Scholar]
- Xu L. Mango R.L. Sands M.S. Haskins M.E. Ellinwood N.M. Ponder K.P. Evaluation of pathological manifestations of disease in mucopolysaccharidosis VII mice after neonatal hepatic gene therapy. Mol. Ther. 2002b;6:745–758. doi: 10.1006/mthe.2002.0809. [DOI] [PubMed] [Google Scholar]
- Xu L. O'Malley T. Sands M.S. Wang B. Meyerrose T. Haskins M.E. Ponder K.P. In vivo transduction of hematopoietic stem cells after neonatal intravenous injection of an amphotropic retroviral vector in mice. Mol. Ther. 2004;10:37–44. doi: 10.1016/j.ymthe.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Zychlinski D. Schambach A. Modlich U. Maetzig T. Meyer J. Grassman E. Mishra A. Baum C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008;16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]