

Abstract
Understanding individual differences in the susceptibility to metabolic side effects as a response to antipsychotic therapy is essential to optimize the treatment of schizophrenia. Here we perform genomewide association studies (GWAS) to search for genetic variation affecting the susceptibility to metabolic side effects. The analysis sample consisted of 738 schizophrenia patients, successfully genotyped for 492K SNPs, from the genomic subsample of the Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE) study. Outcomes included twelve indicators of metabolic side effects, quantifying antipsychotic-induced change in weight, blood lipids, glucose and hemoglobin A1c, blood pressure and heart rate. Our criterion for genomewide significance was a pre-specified threshold that ensures, on average, only 10% of the significant findings are false discoveries. Twenty-one SNPs satisfied this criterion. The top finding indicated a SNP in MEIS2 mediated the effects of risperidone on hip circumference (q =.004). The same SNP was also found to mediate risperidone's effect on waist circumference (q =.055). Genomewide significant finding were also found for SNPs in PRKAR2B, GPR98, FHOD3, RNF144A, ASTN2, SOX5 and ATF7IP2, as well as several intergenic markers. PRKAR2B and MEIS2 both have previous research indicating metabolic involvement and PRKAR2B has previously been shown to mediate antipsychotic response. Although our findings require replication and functional validation, this study demonstrates the potential of GWAS to discover genes and pathways that potentially mediate adverse effects of antipsychotic medication.
Keywords: genomewide association, antipsychotics, pharmacogenomics, personalized medicine, metabolic side effects
Introduction
Antipsychotics are the cornerstone of acute and long-term treatment for schizophrenia1;2. The first generation, sometimes referred to as the “typical” antipsychotics (e.g., haloperidol) was introduced in the 1950s. Despite treatment with these first generation antipsychotics, a substantial proportion of schizophrenia patients do not improve or relapse frequently. Furthermore, these drugs are often associated with significant side effects, including extrapyramidal symptoms (EPS)—involuntary movements that may occur in schizophrenia patients after long-term treatment with antipsychotic medication. Tardive dyskinesia (TD) is a particularly worrisome EPS because of its high annual incidence rates3 and potential irreversibility4
Clozapine was reintroduced in 1989 marking the advent of a second generation of “atypical” antipsychotics5;6. It has enhanced therapeutic effects in patients who respond poorly to treatment and a much lower risk of side effects such as TD. Clozapine has, however, been associated with severe agranulocytosis, necessitating hematologic monitoring and making it unsuitable as a first line drug. Clozapine's success stimulated efforts to develop new antipsychotics, resulting in other second generation drugs such as risperidone and olanzapine7. These newer second generation drugs differ pharmacologically from first generation antipsychotics principally in their lower affinity for the dopamine 2 receptor, relatively greater affinities for other neuroreceptors such as serotonin, and in their ability to modulate glutamate receptor mediated functions and behaviors8. These newer antipsychotics are also associated with a lower incidence of EPS/TD and do not share clozapine's risk of agranulocytosis.
Second generation antipsychotics are, however, associated with a variety of metabolic side effects such as dyslipidemia, elevated glucose levels, and weight gain9;10, with medical consequences ranging from cosmetic concerns to increased rates of cardiovascular disease (e.g., hypertension, coronary artery disease) and diabetes11. Careful monitoring and treatment of metabolic adverse effects is therefore recommended. Furthermore, these side effects are key factors underlying the substantial noncompliance characterizing antipsychotic therapy 12. Clearly, any ability to minimize metabolic side effects by matching individual patients to the optimal drug prior to treatment would have great clinical value.
Genetic factors likely explain a portion of individual differences in susceptibility to metabolic side effects. Studies have suggested associations with, for example, serotonin receptors13. The serotonergic system is involved in the regulation of feeding behavior and satiety control in the central nervous system, and serotonin receptors are expressed in central nervous system areas that are involved in energy balance14. Furthermore, antipsychotics such as olanzapine have high affinity for these receptors and are hypothesized to induce weight gain especially via 5-hydroxytryptamine (serotonin) receptor 2C antagonism15. Histamine receptor H1 antagonism has also been implicated16. However, robust, consistent evidence implicating any specific candidate gene or polymorphism has been rare17. Perhaps more importantly, the selection of candidate genes is restricted to current knowledge about underlying biological mechanisms, which is limited. Methods that systematically screen variants across the whole genome for association with metabolic side effect are therefore critical to discover relevant variants in novel genes. Such genomewide association studies (GWAS) have recently become feasible. Furthermore, the number of new replicated marker-disease associations has increased dramatically since the introduction of GWAS 18 with some initial applications in the context of drug response19-22.
In this paper, we use GWAS to search for genetic variation affecting the susceptibility for antipsychotic-induced metabolic side effects. Our study sample consists of 738 schizophrenia patients from the CATIE study 23;24. After filtering, 492K SNPs were available for analyses25. The analyses were performed on twelve quantitative variables related to weight gain, a blood lipid panel, glucose, hemoglobin A1c, blood pressure and heart rate.
Methods
Study sample
Subjects came from the Clinical Antipsychotic Trial of Intervention Effectiveness (CATIE) study and were diagnosed with schizophrenia using the Structured Clinical Interview for DSM-IV 26. This study sample has been carefully described elsewhere study 23;24. In short, CATIE is a multiphase randomized controlled trial of six antipsychotic medications, including olanzapine, perphenazine, quetiapine, risperidone, ziprasidone and clozapine, which followed patients for up to 18 months. To maximize representativeness, the participants were recruited from 57 clinical settings around the United States. Males constituted 74% of the total sample. The mean age of the participants was 40.9 years (SD=11.0). On average, patients were treated for 16.7 year (SD=11.2) where they received antipsychotic medication for the first time 14.3 (SD=10.8) years ago. All participants or their legal guardians gave written informed consent, including consent for genetic studies. The institutional review board at each site approved the study.
Metabolic and cardiovascular outcome measures
The metabolic measures collected in CATIE have been described in detail previously10;27. To briefly restate, BMI (kg/m2) was calculated in the standard fashion and waist and hip circumferences (inches) were measured at the narrowest and widest points, respectively. Blood pressure (mm Hg) was measured as a single, seated determination, and heart rate (bpm) was measured as resting pulse count in 30 seconds×2.
For assessment of laboratory measures (i.e., glucose (mg/dL), hemoglobin A1c (%), triglycerides (mg/dL), total cholesterol (mg/dL) and HDL(mg/dL)), CATIE subjects were asked to present in a fasting state. However, as information on last meal was collected and suggested a significant range; we explicitly adjusted glucose and triglycerides for fasting time. Specifically, we estimated mixed models predicting glucose and triglycerides with fasting time, and output the residuals as our fasting time-adjusted measures. Additionally, an empirical screen of the influence of fasting time on other laboratory measure indicated that it explained significant amounts of variance in total cholesterol and hemoglobin A1c; thus, fasting time was regressed out of these measures as well. All metabolic laboratory measures were assessed at a single central laboratory.
Descriptive statistics presented in Table 1 confirm that, compared to a matched nationally representative sample from the National Health and Nutrition Examination Survey (NHANES) III, CATIE subjects generally exhibit less favorable metabolic profiles 10;27. For instance, CATIE subjects were characterized by significantly greater waist circumference (mean=40.42), BMI (mean=30.39), diastolic blood pressure (mean =78.85) and triglycerides (mean=209.81), and lower HDL (mean=42.82), than the NHANES III national averages. Heart rate (mean=79.98) was also relatively elevated, while glucose (mean=101.73) and hemoglobin A1c (mean=5.67) were consistent with age-adjusted national averages. Further, columns 3 and 4 of Table 1 indicate that, according to established criteria, the percentage of subjects with problematic levels of metabolic phenotypes increased from baseline for all phenotypes. It is important to note, however, that as different antipsychotics have different metabolic side effect profiles, these aggregate percentages may mask important differences between drugs (see Meyer and colleagues10;27 for further detail).
Table 1.
Descriptive statistics (trial means and SD), percent of subjects experiencing ADRs and total number of assessments per drug
| Descriptive Statistics | ADRs (%) | Total Assessments (Subjects) per Drug | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Phenotype | Mean (SD) | Base | In trial | Olanzapine | Perphenazine | Quetiapine | Risperidone | Ziprasidone | Clozapine |
| BMI (kg/m2) | 30.39 (7.02) | 43.12 | 56.80 | 1225 (283) | 607 (145) | 942 (277) | 1102 (282) | 599 (200) | 175 (52) |
| Waist Circumference (in) | 40.42 (6.54) | 48.47 | 66.93 | 1199 (282) | 598 (144) | 927 (273) | 1089 (280) | 591 (199) | 175 (52) |
| Hip Circumference (in) | 43.12 (5.73) | 35.33 | 54.98 | 1193 (282) | 598 (144) | 927 (273) | 1089 (280) | 591 (199) | 175 (52) |
| Waist-Hip Ratio | 0.94 (0.08) | 29.92 | 54.98 | 1193 (282) | 598 (144) | 927 (273) | 1089 (280) | 591 (199) | 175 (52) |
| Systolic BP (mm Hg) | 123.75 (15.56) | 29.54 | 59.87 | 1227 (282) | 604 (145) | 944 (278) | 1099 (281) | 595 (197) | 174 (52) |
| Diastolic BP (mm Hg) | 78.85 (10.79) | 26.80 | 55.23 | 1227 (282) | 603 (145) | 944 (278) | 1099 (281) | 595 (197) | 174 (52) |
| Heart Rate (bpm) | 79.98 (13.05) | 5.36 | 22.02 | 1226 (282) | 602 (144) | 944 (278) | 1098 (280) | 595 (198) | 174 (52) |
| Glucose (mg/dL) | 101.73 (44.33) | 15.79 | 44.91 | 759 (282) | 357 (142) | 597 (266) | 670 (272) | 381 (195) | 118 (53) |
| Hemoglobin A1c (%) | 5.67 (1.10) | 12.91 | 19.60 | 529 (225) | 241 (94) | 386 (187) | 465 (198) | 224 (116) | 82 (45) |
| Triglycerides (mg/dL) | 209.81 (181.51) | 54.61 | 74.90 | 756 (281) | 357 (142) | 600 (266) | 673 (272) | 383 (194) | 117 (53) |
| HDL Cholesterol (mg/dL) | 42.82 (13.70) | 52.76 | 73.13 | 756 (281) | 357 (142) | 600 (266) | 672 (272) | 383 (194) | 117 (53) |
| Total Cholesterol (mg/dL) | 199.46 (45.41) | 47.31 | 62.82 | 755 (281) | 358 (142) | 600 (266) | 673 (272) | 384 (195) | 117 (53) |
Estimating treatment effects
The right side of Table 1 shows that sample sizes were largest for olanzapine and risperidone and smallest for clozapine. The average number of assessments per subject was about 7.4 observations per patient for variables that did not require phlebotomy (e.g., weight gain-related measures, blood pressure). For variables requiring phlebotomy (e.g., lipids, glucose) there were about 4.9 observations with the exception of hemoglobin A1c, which had a somewhat lower average of 3.5 measurements per patient.
To maximize power for the GWAS, we developed a method to estimate treatment effects from all available information28 using mixed modeling29;30. Our method first determines the optimal functional form of over-time drug response, then screens many possible covariates to select those that improve the precision of the treatment effect estimates, and finally generates the individual treatment effect estimates based on the best fitting model using best linear unbiased predictors (BLUPs)31. As this approach takes advantages of all available information in CATIE, it results in more precise estimates than traditional approaches (e.g., subtracting pre- from post-treatment observations) that estimate treatment effects using only two assessments.
Specifically, to determine the optimal model of over-time drug response for each metabolic outcome we fit a series of models specifying linear change for a given number of days on drug and flat thereafter. This series began with a model assuming that maximal drug response was achieved at day one. Each subsequent model specified an incrementally longer duration until maximal drug response was achieved with the final model assuming that the drug effect did not plateau (i.e., linear change throughout the trial). The model with the best fit of series was selected to determine the average number of days until maximal drug response. This duration varied across metabolic outcomes with triglycerides peaking earliest (20 days on drug), and diastolic blood pressure peaking latest (586 days on drug) (see Supplemental Material A for details).
After determining the optimal functional form of over-time drug response, 36 covariates were screened to identify those that improved the precision of the treatment effect estimates. These covariates consisted of design characteristics, socio-demographic measures, clinical information, confounding medications, and baseline antipsychotic treatment. The number of selected covariates ranged from zero (e.g. for BMI, waist and hip circumference) to four or greater (e.g. blood lipids). Design characteristics, hyperlipidemia treatment (e.g., statins) and baseline olanzapine treatment where the most commonly selected covariates (see Supplemental Material A for details). Finally, treatment effects were generated by employing a unique feature of the mixed model—random effects. To elaborate, the mixed model estimates two types of parameters, coefficients that describe the predictors' average effects for the full sample (i.e., fixed effects) and deviations from the average effects for each subject (i.e., random effects). Thus, for each of the six trial drugs investigated, we were able to output treatment effects as random drug effects. Intuitively, these treatment effects quantify how much each subject's metabolic phenotypes change in response to a given drug, relative to the average effect for all subjects who took the drug. Out of 72 possible treatment effect measures (6 drugs × 12 metabolic outcomes), in 10 instances there were no significant individual differences in drug response (perphenazine-glucose, quetiapine-glucose, ziprasidone-glucose, perphenazine-hemoglobin A1c, quetiapine-hemoglobin A1c, clozapine-hemoglobin A1c, risperidone-triglycerides, ziprasidone-triglycerides, clozapine-HDL cholesterol, risperidone-total cholesterol). These treatment effects were omitted from further analyses.
Genotyping
DNA sampling, genotyping and genotype quality control have been described by Sullivan et al.25. In total, 665,439 SNPs were genotyped using the Affymetrix 500K chipset (Santa Clara, CA, USA) and a custom 164K chip created by Perlegen (Mountain View, CA, USA). After quality control protocols were performed (see Supplemental Materials A), genotypes for 492,900 SNPs from 738 individuals remained for statistical analysis.
Population stratification
Approximately 57% of the CATIE subjects describe themselves as white/European American (EA), 29% as black/African American (AA) and the remaining 14% described themselves as more than one racial category or “other”. It is essential to control for these different ancestral backgrounds, which otherwise can cause false-positive association findings if differences in distribution of genotypes and phenotypes exists within the different strata defined by ancestry. To investigate population stratification we have used the multi-dimensional scaling (MDS) approach implemented in PLINK32. Input data for the MDS approach were the genomewide average proportion of alleles shared identical by state (IBS) between any two individuals. The first MDS dimension from this genetic similarity matrix captures the maximal variance in the genetic similarity; the second dimension must be orthogonal to the first and captures the maximum amount of residual genetic similarity; and so on. The first five dimensions appeared to capture the vast majority of genetic substructure in the CATIE sample and were included in the GWAS analyses.
Association testing, control of false discoveries and cross-outcome comparisons
All GWAS association testing was conducted in PLINK32 using a linear regression model with the five population stratification MDS dimensions as covariates. The Wald test implemented in PLINK was used to test for an additive genetic effects by coding each as a 0, 1, or 2 count of the number of minor alleles.
As argued previously33, we prefer a false discovery rate (FDR) based approach to declare significance because: a) it balances the competing goals of finding true effects versus controlling false discoveries, b) it provides comparable standards across studies because it is much less affected by the number of tests, and c) it is relatively robust to tests of correlated outcomes34-42. We used an FDR threshold<0.1 for declaring genomewide significance33 and a threshold of 0.25 for identifying “potentially interesting” results. These thresholds mean that on average we expect that 10% and 25%, respectively, of the SNPs declared significant will be false discoveries. Operationally these FDR levels were controlled using q-values. Q-values are FDRs calculated using the p-value of the markers as thresholds for declaring significance35;43. Q-values were estimated with the approach outlined by Bukszar et al.44, which is slightly more conservative than conventional q-value approaches and has the desirable property of avoiding blocks of identical q-values despite different p-values. The FDR was controlled for each GWAS separately rather than jointly across all GWAS. This avoids a loss of power for outcomes where the number and size of the effects are relatively larger. Note that performing many GWAS analyses will not only increase the number of false positives but also the number of true positives34. This is because the FDR controls the expected ratio of false to all discoveries.
To avoid an all-or-nothing conclusion about whether a SNP is significant and improve the interpretation of our GWAS results, we also estimated for each SNP the local FDR (lFDR). The lFDR is the (posterior) probability that the SNP has no effect44. The lFDR provides a marker-specific estimate that the GWAS finding is false. This is not the case for the q-value that essentially averages these probabilities across the whole group of markers declared significant. As a result, a marker with a very high probability of being a false discovery may simply have a small q-value because it was tested simultaneously with a marker that has a low probability of being a false positive45.
After indentifying genomewide significant markers, we leverage the multiple metabolic phenotype, multiple drug study design to examine whether genomewide significant markers show association to other, related outcomes. For this examination we extract coefficients and p-values for the genomewide significant SNPs for all outcomes and conduct a t-test to determine if the total number of supporting findings (alternate outcomes with coefficients in the same direction as the genomewide finding and p-value < 0.05) is more than would be expected by chance. We then briefly note some patterns by drug and metabolic phenotypes. Finally, we discuss the top results of this cross-outcome robustness analysis.
Results
Genomewide significant signals
Quantile-Quantile (QQ) plots and p-values for each outcome variable are available for download at www.vipbg.vcu.edu/∼edwin. Figure 1 shows QQ plots for the 4 outcomes with the strongest SNP associations. The plots show that the distribution of the p-values from the GWAS are generally on a straight line, indicating the expected p-value distribution under the null hypothesis assuming no effects of the markers. However, in each of these 4 plots there is also evidence that markers in the right upper corner of the have p-values smaller than would be expected under the null hypothesis, suggesting true association between these markers and the outcome variable. The plots also list the lambda values (i.e., the ratio of the median observed p-value of the distribution to the expected p-value under the null hypothesis), which ideally should equal 1—indicating no systematic test statistic inflation. Of the 62 outcomes analyzed, the mean lambda=0.991 and the maximum lambda=1.019, indicating that population stratification was adequately controlled by the 5 ancestral MDS covariates and not a confounding factor in the GWAS. Also notable, for 11 outcomes, primarily those involving clozapine, lambda values were slightly deflated (0.919<.98), suggesting that results may be conservative in these cases.
Figure 1.
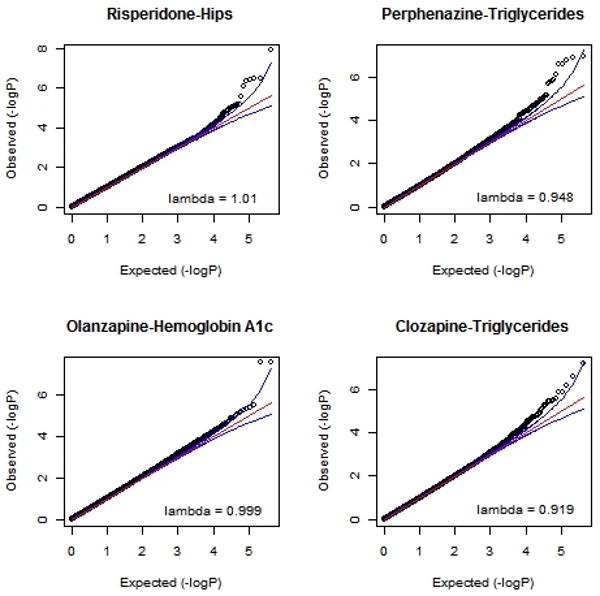
QQ plots for four outcomes showing strongest GWAS results
Table 2 shows the number of significant GWAS results at various FDR thresholds. For concision, the table presents significant results grouped by outcome and drug. Twenty-one SNPs were genomewide significant at our pre-specified threshold for declaring significance in genetic studies using a FDR threshold of 0.133. When grouped by medication, the largest numbers of genomewide significant results were found for risperidone (N=7) and perphenazine (N=7); when grouped by phenotypic outcome, the largest number of findings were found for triglycerides and hip circumference. Using more liberal FDR thresholds of 0.25 and 0.5, we identified 48 and 118 potentially “interesting” results, respectively.
Table 2.
Number of significant GWAS results at various FDR thresholds
| FDR threshold | ||||
|---|---|---|---|---|
| By Antipsychotic | 0.1 | 0.25 | 0.5 | |
| Olanzapine | 2 | 4 | 9 | |
| Perphenazine | 7 | 13 | 26 | |
| Quetiapine | 1 | 2 | 8 | |
| Risperidone | 7 | 13 | 36 | |
| Ziprasidone | 1 | 1 | 3 | |
| Clozapine | 3 | 15 | 36 | |
| Sum | 21 | 48 | 118 | |
| By Metabolic Phenotype | ||||
| BMI | 1 | 1 | 4 | |
| Waist Circumference | 1 | 1 | 2 | |
| Hip Circumference | 6 | 8 | 14 | |
| Waist-Hip Ratio | 0 | 0 | 3 | |
| Systolic BP | 0 | 0 | 3 | |
| Diastolic BP | 0 | 0 | 1 | |
| Heart Rate | 0 | 2 | 9 | |
| Glucose | 0 | 2 | 4 | |
| Hemoglobin A1c | 3 | 7 | 24 | |
| Triglycerides | 9 | 19 | 39 | |
| HDL Cholesterol | 1 | 3 | 7 | |
| Total Cholesterol | 0 | 5 | 8 | |
| Sum | 21 | 48 | 118 | |
Table 3 shows the specific SNPs with q-values smaller than 0.25. The top finding involved rs1568679 (p=1.28E-08 and q=0.004), located in an intron of Meis homeobox 2 (MEIS2) on chromosome 15q14, which was indicated to mediate the effect of risperidone on hip circumference. This same SNP also reached genomewide significance (p=6.82E-08 and q=0.055) for the effect of risperidone on waist circumference. This SNP is polymorphic in both the African American (AA) and the European American (EA) subsamples with MAF of 0.44 and 0.10, respectively. Furthermore, haplotype tests suggested that in both subsamples the same high risk haplotype contributed to the total association signal for both outcome variables. An overall haplotype test did not improve the association signals.
Table 3.
GWAS results with q-values smaller than 0.25
| Outcome | SNP | Gene symbol | Cytogenetic location | MAF | N | Eff | p-value | q-value | lFDR |
|---|---|---|---|---|---|---|---|---|---|
| Perphenazine - HDL | rs11163585 | no | 1p31.1 | 0.241 | 139 | + | 9.32E-07 | 0.249 | 0.614 |
| Perphenazine - Triglycerides | rs17410015 | no | 1p21.2 | 0.063 | 138 | + | 1.52E-06 | 0.117 | 0.429 |
| Perphenazine - Triglycerides | rs6735179 | no | 2p25.3 | 0.346 | 138 | + | 1.34E-07 | 0.018 | 0.08 |
| Perphenazine - Triglycerides | rs6741819 | RNF144A | 2p25.1 | 0.305 | 138 | + | 2.43E-07 | 0.029 | 0.127 |
| Risperidone - Hip Circumference | rs1991126 | no | 2p24.1 | 0.15 | 265 | + | 8.98E-07 | 0.112 | 0.437 |
| Risperidone - Hip Circumference | rs1117324 | no | 2p24.1 | 0.156 | 265 | + | 3.47E-07 | 0.054 | 0.245 |
| Perphenazine - Triglycerides | rs10202231 | no | 2p16.1 | 0.457 | 138 | - | 7.31E-07 | 0.068 | 0.279 |
| Clozapine - Heart Rate | rs399885 | no | 2p12 | 0.327 | 52 | + | 5.11E-07 | 0.135 | 0.356 |
| Clozapine - Heart Rate | rs7570469 | no | 2p12 | 0.418 | 52 | + | 5.50E-07 | 0.141 | 0.369 |
| Clozapine - Triglycerides | rs1534238 | no | 2p11.2 | 0.381 | 52 | - | 2.82E-06 | 0.218 | 0.583 |
| Clozapine - Triglycerides | rs17385675 | no | 2q33.1 | 0.069 | 53 | + | 3.44E-06 | 0.247 | 0.626 |
| Perphenazine - Triglycerides | rs13134954 | no | 4q23 | 0.337 | 135 | + | 2.19E-06 | 0.152 | 0.513 |
| Perphenazine - Triglycerides | rs11735070 | no | 4q23 | 0.338 | 137 | + | 1.30E-06 | 0.105 | 0.395 |
| Ziprasidone - Hip Circumference | rs1405687 | no | 4q23 | 0.088 | 183 | - | 4.72E-08 | 0.038 | 0.181 |
| Olanzapine - Hemoglobin A1c | rs1967256 | GPR98 | 5q14.3 | 0.148 | 207 | + | 2.80E-08 | 0.012 | 0.068 |
| Olanzapine - Hemoglobin A1c | rs11954387 | GPR98 | 5q14.3 | 0.148 | 207 | + | 2.80E-08 | 0.012 | 0.068 |
| Risperidone - Hemoglobin A1c | rs17100498 | no | 5q31.3 | 0.126 | 186 | + | 4.90E-07 | 0.078 | 0.236 |
| Risperidone - Hemoglobin A1c | rs17100506 | no | 5q31.3 | 0.126 | 184 | + | 7.45E-07 | 0.103 | 0.301 |
| Clozapine - Glucose | rs9658108 | PPARD | 6p21.31 | 0.052 | 53 | + | 4.58E-07 | 0.191 | 0.478 |
| Clozapine - Triglycerides | rs1577917 | no | 6q14.3 | 0.21 | 53 | + | 3.48E-06 | 0.248 | 0.628 |
| Clozapine - Total Cholesterol | rs1979096 | no | 7p21.1 | 0.115 | 53 | - | 6.02E-07 | 0.157 | 0.392 |
| Clozapine - Total Cholesterol | rs10499504 | no | 7p21.1 | 0.11 | 52 | - | 4.34E-07 | 0.129 | 0.334 |
| Clozapine - Triglycerides | rs13224682 | PRKAR2B | 7p22.3 | 0.072 | 53 | + | 6.30E-08 | 0.015 | 0.055 |
| Olanzapine - Total Cholesterol | rs977396 | no | 8q22.3 | 0.089 | 261 | + | 3.27E-07 | 0.173 | 0.463 |
| Clozapine - Glucose | rs320209 | no | 9q31.1 | 0.068 | 53 | + | 3.94E-07 | 0.174 | 0.448 |
| Perphenazine - Triglycerides | rs4838255 | ASTN2 | 9q33.1 | 0.139 | 135 | + | 2.62E-07 | 0.031 | 0.135 |
| Clozapine - Triglycerides | rs17661538 | no | 10p12.33 | 0.138 | 51 | + | 1.30E-06 | 0.131 | 0.415 |
| Clozapine - Triglycerides | rs2994684 | no | 10p11.22 | 0.155 | 53 | + | 2.58E-07 | 0.041 | 0.153 |
| Clozapine - Triglycerides | rs1771628 | no | 10p11.22 | 0.191 | 53 | + | 6.66E-07 | 0.082 | 0.286 |
| Risperidone - Hip Circumference | rs7119817 | no | 11q23.1 | 0.371 | 264 | + | 4.51E-07 | 0.067 | 0.292 |
| Risperidone - Hip Circumference | rs7105881 | no | 11q23.1 | 0.367 | 265 | + | 3.27E-07 | 0.052 | 0.235 |
| Risperidone - Hip Circumference | rs7108821 | no | 11q23.1 | 0.367 | 263 | + | 4.21E-07 | 0.063 | 0.279 |
| Clozapine - Triglycerides | rs620875 | KIRREL3 | 11q24.2 | 0.101 | 53 | + | 3.20E-06 | 0.236 | 0.61 |
| Perphenazine - HDL | rs1464500 | SOX5 | 12p12.1 | 0.232 | 138 | + | 1.07E-07 | 0.058 | 0.204 |
| Quetiapine - HDL | rs518590 | no | 13q12.11 | 0.21 | 253 | + | 1.60E-07 | 0.14 | 0.416 |
| Perphenazine - Total Cholesterol | rs1187614 | CLMN | 14q32.13 | 0.313 | 138 | - | 2.37E-07 | 0.23 | 0.541 |
| Risperidone - Hip Circumference | rs1568679 | MEIS2 | 15q14 | 0.103 | 265 | + | 1.28E-08 | 0.004 | 0.018 |
| Risperidone - Waist Circumference | rs1568679 | MEIS2 | 15q14 | 0.103 | 266 | + | 6.82E-08 | 0.055 | 0.244 |
| Risperidone - Hemoglobin A1c | rs13335336 | ATF7IP2 | 16p13.13 | 0.078 | 187 | + | 7.09E-07 | 0.1 | 0.293 |
| Perphenazine - Triglycerides | rs153091 | LOC729993 | 16p13.12 | 0.227 | 138 | + | 1.85E-06 | 0.135 | 0.474 |
| Olanzapine - Total Cholesterol | rs4783227 | no | 16q23.3 | 0.424 | 269 | - | 3.95E-07 | 0.194 | 0.502 |
| Clozapine - Triglycerides | rs17216786 | CDH13 | 16q23.3 | 0.054 | 53 | + | 1.33E-06 | 0.133 | 0.42 |
| Perphenazine - Triglycerides | rs17651157 | FHOD3 | 18q12.2 | 0.068 | 138 | + | 1.07E-07 | 0.016 | 0.067 |
| Perphenazine - Triglycerides | rs10502661 | FHOD3 | 18q12.2 | 0.063 | 138 | + | 1.61E-07 | 0.021 | 0.093 |
| Risperidone - Hemoglobin A1c | rs8092443 | no | 18q22.2 | 0.225 | 186 | - | 9.72E-07 | 0.123 | 0.348 |
| Risperidone - Hemoglobin A1c | rs11663206 | no | 18q22.2 | 0.289 | 187 | - | 2.07E-06 | 0.197 | 0.498 |
| Risperidone - Hip Circumference | rs6092078 | no | 20q13.2 | 0.115 | 264 | - | 2.61E-06 | 0.238 | 0.681 |
Locus information includes gene in region, cytogenetic location, location of SNP, and the minor allele frequency (MAF) calculated in the complete CATIE sample. For each test we report the sample size (N), direction of effect of minor allele frequency (Eff) where a “+” indicates positive association, q values and local FDRs as estimated using the method developed by Bukszar et al.44, and the number of other analyzed outcomes showing significant association to the SNP at p<.05 (#m). Genomewide significant results are indicated in bold. Consecutive shaded rows indicate SNPs in high LD (r2 > 0.8).
The second most significant signal involved rs1967256 and rs11954387 (p=2.80E-08 and q=0.012), which were in complete LD and were associated with the effect of olanzapine on hemoglobin A1c. These SNPs were located in an intron of the G protein-coupled receptor 98 precursor (GPR98) on chromosome 5q14.3. The markers were polymorphic in both EA (MAF=0.08) and AA (MAF=0.24). Haplotype tests did not improve association signals.
For triglycerides, the outcome with the largest number of significant results (Table 2), the top finding involved rs13224682 mediating the effect of clozapine (p=6.30E-08 and q=0.015). This SNP is located in an intron of Homo sapiens protein kinase, cAMP-dependent, regulatory, type II, beta (PRKAR2B) on chromosome 7q22.3. This marker had a very low minor allele frequency (MAF=0.01) in AAs implying that the signal was mainly driven by the EA subsample.
For the combination perphenazine-triglycerides, six genomewide significant SNPs were detected (Table 3). Two of these were rs17651157 (p=1.07E-07 and q=0.016) and rs10502661 (p=1.61E-07 and q=0.021) located 2703 bp apart in an intron of the formin homology 2 domain containing 3 (FHOD3) gene on chromosome 18q12.2. These SNPs were in high LD (in AA r2=1.0 and in EA r2=0.78) and had fairly low minor allele frequencies (MAFs 0.03/0.07 in AA/EA). Haplotype testing improved the signal (p-values of 5.36E-8 and 3.09E-9 for a specific high risk haplotype and overall haplotype tests, respectively) in EAs but not the AA subsample.
As shown in Table 3, four additional genomewide significant SNPs were located within annotated genes and ten genomewide significant SNPs were located in intergenic regions. For the three SNPs on chromosome 11q23.1 mediating the effect of risperidone on hip circumference, the signal improved when conducting an overall versus individual haplotype tests.
Cross-outcome analyses of genomewide significant markers
One advantage of the current analysis is that its inclusion of multiple related phenotypes and various antipsychotic drugs allows an investigation of common mechanisms across outcomes. Specifically, this feature enables searching for patterns of significant SNPs across related phenotypes as a test of robustness; and also across drugs, allowing investigation of common biological pathways. To explore these patterns, we examined if the 48 genomewide significant markers showed association, in the expected direction, to other, related outcomes using a significance threshold of p<.05. On average 4.1 (199/48) secondary associations were detected, which is significantly more than expected by chance (t=2.72; p =0.004). A majority of secondary associations were for initial genomewide significant findings involving risperidone (57) and clozapine (85). In 51% of cases (102/199), secondary associations were for outcomes involving the same drug as the genomewide significant finding. This was particularly true for risperidone (65%; 37/57) and clozapine (52%, 44/85).
Table 4 presents the number of significant secondary associations for all genomewide significant SNPs (q <0.1) and genic SNPs with q-values 0.1-0.25 (for complete list see Supplemental Material B). Results show that secondary associations were common within the weight gain, glucose, and blood pressure phenotype clusters. For instance, the correlation between the number of secondary associations for hip and waist circumferences was 0.73, between diastolic and systolic blood pressure was 0.51, and between glucose and hemoglobin A1c was 0.32. This suggests that the genomewide significant associations observed in the GWAS generally represent robust signals that can be detected across related phenotypes and, in some cases, for different drugs' effects on the same phenotype.
Table 4.
Number of alternate outcome associations (p<.05) to genomewide significant SNPs, by metabolic phenotype
| Weight Gain | Lipids | Glucose | Blood Pressure | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Gene | Genomewide q-value | BMI | Hips | Waist | W/H | TChol | Trig | HDL | Gluc | A1c | Dia | Sys | HR |
| rs1568679 | MEIS2 | 0.004 | 1 | 1* | 1* | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 |
| rs11954387 | GPR98 | 0.012 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1* | 0 | 0 | 1 |
| rs1967256 | GPR98 | 0.012 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1* | 0 | 0 | 1 |
| rs13224682 | PRKAR2B | 0.015 | 1 | 1 | 1 | 0 | 0 | 2* | 0 | 1 | 0 | 0 | 1 | 0 |
| rs17651157 | FHOD3 | 0.016 | 0 | 0 | 0 | 0 | 0 | 1* | 0 | 0 | 0 | 0 | 0 | 0 |
| rs6735179 | no | 0.018 | 1 | 0 | 1 | 1 | 0 | 1* | 0 | 0 | 0 | 0 | 0 | 0 |
| rs10502661 | FHOD3 | 0.021 | 0 | 0 | 0 | 0 | 0 | 1* | 0 | 0 | 0 | 0 | 0 | 0 |
| rs6741819 | RNF144A | 0.029 | 0 | 0 | 1 | 1 | 1 | 1* | 1 | 0 | 2 | 0 | 1 | 0 |
| rs4838255 | ASTN2 | 0.031 | 0 | 0 | 0 | 1 | 0 | 1* | 0 | 0 | 0 | 1 | 1 | 0 |
| rs1405687 | no | 0.038 | 2 | 1* | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| rs2994684 | no | 0.041 | 1 | 2 | 2 | 0 | 0 | 2* | 1 | 0 | 0 | 0 | 0 | 0 |
| rs7105881 | no | 0.052 | 1 | 1* | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 |
| rs1117324 | no | 0.054 | 1 | 1* | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| rs1464500 | SOX5 | 0.058 | 0 | 0 | 0 | 0 | 0 | 1 | 1* | 0 | 0 | 0 | 0 | 0 |
| rs7108821 | no | 0.063 | 1 | 1* | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| rs7119817 | no | 0.067 | 1 | 1* | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 |
| rs10202231 | no | 0.068 | 0 | 0 | 1 | 0 | 0 | 2* | 0 | 0 | 0 | 0 | 0 | 0 |
| rs17100498 | no | 0.078 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1* | 0 | 0 | 0 |
| rs1771628 | no | 0.082 | 1 | 2 | 3 | 0 | 0 | 2* | 1 | 1 | 1 | 0 | 0 | 0 |
| rs13335336 | ATF7IP2 | 0.1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1* | 0 | 0 | 0 |
| rs17216786 | CDH13 | 0.133 | 1 | 1 | 1 | 1 | 0 | 1* | 1 | 1 | 0 | 0 | 0 | 1 |
| rs153091 | LOC729993 | 0.135 | 0 | 0 | 0 | 0 | 0 | 1* | 0 | 0 | 0 | 0 | 0 | 0 |
| rs9658108 | PPARD | 0.191 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1* | 0 | 0 | 0 | 0 |
| rs1187614 | CLMN | 0.23 | 1 | 0 | 1 | 0 | 1* | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| rs620875 | KIRREL3 | 0.236 | 2 | 2 | 1 | 1 | 0 | 1* | 0 | 1 | 1 | 0 | 0 | 0 |
Genomewide significant finding marked with asterisk.
The strongest secondary associations involved rs1568679, in MEIS2, and risperidone-BMI (p=5.02E-06). As discussed above, this SNP was indicated as genomewide significant (q<0.1) in two separate GWAS—risperidone-hip circumference and risperidone-waist circumference. It was also indicated to mediate risperidone's effect on diastolic and systolic blood pressure, as well as olanzapine's effect on glucose in the cross-outcome analysis. Rs13224682, in PRKAR2B, also showed a compelling secondary association pattern. It was implicated to mediate the effects of both clozapine and olanzapine on triglyceride levels, as well as clozapine's effects on a wide range of other metabolic outcomes (BMI, waist circumference, hip circumference and glucose). The two SNPs in complete LD from GPR98 (rs1967256 and rs11954387) showed robust signals for mediating olanzapine's effects on both glucose and hemoglobin A1c, as well as clozapine's effect on heart rate and perphenazine's influence on HDL.
Discussion
Understanding individual differences in the development of metabolic side effects as a response to antipsychotic therapy is essential to individualize the treatment of schizophrenia. In this study we performed GWAS on 12 quantitative metabolic side effect indicators including variables related to weight gain, a blood lipid panel, glucose, hemoglobin A1c, blood pressure and heart rate. We detected 21 SNPs, which, according to our pre-identified criteria (FDR controlled at 0.1 level), can be considered genomewide significant. For each of these markers the estimated posterior probability indicated a reasonable chance of a true finding.
Our top finding involved rs1568679 in MEIS2 reaching genomewide significance mediating the effect of risperidone on both hip and waist circumference and showing secondary associations with BMI, diastolic and systolic blood pressure. There was also some evidence that this SNP mediated olanzapine's effect on glucose. MEIS2 (Meis homeobox 2) is the second member of the human gene family with homology to the murine myeloid ecotropic viral integration site genes, involved in murine myeloid leukemia. The MEIS2 gene encodes a homeobox protein belonging to the TALE (Three Amino acid Loop Extension) family of homeodomain-containing proteins. TALE homeobox proteins are highly conserved transcription regulators and several members have been shown to be essential contributors to many developmental programs46. In addition to critical roles in early development, usually acting as a Hox cofactor, MEIS2 has a transcriptional regulatory function in adults47 and is widely expressed in many tissues48. Of particular note is its role in regulating the activity of PDX1, a transcription factor active in pancreatic β and acinar cells49. It has been shown that MEIS2 switches the activity of PDX1 by forming the trimeric complex PDX1-PBX1b-MEIS250;51. The full trimeric complex is necessary to activate a promoter for ELA1 in pancreatic acinar cells, while unbound PDX1 is necessary to activate insulin-producing β cells. Thus, the transcriptional activity of variants of MEIS2 may be differentially influenced by second generation antipsychotics (particularly risperidone), causing downstream changes in insulin and/or digestive enzyme production. Further, it is also clear that not every function of MEIS2 has yet been determined, as it is a highly complex locus, known to exist as at least 27 distinct splice variants (AceView). Given the robustness of the current association finding across multiple metabolic outcomes and the plausible mechanism suggested by former research, MEIS2 should be considered a promising candidate for further study.
The second and third most significant findings were with GPR98, which were indicated to mediate the effects of olanzapine on hemoglobin A1c levels. GPR98 is a member of the G protein-coupled receptor superfamily of 7 transmembrane domain receptors52. It binds calcium and is expressed in the central nervous system, although it is also expressed in a wide range of other tissues. GPR98 was originally known as VLGR1, or very large G protein-coupled receptor, because it is comprised of over 90 exons that span approximately 600kb, with the largest transcript variant encoding a peptide of 6307 amino acid residues, making it the largest known cell surface protein53. GPR98 has been previously implicated in some forms of epilepsy54 and in Usher syndrome55 (a disorder involving congenital hearing loss and progressive retinitis pigmentosa). There is no current evidence linking it mechanistically with hemoglobin A1c or glucose levels.
SNP rs13224682 in PRKAR2B (Protein kinase, cyclic adensine monophosphate-dependent, regulatory, type II beta) was found to mediate clozapine's and, to a lesser extent, olanzapine's effects on triglyceride levels. The cAMP-dependent protein kinase system controls the cellular effects of cAMP, which acts as a second messenger in many signaling cascades. The kinase holoenzyme consists of two regulatory and two catalytic subunits that dissociate upon binding of cAMP molecules. The free, activated catalytic subunits then phosphorylate downstream proteins, thereby altering their activity or function. PRKAR2B, also known as RII beta, is one of several regulatory subunit proteins56.
RII beta has previously been strongly implicated in metabolic phenotypes and, in separate studies, antipsychotic effects in laboratory animals. First, RII beta knockout mice appear healthy but have markedly diminished white adipose tissue despite normal food intake. They are protected against developing diet-induced obesity and fatty livers57. Furthermore, disruption of RII beta reverses the obesity syndrome of Agouti lethal yellow mice58. One possible mechanism by which RII beta regulates weight is via its known role in the thyroid, where its as an important mediator of thyroid-stimulating hormone (TSH) receptor and cAMP signals to downstream membrane and nuclear substrates59. Another potentially relevant mechanism is suggested by a search with the SLEP bioinformatic tool60, which indicated that the marker is <1 kb from an expression QTL (eQTL) for liver, within the same gene, PRKAR2B61. Second, in relation to antipsychotic effects, mice harboring a targeted disruption of RII beta have a profound deficit in cAMP-stimulated kinase activity in the striatum. When treated with haloperidol, RII beta mutant mice fail to induce either c-fos or neurotensin mRNA and the acute cataleptic response of haloperidol is blocked62. These effects appear to arise because of the importance of cAMP, both in the regulation of metabolism and the transducing of the antipsychotic effect. Our association findings implicating this gene as a mediator of multiple related antipsychotic-induced metabolic outcomes, in addition to ample functional evidence indicating both metabolic function and antipsychotic mediation, make PRKAR2B a compelling candidate for additional investigation.
Two SNPs at FHOD3 were shown to mediate perphenazine's effect on triglycerides at the q<0.1 level. FHOD3 (Formin homology-2 domain-containing protein 3) appears to be expressed in the kidney, heart and brain with little to no expression in other tissues. Its function appears to be as an actin-organizing protein in the cellular cytoskeleton63. Very little else is known of this gene and its function, with only two articles concerning it published to date. Clearly, given this lack of information no firm conclusions about its putative drug-metabolism interaction can be drawn.
A small number of candidate genes have been previously implicated in mediating the metabolic side effects of antipsychotic drugs. These studies have typically focused on weight gain as the outcome measure for second generation antipsychotics. A recent review by Arranz and de Leon64 catalogued previous findings, suggesting positive associations with ADRA2A, CYP2D6, GNB3, HTR2C, LEP and SNAP25. A recent large candidate gene study in CATIE65 also suggestively implicated HTR2A and CHRNA7. As the current study examined the same sample using different methods and a wider SNP panel, we examined the influence of these two candidate genes, as well as the six indicated by Arranz and de Leon. Additionally, we investigated five candidates indicated in a more recent pharmacogenetic study of antipsychotic-induced weight gain in a German sample66, as well as several general metabolic candidates indicated in recent large meta-analyses of non-medicated samples67-73. In total, 1338 SNPs within 45 candidate genes were selected on the basis of either being within the gene boundary or within 50kb flanking either end, leading to a total of 82,956 tests (1338 SNPs×62 outcomes). Numbers of SNPs per gene, a summary of analysis results and QQ plot are given in Supplemental Material A. The most significant findings were rs1962882 in ABCA1, mediating the effects of ziprasidone on waist-hip ratio (p=3.84E-05, q=0.99), followed by rs6449050 at SLC2A9, mediating the effects of olanzapine on glucose levels (p=4.22E-05, q=0.99). These poor q-values, coupled with the fact that none of the genes showed more significant findings than expected by chance, suggests limited support for these as true effects.
While CATIE remains the largest, most comprehensive clinical trial of antipsychotic treatment for schizophrenia with whole genome data, it was not principally designed as a pharmacogenomics study and, consequently, has several limitations in this context. Chief among these is the fact that most subjects were non-naïve to antipsychotic treatment at baseline and were often taking other, potentially confounding, medications with metabolic effects, including antidepressants and mood stabilizers. Moreover, DNA collection took place after the clinical trial and only included a subsample of CATIE participants (51%). As previously described, the genomic subsample had lower symptom severity, less current drug/alcohol abuse/dependence and were less likely to identify as African-American than CATIE subjects not contributing DNA25. Despite statistical adjustment for these factors, inference would be stronger if the trial had a more ideal pharmacogenomics design. Thus, we recommend that future data collection efforts consider the merits of enrolling antipsychotic-naïve subjects, employing stricter recruitment criteria for confounding medications and implementing a more representative sampling frame. Additionally, future research can extend this line of research through incorporating dosage information into the calculation of treatment effects.
As with any genetic associations, our findings will require replication and functional validation. To facilitate this process we provide all p-values (www.vipbg.vcu.edu/∼edwin) as a resource for investigators with the requisite samples to advance this line of research. However, the present study demonstrates the potential of GWAS to discover genes and pathways that mediate adverse effects of antipsychotic medication. A better understanding of these mechanisms and the role of specific polymorphisms may eventually help to personalize antipsychotic medication in order to minimize these adverse drug reactions for patients with schizophrenia.
Supplementary Material
Acknowledgments
The CATIE project was supported by NIMH contract N01 MH90001. Dr Sullivan was supported by R01s MH074027 and MH077139 and Dr van den Oord was supported by R01s MH078069 and HG004240.
Footnotes
Financial Disclosure: Eli Lilly funded the GWAS genotyping done at Perlegen Sciences. Dr Sullivan reports receiving research funding from Eli Lilly in connection with this project. Dr Stroup reports having received research funding from Eli Lilly and consulting fees from Janssen Pharmaceutica, GlaxoSmithKline and Bristol-Myers Squibb. Dr Lieberman reports having received research funding from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Janssen Pharmaceutica and Pfizer, and consulting and educational fees from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly, Forest Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, Novartis, Pfizer and Solvay.
Reference List
- 1.Kane JM, Marder SR. Psychopharmacologic treatment of schizophrenia. Schizophr Bull. 1993;19:287–302. doi: 10.1093/schbul/19.2.287. [DOI] [PubMed] [Google Scholar]
- 2.Kane JM, McGlashan TH. Treatment of schizophrenia. Lancet. 1995;346:820–825. doi: 10.1016/s0140-6736(95)91630-x. [DOI] [PubMed] [Google Scholar]
- 3.Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21:151–156. doi: 10.1097/YCO.0b013e3282f53132. [DOI] [PubMed] [Google Scholar]
- 4.Kane JM, Woerner M, Weinhold P, Kinon B, Lieberman J, Wegner J. Epidemiology of tardive dyskinesia. Clin Neuropharmacol. 1983;6:109–115. doi: 10.1097/00002826-198306000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Pickar D, Litman RE, Hong WW, et al. Clinical response to clozapine in patients with schizophrenia. Arch Gen Psychiatry. 1994;51:159–160. doi: 10.1001/archpsyc.1994.03950020083008. [DOI] [PubMed] [Google Scholar]
- 6.Pickar D, Hsiao JK. Clozapine treatment of schizophrenia. JAMA. 1995;274:981–983. [PubMed] [Google Scholar]
- 7.Pickar D. Prospects for pharmacotherapy of schizophrenia. Lancet. 1995;345:557–562. doi: 10.1016/s0140-6736(95)90469-7. [DOI] [PubMed] [Google Scholar]
- 8.Kinon BJ, Lieberman JA. Mechanisms of action of atypical antipsychotic drugs: a critical analysis. Psychopharmacology (Berl) 1996;124:2–34. doi: 10.1007/BF02245602. [DOI] [PubMed] [Google Scholar]
- 9.Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373:31–41. doi: 10.1016/S0140-6736(08)61764-X. [DOI] [PubMed] [Google Scholar]
- 10.Meyer JM, Davis VG, Goff DC, et al. Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE Schizophrenia Trial: prospective data from phase 1. Schizophr Res. 2008;101:273–286. doi: 10.1016/j.schres.2007.12.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson DC, Doraiswamy PM. Prolactin-related and metabolic adverse effects of atypical antipsychotic agents. J Clin Psychiatry. 2008;69(Suppl 1):32–44. [PubMed] [Google Scholar]
- 12.Bellack AS. Scientific and consumer models of recovery in schizophrenia: concordance, contrasts, and implications. Schizophr Bull. 2006;32:432–442. doi: 10.1093/schbul/sbj044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunes A, Melkersson KI, Scordo MG, Dahl ML. Association between HTR2C and HTR2A polymorphisms and metabolic abnormalities in patients treated with olanzapine or clozapine. J Clin Psychopharmacol. 2009;29:65–68. doi: 10.1097/JCP.0b013e31819302c3. [DOI] [PubMed] [Google Scholar]
- 14.Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 15.Meltzer HY. Treatment of schizophrenia and spectrum disorders: pharmacotherapy, psychosocial treatments, and neurotransmitter interactions. Biol Psychiatry. 1999;46:1321–1327. [PubMed] [Google Scholar]
- 16.Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet. 2005;20:368–378. doi: 10.2133/dmpk.20.368. [DOI] [PubMed] [Google Scholar]
- 17.Malhotra AK, Murphy GM, Jr, Kennedy JL. Pharmacogenetics of psychotropic drug response. Am J Psychiatry. 2004;161:780–796. doi: 10.1176/appi.ajp.161.5.780. [DOI] [PubMed] [Google Scholar]
- 18.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byun E, Caillier SJ, Montalban X, et al. Genome-wide pharmacogenomic analysis of the response to interferon beta therapy in multiple sclerosis. Arch Neurol. 2008;65:337–344. doi: 10.1001/archneurol.2008.47. [DOI] [PubMed] [Google Scholar]
- 20.Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.56. [DOI] [PubMed] [Google Scholar]
- 21.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 22.Liu C, Batliwalla F, Li W, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol Med. 2008;14:575–581. doi: 10.2119/2008-00056.Liu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stroup TS, McEvoy JP, Swartz MS, et al. The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophr Bull. 2003;29:15–31. doi: 10.1093/oxfordjournals.schbul.a006986. [DOI] [PubMed] [Google Scholar]
- 24.Lieberman JA, Stroup TS, Mcevoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan PF, Lin D, Tzeng JY, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.First M, Spitzer R, Gibbon M, Williams J. Structured Clinical Interview for DSM-IV Axis I Disorders--Administration Booklet. Washington D.C.: American Psychiatric Press, Inc.; 1994. [Google Scholar]
- 27.Mcevoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80:19–32. doi: 10.1016/j.schres.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Van den Oord EJCG, Adkins DE, McClay J, Lieberman J, Sullivan PF. A systematic method for estimating individual responses to treatment with antipsychotics in CATIE. Schizophr Res. 2009;107:13–21. doi: 10.1016/j.schres.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein H. Multilevel statistical models. London: Arnold; 1995. [Google Scholar]
- 30.Searle SR, Casella G, McCulloch CE. Variance components. New York: Wiley; 1992. [Google Scholar]
- 31.Pinheiro JC, Bates DM. Mixed-effects models in S and S-plus. New York, NY: Springer; 2000. [Google Scholar]
- 32.Purcell S, Neale B, Todd-Brown K, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Oord EJCG, Sullivan PF. False discoveries and models for gene discovery. Trends Genet. 2003;19:537–542. doi: 10.1016/j.tig.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B. 1995;57:289–300. [Google Scholar]
- 35.Storey J. The positive false discovery rate: A Bayesian interpretation and the q-value. Annals of Statistics. 2003;31:2013–2035. [Google Scholar]
- 36.Brown BW, Russell K. Methods of correcting for multiple testing: operating characteristics. Stat Med. 1997;16:2511–2528. doi: 10.1002/(sici)1097-0258(19971130)16:22<2511::aid-sim693>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Fernando RL, Nettleton D, Southey BR, Dekkers JC, Rothschild MF, Soller M. Controlling the proportion of false positives in multiple dependent tests. Genetics. 2004;166:611–619. doi: 10.1534/genetics.166.1.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van den Oord EJCG, Sullivan PF. A framework for controlling false discovery rates and minimizing the amount of genotyping in the search for disease mutations. Human Heredity. 2003;56:188–199. doi: 10.1159/000076393. [DOI] [PubMed] [Google Scholar]
- 39.Korn EL, Troendle J, McShane L, Simon R. Controlling the number of false discoveries: Application to high-dimensional genomic data. Journal of Statistical Planning and Inference. 2004;124:379–398. [Google Scholar]
- 40.Tsai CA, Hsueh HM, Chen JJ. Estimation of false discovery rates in multiple testing: application to gene microarray data. Biometrics. 2003;59:1071–1081. doi: 10.1111/j.0006-341x.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 41.Van den Oord EJCG. Controlling false discoveries in candidate gene studies. Mol Psychiatry. 2005;10:230–231. doi: 10.1038/sj.mp.4001581. [DOI] [PubMed] [Google Scholar]
- 42.Sabatti C, Service S, Freimer N. False discovery rate in linkage and association genome screens for complex disorders. Genetics. 2003;164:829–833. doi: 10.1093/genetics/164.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storey J, Tibshirani R. Statistical significance for genome-wide studies. Proceedings of the National Academy of Sciences. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bukszar J, McClay JL, van den Oord EJ. Estimating the posterior probability that genomewide association findings are true or false. Bioinformatics. 2009;25:1807–1813. doi: 10.1093/bioinformatics/btp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glonek G, Soloman P. Discussion of resampling-based multiple testing for microarray data analysis by Ge, Dudoit and Speed. Test. 2003;12:1–77. [Google Scholar]
- 46.Moens CB, Selleri L. Hox cofactors in vertebrate development. Dev Biol. 2006;291:193–206. doi: 10.1016/j.ydbio.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Hwang CK, D'Souza UM, Lee SH, Junn E, Mouradian MM. Three-amino acid extension loop homeodomain proteins Meis2 and TGIF differentially regulate transcription. J Biol Chem. 2000;275:20734–20741. doi: 10.1074/jbc.M908382199. [DOI] [PubMed] [Google Scholar]
- 48.Smith JE, Afonja O, Yee HT, Inghirami G, Takeshita K. Chromosomal mapping to 15q14 and expression analysis of the human MEIS2 homeobox gene. Mamm Genome. 1997;8:951–952. doi: 10.1007/s003359900621. [DOI] [PubMed] [Google Scholar]
- 49.Hui H, Perfetti R. Pancreas duodenum homeobox-1 regulates pancreas development during embryogenesis and islet cell function in adulthood. Eur J Endocrinol. 2002;146:129–141. doi: 10.1530/eje.0.1460129. [DOI] [PubMed] [Google Scholar]
- 50.Swift GH, Liu Y, Rose SD, et al. An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2) Mol Cell Biol. 1998;18:5109–5120. doi: 10.1128/mcb.18.9.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, MacDonald RJ, Swift GH. DNA binding and transcriptional activation by a PDX1.PBX1b.MEIS2b trimer and cooperation with a pancreas-specific basic helix-loop-helix complex. J Biol Chem. 2001;276:17985–17993. doi: 10.1074/jbc.M100678200. [DOI] [PubMed] [Google Scholar]
- 52.Nikkila H, McMillan DR, Nunez BS, Pascoe L, Curnow KM, White PC. Sequence similarities between a novel putative G protein-coupled receptor and Na+/Ca2+ exchangers define a cation binding domain. Mol Endocrinol. 2000;14:1351–1364. doi: 10.1210/mend.14.9.0511. [DOI] [PubMed] [Google Scholar]
- 53.McMillan DR, Kayes-Wandover KM, Richardson JA, White PC. Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J Biol Chem. 2002;277:785–792. doi: 10.1074/jbc.M108929200. [DOI] [PubMed] [Google Scholar]
- 54.Scheel H, Tomiuk S, Hofmann K. A common protein interaction domain links two recently identified epilepsy genes. Hum Mol Genet. 2002;11:1757–1762. doi: 10.1093/hmg/11.15.1757. [DOI] [PubMed] [Google Scholar]
- 55.Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levy FO, Oyen O, Sandberg M, et al. Molecular cloning, complementary deoxyribonucleic acid structure and predicted full-length amino acid sequence of the hormone-inducible regulatory subunit of 3′-5′-cyclic adenosine monophosphate-dependent protein kinase from human testis. Mol Endocrinol. 1988;2:1364–1373. doi: 10.1210/mend-2-12-1364. [DOI] [PubMed] [Google Scholar]
- 57.Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 58.Czyzyk TA, Sikorski MA, Yang L, McKnight GS. Disruption of the RIIbeta subunit of PKA reverses the obesity syndrome of Agouti lethal yellow mice. Proc Natl Acad Sci U S A. 2008;105:276–281. doi: 10.1073/pnas.0710607105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porcellini A, Messina S, De Gregorio G, et al. The expression of the thyroid-stimulating hormone (TSH) receptor and the cAMP-dependent protein kinase RII beta regulatory subunit confers TSH-cAMP-dependent growth to mouse fibroblasts. J Biol Chem. 2003;278:40621–40630. doi: 10.1074/jbc.M307501200. [DOI] [PubMed] [Google Scholar]
- 60.Konneker T, Barnes T, Furberg H, Losh M, Bulik CM, Sullivan PF. A searchable database of genetic evidence for psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:671–675. doi: 10.1002/ajmg.b.30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schadt EE, Molony C, Chudin E, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci U S A. 1997;94:12157–12161. doi: 10.1073/pnas.94.22.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanaya H, Takeya R, Takeuchi K, Watanabe N, Jing N, Sumimoto H. Fhos2, a novel formin-related actin-organizing protein, probably associates with the nestin intermediate filament. Genes Cells. 2005;10:665–678. doi: 10.1111/j.1365-2443.2005.00867.x. [DOI] [PubMed] [Google Scholar]
- 64.Arranz MJ, de Leon J. Pharmacogenetics and pharmacogenomics of schizophrenia: a review of last decade of research. Mol Psychiatry. 2007;12:707–747. doi: 10.1038/sj.mp.4002009. [DOI] [PubMed] [Google Scholar]
- 65.Need AC, Keefe RS, Ge D, et al. Pharmacogenetics of antipsychotic response in the CATIE trial: a candidate gene analysis. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Hellard S, Theisen FM, Haberhausen M, et al. Association between the insulin-induced gene 2 (INSIG2) and weight gain in a German sample of antipsychotic-treated schizophrenic patients: perturbation of SREBP-controlled lipogenesis in drug-related metabolic adverse effects? Mol Psychiatry. 2009;14:308–317. doi: 10.1038/sj.mp.4002133. [DOI] [PubMed] [Google Scholar]
- 67.Choquet H, Cavalcanti-Proenca C, Lecoeur C, et al. The T-381C SNP in BNP gene may be modestly associated with type 2 diabetes: an updated meta-analysis in 49 279 subjects. Hum Mol Genet. 2009;18:2495–2501. doi: 10.1093/hmg/ddp169. [DOI] [PubMed] [Google Scholar]
- 68.Lindgren CM, Heid IM, Randall JC, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prokopenko I, Langenberg C, Florez JC, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sandhu MS, Waterworth DM, Debenham SL, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371:483–491. doi: 10.1016/S0140-6736(08)60208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wallace C, Newhouse SJ, Braund P, et al. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia. Am J Hum Genet. 2008;82:139–149. doi: 10.1016/j.ajhg.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.