

Abstract
Aged nonhuman primates accumulate large amounts of human-sequence amyloid β (Aβ) in the brain, yet they do not manifest the full phenotype of Alzheimer’s disease (AD). To assess the biophysical properties of Aβ that might govern its pathogenic potential in humans and nonhuman primates, we incubated the benzothiazole imaging agent Pittsburgh compound B (PIB) with cortical tissue homogenates from normal aged humans, humans with AD, and from aged squirrel monkeys, rhesus monkeys, and chimpanzees with cerebral Aβ-amyloidosis. Relative to humans with AD, high-affinity PIB binding is markedly reduced in cortical extracts from aged nonhuman primates containing levels of insoluble Aβ similar to those in AD. The high-affinity binding of PIB may be selective for a pathologic, human-specific conformation of multimeric Aβ, and thus could be a useful experimental tool for clarifying the unique predisposition of humans to Alzheimer’s disease.
INDEXING TERMS: Alzheimer’s disease, Aβ, benzothiazole, beta-amyloid, cerebral amyloid angiopathy, imaging, nonhuman primates, Pittsburgh Compound B, senile plaques
1. INTRODUCTION
Alzheimer’s disease (AD) is an age-associated neurodegenerative disorder characterized by progressive dementia and the proliferation of senile plaques and neurofibrillary tangles in the brain (Cummings, 2004). While the number of neurofibrillary tangles correlates strongly with clinical measures of dementia (Wilcock and Esiri, 1982, Crystal et al., 1988, Arriagada et al., 1992, Giannakopoulos et al., 2007), genetic, biochemical, and pathologic data support the Aβ-cascade hypothesis, which holds that the intracerebral accumulation of multimeric Aβ is a key early event in the pathogenesis of AD (Hardy and Selkoe, 2002). However, the deposition of Aβ in the human brain is not invariably associated with frank dementia, inasmuch as abundant Aβ lesions sometimes occur in aged humans in the absence of overt cognitive impairment (Crystal et al., 1988, Bennett et al., 2006).
Nonhuman primates and other mammals produce Aβ that is identical in amino acid sequence to human Aβ, and many species naturally accumulate senile plaques and cerebral β-amyloid angiopathy (CAA) with age (Walker and Cork, 1999, LeVine III and Walker, 2006). However, no nonhuman species has been shown to exhibit the full behavioral or pathological characteristics of AD (Rosen et al., 2008). In light of the substantial evidence for a key role of Aβ in the etiology of AD, these findings, along with in vitro and in vivo studies (Petkova et al., 2005, Meyer-Luehmann et al., 2006), can be reconciled by the possibility that aggregated Aβ takes the form of polymorphic molecular strains, some of which are more pathogenic than others (LeVine III and Walker, 2008). The possible existence of functionally heterogeneous protein polymorphs has important implications for understanding the pathogenesis of neurodegenerative disorders such as AD, and for developing specific diagnostic and therapeutic agents. The identification of molecular probes for pathogenic features of Aβ and related molecules will help to address this issue (LeVine III and Walker, 2008).
Pittsburgh compound B (PIB), a synthetic, radiolabeled benzothiazole ligand based on the chemical structure of the amyloid dye Thioflavin-T, has been developed for imaging Aβ deposits in vivo by positron-emission tomography (PET) (Klunk et al., 2004, Nordberg, 2008). Radiolabeled PIB binds with high-affinity and high specificity, at concentrations typically achieved in imaging (~1 nM), to Aβ plaques and CAA (Klunk et al., 2003b, Bacskai et al., 2007, Johnson et al., 2007, Leinonen et al., 2008), but only weakly to neurofibrillary tangles and Lewy Bodies (Klunk et al., 2003b, Fodero-Tavoletti et al., 2007, Ikonomovic et al., 2008, Ye et al., 2008). In vitro studies have identified high- and low-affinity binding sites on Aβ assemblies (Klunk et al., 2005, Ye et al., 2005). The stoichiometry of high-affinity PIB binding in AD brain homogenates indicates that PIB binds directly to Aβ amyloid, with more than 500 binding sites per 1000 molecules of Aβ. However, PIB binds at high-affinity with significantly lower stoichiometry (fewer than one binding site per 1000 Aβ molecules) insynthetic Aβ fibril preparations, as well as in deposit-rich, β-amyloid precursor protein (APP)-transgenic mouse brain (Klunk et al., 2005, Toyama et al., 2005, Maeda et al., 2007, Serdons et al., 2009). With a Kd of 1–2 nM, only the high-affinity PIB binding sites in cerebral Aβ deposits are significantly occupied at the ligand concentrations achieved in PET scans (Mathis et al., 2004).
Several groups have demonstrated only negligible 11C-PIB or 18F-PIB uptake in microPET experiments with APP-transgenic mouse models (Klunk et al., 2005, Toyama et al., 2005, Serdons et al., 2009). Because APP-transgenic mice lack the profound neurodegeneration and cognitive dysfunction seen in AD patients (Dodart and May, 2005, LeVine III and Walker, 2006), these findings suggest that PIB has the potential to differentiate between pathogenic (AD-related) and relatively benign forms of multimeric Aβ, and possibly to reveal structural characteristics that render Aβ especially toxic in the AD brain. Most current transgenic mouse models express artificially high levels of mutant APP, often coexpressed with mutant presenilin (Dodart and May, 2005). The majority of these models produce human-type Aβ in the context of endogenous murine Aβ, which is relatively refractory to aggregation and, when co-aggregated with synthetic human-sequence Aβ, reduces in vitro PIB binding (Otvos, Jr. et al., 1993, Ye et al., 2006). Furthermore, in APP-transgenic mouse models, the Aβ deposits lack many of the post-translational modifications that contribute to the insolubility of the lesions in humans (Kuo et al., 2001). For these reasons, it is important to evaluate PIB binding in longer-lived animal models that naturally form deposits of human-sequence Aβ, such as aged nonhuman primates. We hypothesized that the differential pathogenicity of multimeric Aβ in humans and nonhuman primates is related to structural variations in the molecule that can be distinguished by the high-affinity binding of PIB. Here we present a quantitative analysis of 3H-PIB binding in postmortem cortical homogenates from AD patients, nondemented elderly humans, aged chimpanzees, rhesus macaques, and squirrel monkeys. Despite levels of Aβ that sometimes exceeded those in AD, high-affinity 3H-PIB binding in nonhuman primates was strikingly less than that in humans with AD, suggesting that PIB might serve as a selective probe for human-specific molecular markers of AD.
2. METHODS
2.1. Subjects
We analyzed postmortem brain tissue from 9 rhesus monkeys (Macaca mulatta) (3 females, 6 males), 6 squirrel monkeys (Saimiri sciureus) (1 female, 5 males), 3 female chimpanzees (Pan troglodytes), 9 humans with end-stage AD (6 females, 3 males), and 3 nondemented elderly humans (2 females, 1 male) (Table 1). Human tissues were obtained from the Emory University Alzheimer’s Disease Research Center Brain Bank in accordance with federal and institutional guidelines, and were coded to ensure the anonymity of subjects. Animal tissues were collected in accordance with federal and institutional guidelines for the humane care and use of experimental animals. The Yerkes Center is fully accredited by AAALAC International.
Table 1.
Case list
| Group | Case | Δ Age(y) | Sex | § PMI(h) | Braak Stage | * ApoE |
|---|---|---|---|---|---|---|
| ND Human | E04-46 | 40 | m | 31 | Braak 0 | 3/4 |
| E04-34 | 57 | f | 17 | Braak 0 | 3/3 | |
| OS02-35 | 75 | f | 6 | Braak 0 | 3/3 | |
| Human AD | E04-33 | 57 | f | 20 | Braak V/VI | 3/4 |
| E04-172 | 87 | f | 6 | Braak V/VI | 3/4 | |
| OS03-300 | 75 | f | 12 | Braak V/VI | 4/4 | |
| OS02-159 | 61 | m | 5.5 | Braak V/VI | 3/4 | |
| OS01-128 | 91 | f | 2.5 | Braak V/VI | 3/4 | |
| OS02-106 | 81 | f | 2 | Braak >IV | 3/3 | |
| E05-87 | 61 | m | 4 | Braak V/VI | 3/4 | |
| E08-41 | 84 | m | 4.5 | Braak VI | 3/4 | |
| E05-04 | 64 | f | 4.5 | Braak VI | 3/4 | |
| Chimpanzee | YN06-108Pt | 44 | f | 3 | ||
| YN07-25Pt | 47 | f | 1 | |||
| YN05-400Pt | 41 | f | 1 | |||
| Rhesus macaque | 06-1Mm | 35 | f | <3 | ||
| AM109 | 26.6 | m | 1 | |||
| AM120 | 26.5 | f | 1 | |||
| 554 | 38 | m | <3 | |||
| 1201 | 35 | m | <3 | |||
| 1203 | 33 | f | <3 | |||
| 1210 | 30 | m | <3 | |||
| 1211 | 25 | f | <3 | |||
| 1313 | >20 | m | <3 | |||
| Squirrel monkey | 84L | 21 | m | 1 | ||
| 83GO | 20 | m | 1 | |||
| 86J | 17 | m | 1 | |||
| 06-5Ss | 23 | f | <3 | |||
| 90T | 17 | m | 1 | |||
| 92AH | 15 | m | 1 |
Age (y): Age (years)
PMI (h): Postmortem interval (hours)
ApoE: Apolipoprotein E genotype
2.2. Preparation of tissue samples
For quantitative biochemical analyses, unfixed, fresh-frozen temporal and occipital cortical tissue blocks were weighed, Dounce-homogenized in 5 volumes of homogenization buffer (50mM Tris-HCl and 150mM NaCl, pH 7.5, containing complete protease inhibitor tablets [Santa Cruz Biochemicals, Santa Cruz, CA, USA]), and stored at −80°C until use. For immunohistochemistry, brains were fixed for at least 7 days in 4%, 0.1M Phosphate-buffered saline (PBS)-buffered paraformaldehyde at 4°C. Temporal and occipital cortical blocks from the contralateral hemisphere were paraffin-embedded, sectioned at 8μm thickness and mounted onto silanized slides. For autoradiography and immunohistochemistry, cryosections from unfixed temporal cortical blocks were cut at 10μm, mounted onto silanized slides, and stored in air-tight containers at −80°C until use.
2.3. ELISA quantification of cortical Aβ40 and Aβ42
Cortical homogenates were centrifuged at 100,000g for 60 minutes at 4°C in a TLA 100.4 rotor (Beckman Coulter, Fullerton, CA, USA). The resulting buffer-insoluble pellet was probe-sonicated in 70% formic acid and centrifuged at 16,110g for 60 minutes at 4°C, and the supernatant containing formic acid-solubilized, buffer-insoluble Aβ was retained. Formic acid extracts were neutralized with 1.0M Tris base, pH 11 (1:20 dilution) and diluted in sample buffer. Aβ ending at amino acids 40 or 42 (Aβx-40 and Aβx-42) was measured in each extract by ELISA, using C-terminal specific capture antibodies and an N-terminal specific detection antibody according to the manufacturer’s instructions (The Genetics Company, Schlieren, Switzerland). Aβ content is expressed relative to wet weight of tissue. All samples were assayed in duplicate. After stopping the tetramethylbenzidine-peroxidase reaction with sulfuric acid, plates were read at 450 nm on a Biotek Synergy HT Multidetection plate reader (Biotek, Winooski, VT, USA).
2.4. 3H-PIB binding assay
PIB binding assays were conducted on the same tissue samples as were the Aβ ELISAs. Cortical homogenates were prepared as described above, at an original concentration of 167 mg wet tissue weight/ml. Homogenates were further diluted 1:33.3 in 0.1M PBS (pH 7.4) to a final concentration of 5mg wet weight/ml. In a 96 well polypropylene plate, 20μl of diluted homogenate (100μg wet tissue weight) were added to each of duplicate wells. 200μl of 1 nM 3H-PIB (SA=82 Ci/mmol, custom synthesis, GE Healthcare, UK) were quickly added to each well. Samples were incubated at ambient temperature for 2.5 hours, without shaking, transferred to a 96 well Multiscreen HTS Hi Flow FB filter plate, and filtered with a vacuum manifold (Millipore Corporation, Bedford, MA, USA). Nonspecific binding was defined as the counts retained in the presence of 1 μM unlabeled PIB. Kd and Bmax values were determined for all groups using a competition binding assay with a constant concentration of 1.2nM 3H-PIB and concentrations of unlabeled PIB between 0.1 nM and 1.0 μM. Filters were rapidly washed with 4 × 200μl of PBS at room temperature and dried on the manifold, after which 50μl of MicroScint 20 scintillation fluid (PerkinElmer, Waltham, MA, USA) was added to each well. After 24 hours incubation on an orbital shaker, 3H-PIB binding was quantified in a TopCount scintillation counter (PerkinElmer) and specific binding was calculated by subtracting nonspecific counts per minute (CPM). Using 1 nM 3H-PIB, ligand binding was linear between 25 and 150μg wet weight AD tissue per well. CPMs were converted to femtomoles using an experimentally-determined 15% counting efficiency in the scintillation counter. Similar results were obtained when the filters were placed in vials and counted with scintillation fluid. Finally, to determine whether species-specific molecules might enhance or inhibit 3H-PIB binding, we performed this in vitro binding assay using 1:1 mixtures of AD and nonhuman primate cortical homogenates.
2.5. Immunohistochemistry
The following antibodies were used for immunohistochemistry: Monoclonal antibodies 6E10 and 4G8 to residues 3–8 and 17–24 of the Aβ peptide, respectively (both at 1:5000; Covance Research Products, Denver, PA, USA); rabbit polyclonal antibodies R361 and R398 to C-terminal residues 32–40 and 33–42 of Aβ40 and Aβ42, respectively (both at 1:15,000; provided by Dr. Pankaj Mehta, Institute for Basic Research on Developmental Disabilities, Staten Island, NY, USA); monoclonal antibodies CP13 to phospho-tau 202 (1:10,000), PHF1 to phospho-tau 396/404 (1:10,000), and MC1 to aggregated tau (1:10,000) (all provided by Dr. Peter Davies, Albert Einstein College of Medicine, Bronx, NY, USA); and monoclonal antibody AT8 to phospho-tau 202/205 (1:5000; Covance) (Rosen et al., 2008). Vectastain Elite kits (Vector Laboratories, Burlingame, CA, USA) were used for avidin-biotin complex (ABC)-based horseradish peroxidase immunodetection of antigen-antibody complexes.
Endogenous peroxidase was inactivated with 3% H2O2 in methanol, and nonspecific reagent binding was blocked with 2% normal horse serum (for monoclonals) or normal goat serum (for polyclonals) in 0.2% Tween 20 in PBS (blocking solution) for one hour at room temperature. For Aβ-immunodetection, sections were pretreated for 10 minutes with 100% formic acid to expose antigenic sites and then incubated in primary antibody (diluted in blocking solution) overnight at 4°C. After rinsing, sections were placed for one hour at room temperature in biotinylated secondary antibody (1:200 in blocking solution), rinsed, immersed for 30 minutes in avidin-biotin complex, and then developed with 3,3′-diaminobenzidine (DAB) (Vector Laboratories). Tissue from pathologically verified human AD cases was used as positive control material, and non-immune mouse IgG or rabbit serum was used in place of the primary antibodies as negative controls. In some instances, a hematoxylin counterstain was applied after immunostaining.
Aβ load was assessed histopathologically in 6E10-immunostained sections of temporal and occipital cortices. For each section, two researchers rated the levels of diffuse plaques, compact plaques, and CAA in capillaries and large vessels (+++ frequent, ++ moderate, + rare, − absent) (Lockhart et al., 2007).
2.6. 3H-PIB Autoradiography and Immunohistochemistry
Cryosections were thawed for 10 minutes, immersed in 10% ethanol for 20 minutes, and incubated for 1 hour in 1.0nM 3H-PIB in 5% ethanol/PBS, at room temperature. Nonspecific binding was determined in the presence of 1.0μM unlabeled PIB. Sections were then quickly rinsed twice with 10% ethanol and twice with deionized water, both at 4°C. After drying under a cold air stream, sections were directly apposed to Hyperfilm 3H (GE Amersham, UK). After a 2-week exposure, film was developed with D19 developer solution (Kodak, New Haven, CT) and images were captured with a QICAM digital camera (QImaging, BC, Canada). Except for size cropping, images were not digitally manipulated prior to publication.
For confirmation of Aβ deposition in the regions analyzed by autoradiography, adjacent cryosections were fixed for 30 minutes in 70% ethanol at room temperature and immunohistochemistry with antibody 6E10 was performed as described above, except that the sections were incubated in 100% formic acid for 2 minutes.
2.7. Statistical analysis
Analysis of variance (ANOVA) was used to determine group differences in insoluble Aβ levels between AD and the three nonhuman primate groups. ANOVA also was used to assess potential group differences in PIB binding among the nonhuman primates. Because PIB binding did not differ significantly among the three nonhuman primate species in either the temporal or occipital cortical samples, and because of the small number of samples in some groups, we combined the nonhuman primate cases for subsequent analyses, in which we conducted nonparametric Mann-Whitney U tests (two-tailed, CI=95%) to compare PIB binding between humans with AD and nonhuman primates. We also employed nonparametric t-tests (Mann-Whitney) to compare PIB binding between AD and nondemented human cases and between nondemented human cases and nonhuman primates. For the determination of PIB binding site characteristics in cortical homogenates, we conducted a curve-fit analysis of the displacement binding data using a nonlinear homologous competition equation. To assess the relationship between PIB binding and insoluble Aβ in the temporal and occipital cortices of AD cases, we applied the two-tailed Pearson product-moment correlation coefficient (Pearson’s r, CI=95%). For the homogenate “mixing experiments”, we used ANOVA followed by post-hoc t-tests to detect possible synergistic effects of mixing cortical homogenates on PIB binding levels.
3. RESULTS
3.1. Aβ and tau pathology in human and nonhuman primate brain
Immunohistochemistry w0ith antibodies to Aβ revealed species-typical Aβ-immunoreactivity patterns in cortical sections from every AD and aged nonhuman primate subject examined in this study, whereas the nondemented humans were largely devoid of Aβ-lesions (Table 2). Humans with AD showed abundant parenchymal Aβ deposits, as well as occasional CAA, whereas the aged nonhuman primates displayed relatively more CAA accompanied by parenchymal Aβ deposits. Immunohistochemistry using antibodies to pathological forms of tau revealed widespread neurofibrillary tangles in all AD cases examined. We detected little or no aberrant tau immunoreactivity in the nondemented humans or in the nonhuman primates, with the exception of one aged chimpanzee (Y05-400Pt) that presented with tauopathy in addition to moderate CAA, as previously described (Rosen et al., 2008).
Table 2.
Histopathological assessment of Aβ-plaques and cerebral amyloid angiopathy
| Temporal Cortex |
Occipital Cortex |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group | Case | DP | CP | CaAA | LVAA | DP | CP | CaAA | LVAA |
| ND Human | E04-46 | − | − | − | − | − | − | − | + |
| E04-34 | − | − | − | − | − | − | − | − | |
| OS02-35 | − | − | − | − | − | − | − | − | |
| Human AD | E04-33 | ++ | +++ | ++ | + | ++ | +++ | + | + |
| E04-172 | +++ | +++ | ++ | ++ | ++ | +++ | + | + | |
| OS03-300 | +++ | +++ | + | ++ | +++ | +++ | ++ | ++ | |
| OS02-159 | +++ | +++ | ++ | ++ | +++ | +++ | ++ | ++ | |
| OS01-128 | +++ | + | + | − | +++ | +++ | ++ | ++ | |
| OS02-106 | ++ | ++ | + | + | ++ | ++ | ++ | ++ | |
| E05-87 | +++ | +++ | + | ++ | n/a | n/a | n/a | n/a | |
| E08-41 | +++ | +++ | + | + | n/a | n/a | n/a | n/a | |
| E05-04 | ++ | +++ | − | + | n/a | n/a | n/a | n/a | |
| Chimpanzee | YN06-108Pt | − | − | + | ++ | − | − | ++ | ++ |
| YN07-25Pt | − | + | + | + | n/a | n/a | n/a | n/a | |
| YN05-400Pt | − | + | ++ | + | n/a | n/a | n/a | n/a | |
| Rhesus macaque | 06-1Mm | ++ | + | + | + | +++ | ++ | +++ | ++ |
| AM109 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | |
| AM120 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | |
| 554 | n/a | n/a | n/a | n/a | ++ | − | + | − | |
| 1201 | n/a | n/a | n/a | n/a | +++ | ++ | ++ | ++ | |
| 1203 | n/a | n/a | n/a | n/a | ++ | + | ++ | ++ | |
| 1210 | n/a | n/a | n/a | n/a | + | − | − | + | |
| 1211 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | |
| 1313 | n/a | n/a | n/a | n/a | + | − | ++ | − | |
| Squirrel monkey | 84L | ++ | ++ | +++ | + | − | − | + | ++ |
| 83GO | − | ++ | ++ | ++ | ++ | + | ++ | +++ | |
| 86J | +++ | ++ | ++ | + | n/a | n/a | n/a | n/a | |
| 06-5Ss | +++ | ++ | ++ | + | − | − | ++ | + | |
| 90T | ++ | +++ | ++ | ++ | ++ | − | + | ++ | |
| 92AH | ++ | + | ++ | + | − | − | − | + | |
DP: Diffuse plaques
CP: Compact plaques
CaAA: Capillary amyloid angiopathy
LVAA: Large vessel amyloid angiopathy
n/a: Not available
3.2. Quantification of Aβ40 and Aβ42
Because previous studies have shown that PIB binding positively correlates with the levels of buffer-insoluble, but not soluble Aβ (Klunk et al., 2003a, Klunk et al., 2005, Bacskai et al., 2007, Ikonomovic et al., 2008, Svedberg et al., 2008), our quantitative analysis focused on insoluble Aβ, which constitutes the great majority of the protein in senile plaque cores and CAA. We found that Aβ40 and Aβ42 accumulate in the aged nonhuman primate brain to similar or higher levels than in the end-stage AD brain; by ELISA of temporal cortical samples, aged squirrel monkeys had higher mean levels of total insoluble Aβ than did the AD cases (365.6 fmol/100μg wet tissue [n=6, SD=272.48 fmol] vs. 253.58 fmol/100μg [n=9, SD=172.91 fmol], respectively). Mean levels of total Aβ in nondemented human (n=3), rhesus macaque (n=1) and chimpanzee (n=3) temporal cortex were 0.41 fmol/100μg wet tissue (SD=0.51 fmol), 349.95 fmol/100μg, and 70.43 fmol/100μg (SD=42.76 fmol), respectively (Figure 1). In the occipital cortex, total insoluble Aβ levels in nonhuman primates were consistently higher than in AD (AD mean=194.49 fmol/100μg wet tissue [n=6, SD=81.02 fmol], chimpanzee mean=245.62 fmol/100μg [n=2, SD=257.7 fmol], rhesus macaque mean=235.06 fmol/100μg [n=9, SD=417.68 fmol], squirrel monkey mean=573.01 fmol/100μg [n=6, SD=624.28 fmol]) (Figure 1). Statistically, however, Aβ levels did not differ significantly between the AD cases and the nonhuman primates in either cortical region, probably because of the high inter-individual variability in the groups.
Figure 1. ELISA quantitation of insoluble Aβx-40 and Aβx-42 in temporal (A) and occipital (B) cortical homogenates from aged humans and nonhuman primates.

A: Insoluble Aβ in temporal cortex (ND, n=3; AD, n=9; Chimpanzee, n=3; Rhesus macaque, n=1; Squirrel monkey, n=6). Mean Aβ42 levels were higher than Aβ40 levels in the temporal cortex of all 4 groups. B: Insoluble Aβ in occipital cortex (ND, n=3; AD, n=6; Chimpanzee, n=2; Rhesus macaque, n=9; Squirrel monkey, n=6). In nonhuman primates, the occipital cortical Aβ40:Aβ42 ratio was somewhat higher than in AD. However, no statistically significant differences in Aβ levels were detected between AD and nonhuman primate groups in either cortical region. ND: Nondemented human; AD: Alzheimer’s disease; Pt: Pan troglodytes (Chimpanzee); Mm: Macaca mulatta (Rhesus macaque); Ss: Saimiri sciureus (Squirrel monkey). Bars = standard deviations.
Because a predominance of the Aβ42 isoform has been implicated in the toxicity and aggregability of the peptide in AD brain (Hasegawa et al., 1999, McGowan et al., 2005), we calculated the Aβ40:Aβ42ratios for each case and cortical region. Overall, Aβ42 was more abundant than was Aβ40 in both cortical regions of the humans and nonhuman primates, although the relative levels of Aβ40 in the occipital cortex were somewhat higher in the nonhuman primates (Figure 1). The Aβ40:Aβ42 ratio was less than 1.0 in every AD case examined, and exceeded 1.0 in at least one cortical region of only four nonhuman primates (YN06-108Pt, 1201, 84L and 83G0; see Table 3).
Table 3.
Insoluble Aβ levels and PIB binding(fmol/100μg wet weight tissue)
| Temporal Cortex |
Occipital Cortex |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group | Case | Aβ40 | Aβ42 | Total Aβ | PIB binding | Aβ40 | Aβ42 | Total Aβ | PIB binding |
| ND Human | E04-46 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 |
| E04-34 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |
| OS02-35 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | |
| Human AD | E04-33 | 3 | 83 | 86 | 17 | 2 | 211 | 213 | 31 |
| E04-172 | 8 | 217 | 225 | 17 | 11 | 176 | 187 | 18 | |
| OS03-300 | 62 | 98 | 160 | 26 | 5 | 187 | 192 | 34 | |
| OS02-159 | 40 | 175 | 215 | 23 | 15 | 298 | 313 | 43 | |
| OS01-128 | 18 | 196 | 214 | 35 | 10 | 191 | 202 | 24 | |
| OS02-106 | 1 | 146 | 146 | 10 | 0 | 59 | 60 | 24 | |
| E05-87 | 275 | 353 | 628 | 26 | n/a | n/a | n/a | n/a | |
| E08-41 | 4 | 156 | 160 | 25 | n/a | n/a | n/a | n/a | |
| E05-04 | 4 | 445 | 449 | 22 | n/a | n/a | n/a | n/a | |
| Chimpanzee | YN06-108Pt | 15 | 93 | 109 | 3 | 249 | 179 | 428 | 0 |
| YN07-25Pt | 0 | 24 | 24 | 1 | 1 | 62 | 63 | 0 | |
| YN05-400Pt | 10 | 69 | 79 | 0 | n/a | n/a | n/a | n/a | |
| Rhesus macaque | 06-1Mm | 100 | 250 | 350 | 2 | 15 | 190 | 205 | 2 |
| AM109 | n/a | n/a | n/a | n/a | 0 | 29 | 29 | 2 | |
| AM120 | n/a | n/a | n/a | n/a | 0 | 18 | 19 | 2 | |
| 554 | n/a | n/a | n/a | n/a | 7 | 24 | 31 | 0 | |
| 1201 | n/a | n/a | n/a | n/a | 843 | 487 | 1330 | 0 | |
| 1203 | n/a | n/a | n/a | n/a | 10 | 228 | 238 | 2 | |
| 1210 | n/a | n/a | n/a | n/a | 0 | 104 | 105 | 2 | |
| 1211 | n/a | n/a | n/a | n/a | 2 | 90 | 93 | 0 | |
| 1313 | n/a | n/a | n/a | n/a | 0 | 67 | 67 | 0 | |
| Squirrel monkey | 84L | 367 | 230 | 597 | 1 | 396 | 618 | 1014 | 3 |
| 83GO | 497 | 252 | 750 | 0 | 1121 | 508 | 1629 | 3 | |
| 86J | 8 | 91 | 99 | 3 | 1 | 174 | 176 | 3 | |
| 06-5Ss | 19 | 318 | 337 | 1 | 26 | 370 | 396 | 1 | |
| 90T | 3 | 357 | 360 | 1 | 43 | 178 | 221 | 0 | |
| 92AH | 0 | 51 | 52 | 1 | 0 | 2 | 2 | 3 | |
n/a: Not available
3.3. In vitro 3H-PIB binding
The binding of one nanomolar 3H-PIB was strikingly less in cortical homogenates from all nonhuman primates compared to samples from the AD cases (temporal cortex: p<0.0001; occipital cortex: p=0.0004)(Figure 2). We found high levels of specific PIB binding in the AD cases (temporal cortex: mean = 22.48 fmol/100μg wet tissue [SD=6.91], occipital cortex: mean = 29.01 fmol/100μg wet tissue [SD=8.93]) that correlated positively, but not significantly, with the amount of Aβ measured by ELISA (temporal cortex, r=0.3417, p=0.3682; occipital cortex, r=0.7088, p=0.1149) (Figure 3). PIB binding in the AD cases was consistently greater in occipital cortex than in temporal cortex, despite similar or lower levels of Aβ in this region by ELISA (Figures 1, 2). Nonlinear regression analysis of the homologous competition binding data reveals a Kd of 3.0 nM and Bmax of 209.28 fmol/100ug wet tissue for high-affinity PIB binding sites in AD temporal cortex, and a Kd of 3.0nM and Bmax of 234.48 fmol/100ug wet tissue for the ligand in AD occipital cortex (Figure 4). We were unable to detect a high-affinity binding site in cortical homogenates from any of the three nonhuman primate groups.
Figure 2. Postmortem quantification of 3H-PIB binding in cortical homogenates from aged humans and nonhuman primates.

Despite comparable mean levels of insoluble Aβ, 3H-PIB (1 nM) binding is low to undetectable in aged nonhuman primate temporal (A) and occipital (B) cortices compared to the same regions from humans with Alzheimer’s disease. (AD vs. nonhuman primates, temporal cortex: p<0.0001, occipital cortex: p=0.0004). Note also the low binding of PIB in nondemented control humans (ND vs. AD, temporal cortex: p=0.0091, occipital cortex: p=0.0238). No significant difference in PIB binding was detected between nondemented humans and nonhuman primates. [Temporal cortex: ND, n= 3; AD, n=9; Chimpanzee, n=3; Rhesus macaque, n=1; Squirrel monkey, n=6. Occipital cortex: ND, n=3; AD, n=6; Chimpanzee, n=2; Rhesus macaque, n=9; Squirrel monkey, n=6]. ND: Nondemented human; AD: Alzheimer’s disease; Pt: Pan troglodytes (Chimpanzee); Mm: Macaca mulatta (Rhesus macaque); Ss: Saimiri sciureus (Squirrel monkey). Bars = means.
Figure 3. Cortical homogenate mixing experiments do not indicate a species-specific factor that modulates 3H-PIB binding in aged humans or nonhuman primates.

Homogenates from AD temporal (A) and occipital (B) cortices (100μg tissue) were incubated with temporal and occipital cortical homogenates (100μg tissue) from nondemented humans, chimpanzees, rhesus macaques, and squirrel monkeys in an in vitro 3H-PIB binding assay. The amount of ligand binding to homogenate mixtures approximately equaled the sum of 3H-PIB binding to each mixture component, indicating an absence of species-specific, auxiliary factors that enhance or suppress PIB binding. ND: Nondemented human; AD: Alzheimer’s disease; Pt: Pan troglodytes (Chimpanzee); Mm: Macaca mulatta (Rhesus macaque); Ss: Saimiri sciureus (Squirrel monkey). Bars = Standard deviations.
Figure 4. Relationship between insoluble Aβ levels and 3H-PIB binding in AD cortical homogenates.

Total levels of insoluble Aβ (Aβ40 and Aβ42) correlate positively with 3H-PIB binding to AD temporal and occipital cortical homogenates. The correlation was not statistically significant in either cortical region, however (temporal cortex, r=0.3417, p=0.3682; occipital cortex, r=0.7088, p=0.1149).
In the nonhuman primates, only background PIB binding was observed in temporal and occipital cortices of all subjects examined (Table 2; temporal cortex: mean = 1.41 fmol/100μg wet tissue [SD=0.93], occipital cortex: mean = 1.33 fmol/100μg [SD=1.15]), even though the levels of total Aβ in most simians were at least as high as those in the AD brain (Figure 1). In the cerebellar cortex, which is relatively unaffected by Aβ plaque deposition or neurodegeneration, only background levels of 3H-PIB binding were detected in the AD cases (data not shown).
To address the possibility that species-specific molecules might enhance or inhibit 3H-PIB binding, we performed the same in vitro binding assay with 1:1 mixtures of AD and nonhuman primate cortical homogenates. There was only an additive effect of combining homogenates from AD cases with those from the nonhuman primates (Figure 5), indicating an absence of species-specific binding-modulatory factors.
Figure 5. Analysis of high-affinity 3H-PIB binding in AD and chimpanzee cortical homogenate.
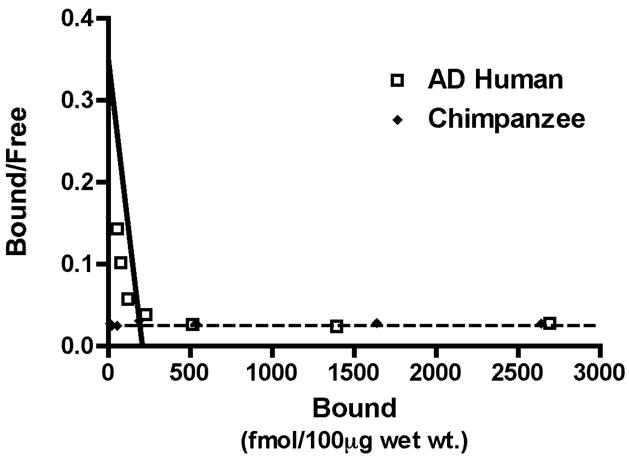
A homologous competition binding analysis with 1.2nM 3H-PIB and unlabeled PIB between 1.0 nM and 1.0μM reveals a high-affinity PIB binding component in AD temporal cortical homogenate (Kd = 3.0nM, Bmax = 209.28 fmol/100μg wet tissue) but no detectable high-affinity PIB binding components in chimpanzee temporal cortical homogenate (AD human: open squares, Chimpanzee: filled diamonds). Similar results were seen with rhesus macaque and squirrel monkey cortical homogenates (data not shown).
3.4. 3H-PIB autoradiography
To determine if 3H-PIB binds to a particular subset of amyloid lesions in AD or nonhuman primate brain, we performed 3H-PIB autoradiography on unfixed cryosections from the superior temporal gyrus of nondemented human, AD, and squirrel monkey subjects (n=2 for all groups). At the same ligand concentration used in the cortical homogenate binding assays (1.0 nM), we detected strong, specific binding to a broad morphological range of senile plaques in the grey matter of the AD superior temporal gyrus (Figure 6) but not to neurofibrillary tangles (data not shown). PIB also bound to a smaller number of lesions in the subcortical white matter (Figure 6). In the two squirrel monkey cases examined, we did not detect specific 3H-PIB binding to any Aβ lesions, despite heavy cortical and vascular amyloid deposition in both cases, as shown by Aβ immunohistochemistry of adjacent cryosections (Figure 6). No 3H-PIB binding was detected in cortical sections from nondemented human subjects who lacked Aβ deposition (data not shown).
Figure 6. 3H-PIB Autoradiography in AD and aged squirrel monkey cortical tissue sections.

1.0nM 3H-PIB binds to Aβ lesions throughout the cortex and, to a lesser extent, in the white matter of AD temporal cortical cryosections (A). This binding is mostly, but not entirely, blocked by 1μM unlabeled PIB (B), and corresponds to 6E10-immunoreactive Aβ lesions in adjacent cryosections (arrows in A and C). In cryosections from squirrel monkey temporal cortex, negligible 3H-PIB binding is detected (D), despite the presence of heavy vascular and parenchymal Aβ deposition, as shown with 6E10 immunohistochemistry in adjacent cryosections (F) (asterisks denote the same large blood vessel in panels D–F). Note that the Aβ-positive lesions in squirrel monkeys consist largely of microvascular deposits and small parenchymal plaques (inset in F), as previously described (Elfenbein et al., 2007). When 3H-PIB is incubated in the presence of 1μM unlabeled PIB, binding is mostly blocked (E); however, as in the AD cases, 3H-PIB binding was mostly, but not entirely eliminated by unlabeled PIB. Bar in F = 1mm for panels A–F; bar in the inset = 50μm.
4. DISCUSSION
We describe the first evidence that Pittsburgh compound B (PIB), a high-affinity molecular probe for Aβ deposits in the AD brain, binds at substantially diminished stoichiometry to cortical homogenates from aged nonhuman primates, despite total Aβ levels equal to, or greater than, the levels measured in AD brains. The virtual absence of PIB binding in monkeys and chimpanzees under the conditions that we employed suggests a paucity of high-affinity PIB binding sites on Aβ multimers in nonhuman primates, similar to that reported for APP/presenilin-1-transgenic mice (Klunk et al., 2005). Klunk, Lockhart, and colleagues have provided evidence for high-and low-affinity PIB binding sites located within the fibrillar Aβ assembly (Klunk et al., 2003a, Lockhart et al., 2005). The AD brain contains approximately 500-fold more high-affinity PIB binding sites per Aβ molecule than does synthetic Aβor Aβ from transgenic mouse brains (Klunk et al., 2005). In the present study, PIB binding to nonhuman primate cortical homogenates was negligible at a 1 nM ligand concentration, which is selective for the high-affinity PIB binding sites that are the primary target in PET studies (Klunk et al., 2003b). To the best of our knowledge, this is the first report of deficient PIB binding to cortical tissue containing profuse, naturally-occurring Aβ plaques and cerebral β-amyloid angiopathy. Our findings suggest that, unless ultra-high specific-activity PIB is used (Maeda et al., 2007), PIB would not be an effective β-amyloid imaging agent in nonhuman primates (Noda et al., 2008), even in aged animals with heavy Aβ deposition.
The dearth of high-affinity cortical PIB binding sites in simians may be attributable to structural variants of multimeric Aβ. In vitro (Petkova et al., 2005) and in vivo (Meyer-Luehmann et al., 2006) evidence increasingly favors the existence of polymorphic Aβ multimers, analogous to structurally and functionally diverse prion strains (Chien et al., 2004, Collinge and Clarke, 2007, Makarava and Baskakov, 2008). Under different in vitro conditions, distinct Aβ strains, distinguishable by biophysical and/or imaging techniques, can be generated from the identical starting peptide (Petkova et al., 2005). In vivo, morphologically distinct Aβ-deposits can be seeded in the brains of transgenic mice by Aβ-rich cortical extracts from different types of APP-transgenic mice (Meyer-Luehmann et al., 2006). Our findings support the hypothesis that Aβ can form polymorphic molecular assemblies that are distinguishable by their PIB-binding characteristics.
Histological observations using the fluorescent PIB analog, 6-CN-PIB, further indicate that PIB binds selectively to a structural motif within fibrillar Aβ assemblies. Denaturation of proteins in cortical tissue sections from AD cases abolishes the binding of 6-CN-PIB (Ikonomovic et al., 2008), indicating a conformation-sensitive binding substrate for PIB. Diffuse Aβ plaques in the cerebellum (a region of the brain that usually shows little degenerative change in AD) do not bind 6-CN-PIB or unlabeled PIB in AD cases (Ikonomovic et al., 2008, Svedberg et al., 2008), suggesting variations in the molecular structure of these lesions even within the same case. 6-CN-PIB binds to a subset of extracellular neurofibrillary tangles in AD tissue sections (Ikonomovic et al., 2008), but these ‘ghost’ tangles also can be immunoreactive for Aβ (Walker et al., 2000), and the 6-CN-PIB concentrations used for histostaining are 100- to 10,000-fold higher than the brain concentrations attained in PET studies. This and other autoradiographic studies show that 3H-PIB does not bind to neurofibrillary tangles at a 1-nanomolar concentration (Lockhart et al., 2007). The high selectivity of PIB for aggregated Aβ, as opposed to tau (neurofibrillary tangles) or α-synuclein (Lewy Bodies) (Klunk et al., 2003a, Fodero-Tavoletti et al., 2007, Ikonomovic et al., 2008), makes it unlikely that the strong PIB signal in human AD brain is due to the presence of such lesions, which are rare or nonexistent in most nonhuman primates. Furthermore, one aged chimpanzee that we examined (YN05-400Pt) had significant tauopathy (Rosen et al., 2008), but the 3H-PIB binding in this case was very low, similar to that in nonhuman primates that were mostly devoid of tauopathy (Table 3).
In the present study, PIB binding to AD brain homogenates correlated positively with total insoluble Aβ, as previously reported (Klunk et al., 2005, Ikonomovic et al., 2008, Svedberg et al., 2008). The variability in published reports of PIB binding levels and insoluble Aβ levels in AD cortical homogenates may be attributable to the heterogeneity of Aβ deposition among brain regions and among individuals (Vinters et al., 1996), and/or to minor differences in the assay conditions in different laboratories. In these four studies, quantitative Aβ ELISAs were performed with different capture and detection antibody pairs, on insoluble Aβ samples prepared with distinct buffers and extraction techniques. Further, our 3H-PIB assay employed a scaled-down microplate version of the cell harvester protocols utilized in the other studies. Importantly, the positive correlation between PIB binding and Aβ levels in our study confirms previous findings, and supports the view that PIB is a useful probe for the pathologic accumulation of Aβ in the human brain.
In autoradiographic experiments using the same tracer levels and specific activity employed in our binding assays, 3H-PIB specifically labels a wide morphological variety of Aβ deposits in cortex and subcortical white matter of AD cases, but not of squirrel monkeys. The intensity of binding is not explained by the unique presence of a particular fragment of Aβ, since previous MALDI-TOF mass-spectrometric studies indicate that all Aβ fragments, including pyroglutamate Aβ pE3-40 and pE3-42, are present in the neocortices of humans with AD and aged nonhuman primates (Rosen et al., 2006). The ratio of Aβ40:Aβ42 could influence PIB binding (Klunk et al., 2005), but our ELISA data indicate that the relative amounts of Aβ peptides ending at amino acids 40 and 42 are largely similar in human and nonhuman primates, especially in the temporal neocortex. Together, these data strengthen the hypothesis that high-affinity PIB binding is dependent upon quaternary structural motifs of assembled fibrillar Aβ, and thus the high-affinity binding element may not be quantifiable with techniques that detect only histologic or primary structural characteristics of the Aβ peptide. Finally, our homogenate-mixing experiments (as well as those of Klunk et al, 2005) do not support the existence of a species-specific factor that modulates PIB binding.
In summary, we show that high-affinity PIB binding sites are significantly deficient in Aβ-rich cortical extracts from aged nonhuman primates compared to extracts from humans with Alzheimer’s disease. Considerable evidence supports a central role for Aβ in AD neurodegeneration (Hardy and Selkoe, 2002), yet AD-like dementia has not been reported in any nonhuman primate (Walker and Cork, 1999). PIB thus could be a useful experimental tool for clarifying the molecular underpinnings of the uniquely human predisposition to Alzheimer’s disease.
Acknowledgments
We thank Brian Ciliax, M. Paul Murphy and Jorge Ghiso for helpful discussions and technical expertise, Marla Gearing, Todd Preuss, Mary Lou Voytko, Amy Arnsten, Douglas Rosene and Daniel Anderson for generously providing tissue, and Jeromy Dooyema, Aaron Farberg and Carolyn Suwyn for excellent technical assistance. Funding was provided by the University Research Committee of Emory University, RR-00165, PO1AG026423, P50AG025688 and the Woodruff Foundation.
Footnotes
Disclosure Statement. The authors declare that they have no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, Irizarry MC, Klunk WE, Mathis CA, Dekosky ST, Greenberg SM, Hyman BT, Growdon JH. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem. 2004;73:617–656. doi: 10.1146/annurev.biochem.72.121801.161837. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38:1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- Dodart JC, May P. Overview on rodent models of Alzheimer’s disease. Curr Protoc Neurosci. 2005 doi: 10.1002/0471142301.ns0922s33. Chapter 9, Unit 9.22. [DOI] [PubMed] [Google Scholar]
- Elfenbein HA, Rosen RF, Stephens SL, Switzer RC, Smith Y, Pare J, Mehta PD, Warzok R, Walker LC. Cerebral beta-amyloid angiopathy in aged squirrel monkeys. Histol Histopathol. 2007;22:155–167. doi: 10.14670/HH-22.155. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Smith DP, McLean CA, Adlard PA, Barnham KJ, Foster LE, Leone L, Perez K, Cortes M, Culvenor JG, Li Q-X, Laughton KM, Rowe CC, Masters CL, Cappai R, Villemagne VL. In vitro characterization of Pittsburgh Compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P, Gold G, Kovari E, von Gunten A, Imhof A, Bouras C, Hof PR. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol (Berl) 2007;113:1–12. doi: 10.1007/s00401-006-0144-y. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Yamaguchi I, Omata S, Gejyo F, Naiki H. Interaction between A beta(1–42) and A beta(1–40) in Alzheimer’s beta-amyloid fibril formation in vitro. Biochemistry. 1999;38:15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PIB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003a;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Bacskai BJ, Wang Y, Price JC, Bergstrom M, Hyman BT, Langstrom B, Mathis CA. Imaging the pathology of Alzheimer’s disease: amyloid-imaging with positron emission tomography. Neuroimaging Clin N Am. 2003b;13:781–789. ix. doi: 10.1016/s1052-5149(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Kuo Y-M, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276:12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, Tapiola T, Pirttila T, Rinne J, Jaaskelainen JE, Soininen H, Rinne JO. Assessment of {beta}-Amyloid in a Frontal Cortical Brain Biopsy Specimen and by Positron Emission Tomography With Carbon 11-Labeled Pittsburgh Compound B. Arch Neurol. 2008 doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- LeVine H, III, Walker LC. Models of Alzheimer’s disease. In: Conn PM, editor. Handbook of Models for Human Aging. Academic Press; Burlington: 2006. pp. 121–134. [Google Scholar]
- LeVine H, III, Walker LC. Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong G, Gee AD, Brown J. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. J Biol Chem. 2005;280:7677–7684. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, Suzuki K, Mori H, Higuchi M, Suhara T. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci. 2007;27:10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarava N, Baskakov IV. The same primary structure of the prion protein yields two distinct self-propagating states. J Biol Chem. 2008;283:15988–15996. doi: 10.1074/jbc.M800562200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Klunk WE. Imaging beta-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr Pharm Des. 2004;10:1469–1492. doi: 10.2174/1381612043384772. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Noda A, Murakami Y, Nishiyama S, Fukumoto D, Miyoshi S, Tsukada H, Nishimura S. Amyloid imaging in aged and young macaques with [11C]PIB and [18F]FDDNP. Synapse. 2008;62:472–475. doi: 10.1002/syn.20508. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Amyloid plaque imaging in vivo: current achievement and future prospects. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S46–50. doi: 10.1007/s00259-007-0700-2. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Szendrei GI, Lee VMY, Mantsch HH. Human and rodent Alzheimer beta-amyloid peptides acquire distinct conformations in membrane-mimicking solvents. European Journal of Biochemistry. 1993;211:249–257. doi: 10.1111/j.1432-1033.1993.tb19893.x. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Rosen RF, III, LeVine H, Pohl J, Preuss TM, Reed M, Tomidokoro Y, Farberg A, Rosene DL, Voytko ML, Lah JJ, Murphy MP, Gearing M, Ghiso JA, Walker LC. 2006 Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience; 2006. Mass spectrometric detection of modified cerebral Abeta peptides in Alzheimer’s disease, aged humans, and aged nonhuman primates. Program # 170.22. Program No. 170.22. [Google Scholar]
- Rosen RF, Farberg AS, Gearing M, Dooyema J, Long PM, Anderson DC, Davis-Turak J, Coppola G, Geschwind DH, Pare JF, Duong TQ, Hopkins WD, Preuss TM, Walker LC. Tauopathy with paired helical filaments in an aged chimpanzee. J Comp Neurol. 2008;509:259–270. doi: 10.1002/cne.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serdons K, Verduyckt T, Vanderghinste D, Cleynhens J, Borghgraef P, Vermaelen P, Terwinghe C, Leuven FV, Laere KV, Kung H, Bormans G, Verbruggen A. Synthesis of 18F-labelled 2-(4′-fluorophenyl)-1,3-benzothiazole and evaluation as amyloid imaging agent in comparison with [11C]PIB. Bioorganic & Medicinal Chemistry Letters. 2009;19:602–605. doi: 10.1016/j.bmcl.2008.12.069. [DOI] [PubMed] [Google Scholar]
- Svedberg MM, Hall H, Hellstrom-Lindahl E, Estrada S, Guan Z, Nordberg A, Langstrom B. [(11)C]PIB-amyloid binding and levels of Abeta40 and Abeta42 in postmortem brain tissue from Alzheimer patients. Neurochem Int. 2008 doi: 10.1016/j.neuint.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Toyama H, Ye D, Ichise M, Liow JS, Cai L, Jacobowitz D, Musachio JL, Hong J, Crescenzo M, Tipre D, Lu JQ, Zoghbi S, Vines DC, Seidel J, Katada K, Green MV, Pike VW, Cohen RM, Innis RB. PET imaging of brain with the beta-amyloid probe, [11C]6-OH-BTA-1, in a transgenic mouse model of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2005;32:593–600. doi: 10.1007/s00259-005-1780-5. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Wang ZZ, Secor DL. Brain Parenchymal and Microvascular Amyloid in Alzheimer’s Disease. Brain Pathology. 1996;6:179–195. doi: 10.1111/j.1750-3639.1996.tb00799.x. [DOI] [PubMed] [Google Scholar]
- Walker LC, Cork LC. The Neurobiology of Aging in Nonhuman Primates. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer Disease. 2. Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 233–243. [Google Scholar]
- Walker LC, Pahnke J, Madauss M, Vogelgesang S, Pahnke A, Herbst EW, Stausske D, Walther R, Kessler C, Warzok RW. Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol. 2000;100:36–42. doi: 10.1007/s004010051190. [DOI] [PubMed] [Google Scholar]
- Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- Ye L, Morgenstern JL, Lamb JR, Lockhart A. Characterisation of the binding of amyloid imaging tracers to rodent Abeta fibrils and rodent-human Abeta co-polymers. Biochem Biophys Res Commun. 2006;347:669–677. doi: 10.1016/j.bbrc.2006.06.126. [DOI] [PubMed] [Google Scholar]
- Ye L, Morgenstern JL, Gee AD, Hong G, Brown J, Lockhart A. Delineation of positron emission tomography imaging agent binding sites on beta-amyloid peptide fibrils. J Biol Chem. 2005;280:23599–23604. doi: 10.1074/jbc.M501285200. [DOI] [PubMed] [Google Scholar]
- Ye L, Velasco A, Fraser G, Beach TG, Sue L, Osredkar T, Libri V, Spillantini MG, Goedert M, Lockhart A. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem. 2008;105:1428–1437. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]