

Abstract
Pigtail macaques (PTM) rapidly progress to AIDS after SIV infection. Given the strong association between HIV/SIV disease progression and microbial translocation and immune activation, we assessed whether high basal levels of immune activation and microbial translocation exist in PTM. We found that prior to SIV infection, PTM had high levels of microbial translocation that correlated with significant damage to the structural barrier of the GI tract. Moreover, this increased microbial translocation correlated with high levels of immune activation and was associated with high frequencies of IL-17-producing T cells. These data highlight the relationship between mucosal damage, microbial translocation and systemic immune activation in the absence of HIV/SIV replication and underscore the importance of microbial translocation in the rapid course of disease progression in SIV-infected PTM. Furthermore, these data suggest that PTM may be an ideal model to study therapeutic interventions aimed at decreasing microbial translocation-induced immune activation.
Introduction
The mucosal immune system is intricate and complex and relies on a balance of multiple cell types to maintain homeostasis and to function appropriately1, 2. The tight epithelial barrier of the gastrointestinal (GI) tract functions to prevent commensal organisms and pathogens crossing from the intestinal lumen into circulation, while allowing nutrients to be absorbed3. An important role for mucosal T cells in maintaining GI tract function has recently been highlighted by numerous studies dedicated to a newly identified subset of CD4+ T cells, Th17 cells, which are found most prominently in the GI tract4-10. These T cells produce IL-17, which is important in adaptive immunity against extracellular bacteria and fungi, recruite neutrophils, induce defensin production, promote enterocyte homeostasis, and are important for maintenance of epithelial tight junctions in mucosal tissues 11-16. However, Th17 cells have also been implicated as potent inducers of tissue inflammation in several GI disorders such as Crohn's disease and ulcerative colitis, which result from a chronic inflammatory state associated with infiltration of inflammatory immune cells and damage to the gut epithelium17-21. Specifically, an increased frequency of IL-17 producing cells in the GI tract has been directly associated with inflamed mucosa in inflammatory bowel diseases13, 20, 22-24. Thus, dysregulation of mucosal associated lymphoid tissues (MALT) can lead to pathogenic consequences.
Dysfunction of the mucosal immune system has also been demonstrated in chronic viral infections, particularly during HIV/SIV pathogenesis. HIV and SIV infections are characterized by chronic virus replication and depletion of CD4+ T cells, ultimately resulting in opportunistic infections and progression to AIDS. Because HIV/SIV preferentially infects CD4+CCR5+ T cells, which are enriched in mucosal tissues, there is a rapid and severe depletion of CD4+ T cells in MALT during acute infection 25-29. However, CD4 depletion alone is not sufficient to cause AIDS27, 30-32. Rather, the strongest predictor of disease progression is the extent of chronic, systemic immune activation associated with HIV pathogenesis33. This systemic immune activation is characterized by increased cell proliferation, high rates of lymphocyte apoptosis, cell cycle dysregulation, and increased levels of proinflammatory cytokines34-36. Massive infection of CD4+ T cells in MALT early in HIV/SIV infections is directly associated with inflammation and a breakdown of the mucosal integrity; this allows microbial products to translocate from the lumen of the GI tract into the peripheral circulation26, 37-43. Translocation of microbial products during HIV/SIV infections, demonstrated by an increase in plasma lipopolysacharide (LPS) and bacterial DNA levels, is associated with systemic immune activation26, 37-42. The gastrointestinal pathology associated with HIV/SIV infections includes significant enterocyte apoptosis, enteropathy of the MALT, as well as increased levels of inflammation and decreased levels of mucosal repair and regeneration44-46. However the mechanisms underlying the damage to the GI tract are not well understood and clarifying to what extent structural and/or immunolgical damage to the GI tract and microbial translocation underlies immune activation is vital to characterizing the interactions between the MALT and peripheral immune system.
Non-human primate (NHP) models are essential to better understand how and to what extent dysfunction and damage to the mucosal immune system affects systemic immune activation in vivo. Pathogenic SIV infection of Asian rhesus macaques (RM; Macaca mulatta) is the most widely studied NHP model for HIV pathogenesis to date. In this model, infection of RM with SIVmac strains (derived from SIVsmm) recapitulates many key features of HIV infection in humans47-49, including rapid and severe depletion of CD4+ T cells in mucosal tissues during acute infection, progressive loss of CD4+ T cells in periphery during chronic infection, high viral load, high levels of immune activation, and microbial translocation34, 37, 47, 48, 50, 51. Another important model for HIV infection is SIV infection of pigtail macaques (PTM; Macaca nemestrina). Like RM, after SIV infection, PTM lose CD4+ T cells, have high levels of immune activation, and progress to AIDS52-54. These animals are of particular interest, in that PTM typically progress to AIDS more rapidly than RM55, 56. After infection with SIVsmE543, the majority of PTM progress to AIDS within 6 months after infection (as opposed to approximately 2 years for RM)55, 56. This rapid disease progression observed in PTM is not associated with virus inoculation, but is likely due to host factors56-58. Interestingly, uninfected PTM in captivity have an increased incidence of diarrhea and GI diseases, and older animals frequenctly present with systemic amyloidosis59, 60. Indeed, in 47 SIV-uninfected PTM in our animal facility that have been followed for a minimum of five years, 24 animals (51.1%) have had histories of gastrointestinal disorders as defined by at least 2 bouts of diarrhea requiring treatment. In contrast, only 12 of 103 (11.6%) SIV-uninfected RM in our facility have had such disorders (P<0.0001). Similar observations have also been made at two separate animal facilites across the world (Shiu-Lok Hu, University of Washington, and Stephen J. Kent, University of Melbourne; personal communication). Moreover, after certain SIV infections such as SIVPBj-14 infection, PTM are more susceptible than RM to death resulting from gastrointestinal distress61. We hypothesized that the rapid disease progression observed in PTM after SIV infection may, in part, be due to pre-existing conditions that result in dysfunction and damage of the mucosal immune system leading to increased microbial translocation and consequent immune activation. To address this hypothesis, in this study we assessed the T cell immunophenotype, function and activation in multiple anatomical sites including blood, lymph nodes (LN), and tissues of the GI tract. We also determined the level of mucosal integrity, microbial translocation and immune activation in SIV-uninfected PTM in comparison to RM.
Results
Uninfected PTM have a higher frequency of proliferating and memory-effector T cells than RM
We initially performed flow cytometric analysis to study cellular markers associated with activation and differentiation in a cohort of SIV-uninfected PTM and SIV-uninfected RM. We found that PTM had significantly increased frequencies of proliferating CD4+Ki67+ T cells in blood compared to RM (10.62% ± 4.5 compared to 4.59% ± 1.7, P=0.0006, Figure 1a), as well as increased CD8+Ki67+ T cells in blood from PTM compared to RM (10.06% ± 5.9 compared to 5.76% ± 1.8, P=0.0637, Figure 1b). Accordingly, PTM also had a significantly greater frequency of memory and effector T cells in blood as compared to RM as measured by CD28+CD95+ and CD28-. On average, 72.02% (± 12.0) of CD4+ T cells were memory-effector phenotype in PTM, as compared to 47.97% (± 18.9) of RM CD4+ T cells being memory-effector cells, P=0.0013 (Figure 1c). A similar finding was observed for CD8+ T cells, as PTM had an average of 80.34% (± 16.3) of CD8+ memory T cells, as compared to 66.34% (± 14.5) of RM CD8+ T cells having a memory-effector phenotype, P=0.0112 (Figure 1d).
Figure 1. Uninfected pigtail macaques have high levels of immune activation and memory-effector T cells compared to rhesus macaques and humans.

(a) Frequencies of CD4+Ki67+ T cells in blood, (b) Frequencies of CD8+Ki67+ T cells in blood, (c) Frequencies of CD4+ memory-effector T cells in blood and necropsy tissues, (d) Frequencies of CD8+ memory-effector T cells in blood and necropsy tissues. PTM (circles), RM (triangles). Memory-effector T cells were determined by gating on both CD28+C95+ and CD28- subsets as measured by flow cytometry. Horizontal bars indicate median. P values calculated from Mann-Whitney U test.
High frequencies of activated memory-effector T cells in peripheral blood may not reflect frequencies of T cells in all anatomical sites44, 62. Therefore we measured these cells by flow cytometry on mononuclear cells isolated from multiple tissues from sacrificed, SIV-uninfected, PTM and RM. We found high frequencies of memory CD4+ T cells in multiple PTM tissues (Figure 1c), with an average of 87.74% (± 7.8) in spleen, 78.77% (± 12.4) in axillary LN, 78.15% (± 9.9) in inguinal LN, 73.97% (± 13.8) in mesenteric LN, 85.36% (± 18.2) in duodenum, 93.38% (± 4.3) in jejunum, 85.02% (± 12.4) in ileum, 91.57% (± 5.0) in cecum, and 88.63% (± 10.2) in colon. In comparison, RM had lower levels of CD4+ memory-effector T cells in many tissues (Figure 1c), with an average of 77.23% (± 13.4) in spleen, 49.28% (± 7.8) in axillary LN, 50.42% (± 6.3) in inguinal LN, 36.75% (± 10.23) in mesenteric LN, 97.47% (± 4.3) in duodenum, 91.16% (± 7.9) in jejunum, 63.92% (± 10.6) in ileum, 66.32% (± 6.0) in cecum, and 62.52% (± 10.1) in colon. Indeed, the frequency of memory-effector CD4+ T cells in PTM was significantly higher than in multiple tissues from RM (Figure 1c), including spleen (P=0.0218), axillary LN (P=0.0010), inguinal LN (P=0.0004), mesenteric LN (P=0.0026), ileum (P=0.0076), cecum (P=0.0007), and colon (P=0.0016). We also found high frequencies of memory-effector CD8+ T cells in the same PTM tissues (Figure 1d), with an average of 88.1% (± 12.9) in spleen, 72.44% (± 21.6) in axillary LN, 64.61% (± 22.4) in inguinal LN, 61.27% (± 23.6) in mesenteric LN, 93.16% (± 14.7) in duodenum, 98.42% (± 1.3) in jejunum, 79.79% (± 20.0) in ileum, 86.51% (± 14.2) in cecum, and 85.01% (± 20.4) in colon. In comparison, the frequency of CD8+ memory-effector T cells in RM was on average 82.77% (± 13.6) in spleen, 35.67% (± 9.9) in axillary LN, 38.5% (± 13.6) in inguinal LN, 22.52% (± 7.4) in mesenteric LN, 95.67 (± 2.7) in duodenum, 91.16% (± 8.3) in jejunum, 41.4% (± 14.0) in ileum, 45.1% (± 7.3) in cecum and 49.35% (± 13.7) in colon. The frequency of memory-effector CD8+ T cells was also significantly higher in many tissues from PTM compared to RM (Figure 1d), including in axillary LN (P=0.0017), inguinal LN (P=0.0312), mesenteric LN (P=0.0010), ileum (P=0.0080), cecum (P=0.0010) and colon (P=0.0075). These data are of particular interest, since it is expected that effector tissues of the GI tract have high frequencies of memory and effector T cells, as previously described in RMs and humans63, 64. However, lymphoid tissues such as spleen and lymph nodes typically have much lower frequencies of memory T cells, and higher frequencies of naïve T cells, as demonstrated by the low level of memory-effector T cells in RM (Figure 1c-d). Here we describe that PTM have very high frequencies of memory T cells (and thus low frequencies of naïve T cells) in all sites examined. Moreover, these results highlight anatomically restricted differences in the phenotypic distribution of T cells. Even across different regions of the GI tract, T cell subsets are not uniform with differing frequencies of memory and effector T cells residing in different locations (Figures 1c-d, Supplementary Tables 1-2). Of note, the increased proliferating and memory-effector T cells observed is not due to a transient infection, as longitudinal sampling demonstrated that these cells remained at high frequencies over time (data not shown). These data suggest that one of the mechanisms underlying the increased SIV disease progression in PTM may involve the high baseline frequency of activated targets for the virus prior to SIV infection.
Uninfected PTM have a higher frequency of CD4+CCR5+ T cells as compared to RM, with high levels in GI tissues
Because HIV and SIV preferentially infects activated CCR5+ memory CD4+ T cells65, we next determined if the increased frequency of memory and effector T cells observed in PTM translated to an increased frequency of CCR5+ T cells. Thus we measured the frequency of CCR5+ T cells in peripheral blood and multiple tissues from necropsies of both SIV-uninfected PTM and RM. We found that PTM had significantly higher frequencies of CD4+CCR5+ T cells in blood as compared to RM (14.54% ± 6.3 compared to 7.76% ±4.8, P=0.0008, Figure 2), as well as a higher frequency of CD8+ CCR5+ T cells (data not shown). The frequency of CD4+CCR5+ T cells was elevated in multiple anatomical sites of PTM (Figure 2), with an average of 14.17% (± 3.7) in spleen, 6.41% (± 1.9) in axillary LN, 6.54% (± 2.8) in inguinal LN, 5.97% (± 2.6) in mesenteric LN, 59.4% (± 28.2) in duodenum, 47.18% (± 22.7) in jejunum, 17.17% (± 13.8) in ileum, 25.53% (± 13.8) in cecum, and 27.86% (± 21.5) in the colon of PTM (Figure 2b). In comparison, the average frequency of CD4+CCR5+ T cells in RM was 16.04% (± 8.5) in spleen, 5.43% (± 3.0) in axillary LN, 5.34% (± 2.5) in inguinal LN, 3.15% (± 1.7) in mesenteric LN, 16.42% (± 12.8) in duodenum, 27.43% (± 17.7) in jejunum, 8.38% (± 5.6) in ileum, 7.24% (± 1.1) in cecum, and 8.98% (± 2.7) in the colon (Figure 2). In addition to blood, the frequency of CD4+CCR5+ T cells was significantly higher in duodenum (P=0.0334) and cecum (P=0.0080) of PTM compared to RM (Figure 2). We also investigated whether CD4+CCR5+ T cells had higher levels of CCR5 per cell, and found that though PTM had a slightly increased mean fluorescence intensity of CCR5 on CD4+ T cells, the difference was not significant between PTM and RM (P=0.1008, data not shown). Not all tissues had significantly higher frequencies of CD4+CCR5+ T cells in PTM compared to RM, likely due to the low amount of RMs available for analysis, but Figure 2 demonstrates that on average, PTM have significantly more CD4+CCR5+ T cells than RM. Of note, there was a greater frequency of CD4+CCR5+ T cells in the duodenum and jejunum compared to other tissues, including the large intestine (Supplementary Table 3). These data demonstrate that prior to SIV infection, PTM have an increased frequency of activated targets for SIV infection, which may, in part, explain the rapid disease progression observed after SIV infection, as increased targets may result in more rapid CD4+ T cell depletion.
Figure 2. Uninfected pigtail macaques have high frequencies of CD4+CCR5+T cells compared to rhesus macaques.

Frequencies of CD4+CCR5+ T cells in blood and necropsy tissues. PTM (circles), RM (triangles). Horizontal bars indicate median. P values from Mann-Whitney U test.
Uninfected PTM T cells are enriched for IL-17, but not IFNγ production when compared to RM, and IL-17+ T cells are found in all tissues of PTM
To determine the functionality of T cells in PTM and RM and to attempt to understand the mechanisms underlying the increased frequencies of activated and memory T cells in PTM, we stimulated PBMCs with the mitogens phorbol myrsil acetate (PMA) and ionomycin and measured IFNγ and IL-17 production by flow cytometry. IFNγ can be produced by T cells in all tissues, typically in response to intracellular pathogens such as viruses. In contrast, IL-17, is produced by T cells in response to extracellular bacteria and fungi, and IL-17 producing T cells are found at high frequencies in mucosal tissues where these antigens predominate66. Surprisingly, we found that PTM had significantly lower frequencies of CD4+ T cells that produce IFNγ in peripheral blood compared to RM (15.1% ± 7.7 compared to 38.93% ± 21.3, P=0.0095, Figure 3a). However, there was no difference between the frequencies of IFNγ producing CD8+ T cells (44.64% ± 24.3 compared to 39.4% ± 18.2, P=0.6, Figure 3b). Interestingly, PTM had higher frequencies of peripheral blood CD4+ T cells that produced IL-17 (5.92% ± 3.3 compared to 1.91% ± 1.8, P=0.0081, Figure 3c). Perhaps most intriguingly, PTM also had higher fequencies of CD8+ T cells that produced IL-17 (3.33% ± 1.8 compared to 0.11% ± 0.2, P<0.0001, Figure 3d). Furthermore, we found that there is a significant, direct correlation between the frequency of IL-17 production between CD4+ and CD8+ T cells in PTM (Spearman r=0.4824, P<0.0001, Figure 3e). These data suggest that the same antigens may drive expansion of both IL-17 producing CD4+ and CD8+ T cells in PTM. Given the known antigen-specificity of IL-17-producing CD4 T cells66, these observations raise the possibility that response to bacterial and fungal antigen may be one of the underlying factors involved in the high frequencies of IL-17-producing, activated, memory T cells in PTM.
Figure 3. High frequencies of T cells from uninfected pigtail macaques produce IL-17 compared to rhesus macaques.
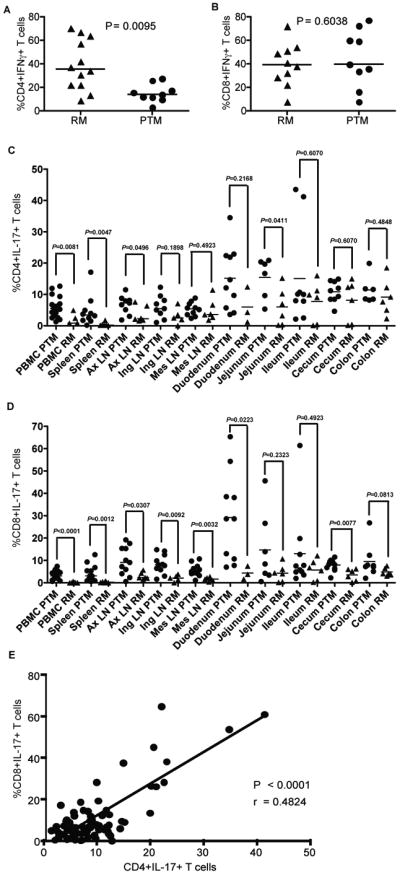
(a) Frequencies of CD4+ IFNγ producing T cells in blood, (b) Frequencies of CD8+ IFNγ producing T cells in blood, (c) Frequencies of CD4+ IL-17 producing T cells in blood and necropsy tissues, (d) Frequencies of CD8+ IL-17 producing T cells in blood and necropsy tissues, (e) Frequencies of CD4+ vs. CD8+ IL-17 producing T cells in blood of PTM. Correlation (r) determined by Spearman's rank correlation. All samples were stimulated with PMA and Ionomycin. PTM (circles) and RM (triangles). Horizontal bars indicate median. P values from Mann-Whitney U test.
After observing the surprisingly high frequency of IL-17 producing T cells in peripheral blood, we determined the frequency of IL-17 producing T cells in multiple tissues from PTM and RM. We found CD4+IL-17+ T cells in all tissues of PTM, with an average of 4.67% (± 4.9) in spleen, 6.7% (± 3.0) in axillary LN, 6.03% (± 3.6) in inguinal LN, 5.33% (± 2.1) in mesenteric LN, 15.16% (± 10.1) in duodenum, 14.45% (± 6.5) in jejunum, 15.45% (± 6.5) in ileum, 10.83% (± 3.4) in cecum, and 11.423% (± 4.5) in colon (Figure 3c). In comparison, the average frequency of IL-17 producing CD4+ T cells in RM was 0.47% (± 0.6) in spleen, 3.63% (± 2.62) in axillary LN, 3.01% (± 2.6) in inguinal LN, 4.96% (± 3.6) in mesenteric LN, 5.96% (± 5.8) in duodenum, 6.02% (± 6.0) in jejunum, 7.77% (± 6.3) in ileum, 8.12% (± 6.5) in cecum, and 9.13% (± 5.7) in colon (Figure 3c). Though the only significant difference between CD4+IL-17+ T cells in gut tissues between PTM and RM was in jejunum (P=0.0411), significant differences were observed in spleen (P=0.0047) and axillary LN (P=0.0496), which indicates that bacteria specific IL-17 producing CD4+ T cells are found at higher levels in the periphery than typically observed in RM or human (figure 3c)9, 22, 66, 67.
Similarly high frequencies of IL-17 producing CD8+ T cells were observed in PTM, with an average of 4.27% (± 3.8) in spleen, 9.72% (± 6.4) in axillary LN, 8.38% (± 4.2) in inguinal LN, 5.59% (± 2.7) in mesenteric LN, 29.19% (± 20.2) in duodenum, 14.68% (± 16.2) in jejunum, 13.03% (± 17.8) in ileum, 7.97% (± 2.6) in cecum, and 9.53% (± 7.5) in colon (Figure 3d). In comparison, RM had very low levels of IL-17 producing CD8+ T cells, with an average of 0.30% (± 0.3) in spleen, 3.07% (± 1.6) in axillary LN, 2.07% (± 1.5) in inguinal LN, 1.62% (± 0.8) in mesenteric LN, 4.32% (± 2.9) in duodenum, 4.32% (± 3.9) in jejunum, 5.74% (± 4.6) in ileum, 3.54% (± 2.6) in cecum, and 4.83% (± 1.8) in colon (figure 3d). As expected given the low levels of CD8+IL-17+ T cells in RM, there was a significant difference between PTM and RM for many tissues in addition to blood, including spleen (P=0.0012), axillary LN (P=0.0307), inguinal LN (P=0.0092), mesenteric LN (P=0.0032), duodenum (P=0.0223), and cecum (P=0.0077), again with the most differences being observed in peripheral and lymphoid tissues (figure 3d). Of note, similar to CD4+CCR5+ T cells (Figure 2), the small intestine (duodenum, jejunum and some animals in ileum) had the highest frequency of IL-17 producing T cells in PTM (Supplementary Tables 4-5). Taken together, these data highlight the differences between anatomical sites of the GI tract and demonstrate the importance of studying multiple tissues to understand more completely the dynamic nature of mucosal immune compartments.
Uninfected PTM have high levels of LPS in plasma and increased permeability of the small intestine in comparison to RM and humans
Given the antigens (i.e. bacteria and fungi) typically targeted by Th17 cells, and the increased frequency of IL-17 producing T cells in PTM, we hypothesized that a contributing cause of the increased T cell activation and high frequencies of IL-17-producing T cells may be microbial translocation. To evaluate potential systemic dissemination of translocated microbial products in PTM, we measured plasma levels of lipopolysaccharide (LPS), which is used as a quantitative indicator of microbial translocation26, 37-42. We found that PTM had higher levels of plasma LPS, with an average of 45.33 pg/mL of LPS in the plasma (± 12.6), as compared to RM with an average of 19.21 pg/mL of LPS (± 13.3), P=0.0013 or humans, with an average of 20.32 pg/mL of LPS (± 15.9), P=0.0014 (Figure 4a). To assess whether the increased LPS was associated with damage to the GI tract, we measured small intestine permeability using a clinically employed test based on saccharide permeability, which measures the ratio of lactulose to mannitol following saccharide solution ingestion68, 69. PTM demonstrated higher gut permeability, with an average lactulose/mannitol ratio of 0.088 (± 0.03) compared to humans, with an average lactulose/mannitol ratio of 0.017 (± 0.008), P=0.0005 (Figure 4b). Hence one of the potential causes of the increased frequencies of activated and IL-17 producing T cells in PTM may be microbial products that translocate through a permeable GI tract.
Figure 4. Plasma LPS and Lactulose to Mannitol ratio indicate microbial translocation and gut permeability in uninfected pigtail macaques.

(a) Plasma LPS levels. (b) Lactulose to manitol ratio. PTM (circles), RM (triangles), humans (squares). Horizontal bars indicate median. P values from Mann-Whitney U test.
Uninfected PTM have increased damage to the gut epithelia and microbial translocation compared to RM
In order to determine the mechanism underlying the permeability of the GI tract and consequent microbial translocation in PTM, both PTM and RM were euthanized and we performed immunohistochemical (IHC) studies on sections of the GI tract taken at necropsy. Initially we stained GI tract tissues with antibodies against the tight junction protein claudin-3 to measure the continuity of the structural barrier of the gut epithelia. This analysis allowed us to determine to what extent breaches and ulcerations in the tight epithelial barrier occur in PTM. We found that PTM (Figure 5a) had a much higher frequency of breaches in the epithelium of the colon as compared to RM (Figure 5b). To confirm these findings we used quantitative image analysis to determine the percentage of damaged GI barrier (breach to intact ratio) by measuring the length of the tight epithelial barrier that was not intact (not stained for claudin) over the length of the colon barrier that was intact (stained for claudin). On average, the breach to intact ratio for PTM was 0.303 (± 0.21), compared to 0.017 (± 0.01) for RM, P=0.0264 (Figure 5c), hence a putative mechanism for the observed increase in frequency and extent of diarrhea, intestinal permeability, and microbial translocation in PTM may be the relatively increased pre-existing damage to the structural barrier of the GI tract.
Figure 5. Uninfected pigtail macaques have increased damage to the tight epithelial barrier and high levels of LPS in the lamina propria of the colon.

(a) Representative claudin-3 staining (brown) in colon of PTM, (b) Representative claudin-3 staining (brown) in colon of RM, (c) Ratio of measurements of breached epithelial (no claudin) to intact epithelial (claudin) in either PTM and RM, (d) Representative LPS staining (brown) in colon of PTM, (e) Representative LPS staining (brown) in colon of RM, (f) Area of colon lamina propria LPS measured in PTM and RM. PTM (circles), RM (triangles). Horizontal bars indicate median. P values from Mann-Whitney U test.
In order to determine if these breaches in the tight junctions of the epithelium correlated with the observed increase in microbial translocation, we studied colon sections by IHC using an antibody for LPS core antigen by IHC to measure bacterial products directly within the lamina propria. We found that PTM had increased levels of LPS in the lamina propria of the colon (Figure 5d) as compared to RM (Figure 5e). Using quantitative image analysis we determined the percent area of colonic lamina propria which contained LPS, and found that on average, LPS accounted for 13.00% (± 10.01) of the lamina propria area in PTM, while only 0.274% (± 0.20) of the lamina propria was occupied by LPS in RM, P<0.0001 (Figure 5f). These data strongly suggest that a mechanism underlying the increased microbial translocation involves structural damage to the gut epithelium in PTM. Indeed, we found a significant positive correlation between the breach/intact ratio and the area fraction of colon LPS in PTM (Spearman r=0.6424, P=0.0368, Figure 6a).
Figure 6. Breaches in the colon correlates with microbial translocation, and microbial translocation correlates with immune activation in the colon of uninfected pigtail macaques.

(a) The breach/intact ratio of the colon as measured by claudin significantly correlates with the amount of microbial tranlsocation into the lamina propria of the colon of PTM (b) The amount of MxA (a specific type I IFN responsive gene) in the colon as measured by IHC staining significantly correlates with the amount of microbial translocation into the colon of PTM. Correlations determined by Spearman's rank correlation. Lines indicate linear regression.
Damage to gut epithelium in uninfected PTM results in systemic microbial translocation that correlates with immune activation
To determine the significance of microbial translocation contributing directly to immune activation, we also quantified the amount of immune activation in the colon of PTM by performing quantitative image analysis of colon sections that were stained with a monoclonal antibody against MxA, a protein made in response to recent production of type one interferons70, 71. We found that on average, 11.55% (± 11.29) of the cells in the lamina propria of the colon of PTM were stained for MxA. The extent of microbial translocation positively correlated with local immune activation in the colon, based on analysis of the fraction of colon lamina propria tissue sections staining for LPS and MxA (Spearman correlation: r=0.7133, P=0.0118, Figure 6b). In an attempt to understand the molecular mechanism(s)/events leading to GI epithelial barrier damage in uninfected PTM, we reasoned that increased enterocyte apoptosis could lead to increased microbial translocation in these animals by altering epithelial barrier homeostasis and function. Thus we investigated whether the increased damage to the epithelial barrier and resulting microbial translocation we observe in the colon of PTM may be a result of increased enterocyte cell death by apoptosis. To test this hypothesis, we measured active caspase-3 by IHC in the colon of PTM and RM, and found that, indeed, where colonic epithelial cells were still present there were higher levels of apoptosis in the enterocytes along the colon cryptal tips in PTM compared to RM (data not shown).
In order to determine if the microbial translocation and immune activation that was observed in the colon resulted in systemic consequences, we also used IHC to measure LPS and MxA in mesenteric lymph nodes, which drain the GI tract, as well as in peripheral, axillary lymph nodes. With IHC and quantitative image analysis we found a significant, positive, correlation between the fractional area of tissue sections staining for LPS in the colon and in the mesenteric LN (Spearman r=0.5604, P=0.0463, Figure 7a), as well as between the area staining for MxA in the colon and in the mesenteric LN (Spearman r=0.6993, P=0.0142, Figure 7b). Furthermore, there was a significant, direct correlation between LPS in mesenteric LNs and LPS in peripheral, axillary LNs (Spearman r=0.6648, P=0.0132, Figure 7c) as well as between MxA in mesenteric LNs and MxA in axillary LNs (Spearman r=0.6868, P=0.0095, Figure 7d). These data indicate that translocation of microbial products and the resulting immune activation have not only local consequences within the GI tract, but are also associated with systemic dissemination of LPS and systemic immune activation. We established further evidence of systemic microbial translocation by measuring plasma levels of soluble CD14 (sCD14), which is produced by CD14+ monocytes and macrophages upon direct LPS stimulation in vivo37. We found an association between LPS in the colon and sCD14 in the plasma (Spearman r=0.5000, P=0.0819, Figure 7e), and there is a significant, direct correlation between MxA in the colon and sCD14 (Spearman r=0.7203, P=0.0106, Figure 7f). Taken together, these data demonstrate that when microbial products translocate from the lumen of the colon into the lamina propria, it can result in local and systemic immune activation. Moreover, these data define the degree to which microbial translocation can stimulate the immune system, both locally and systemically, in the absence of ongoing viral replication.
Figure 7. Local microbial translocation and immune activation in the colon results in systemic microbial translocation and immune activation in uninfected pigtail macaques.

(a) The amount of LPS measured in the colon significantly correlates with LPS in the mesenteric LN. (b) The amount of MxA in the colon significantly correlates with the amount of MxA in the mesenteric LN. (c) LPS in the mesenteric LN significantly correlates with LPS in the axillary LN. (d) MxA in the mesenteric LN significantly correlates with MxA in the axillary LN. (e) LPS in the colon is associated with sCD14 in plasma, (f) MxA in the colon significantly correlates with sCD14 in plasma. Correlations determined by Spearman's rank correlation. Lines indicate linear regression.
Discussion
Microbial translocation has been implicated in the deleterious immune activation that characterizes the chronic phase of HIV infection of humans and SIV infection of Asian macaques. However, the degree to which microbial products alone, relative to persistent viral replication, proinflammatory mediators such as cytokines and chemokines, and/or other unknown factors, contribute to immune activation has remained unclear. Using cells and tissue samples from pigtail macaques, animals that have recurring diarrhea and progress to AIDS quickly after SIV infection, we have shown that in the absence of SIV infection, PTM have damage to their GI tract and increased basal levels of microbial translocation and immune activation. Specifically, we have demonstrated that: i) uninfected PTM have damage to the tight epithelial barrier of the GI tract; ii) this damage correlates with translocation of microbial products both locally within the GI tract and systemically in LN and blood; iii) the microbial translocation that occurs correlates with immune activation both locally and systemically; and iv) uninfected PTM also have higher frequencies of activated and memory T cells producing IL-17 than do RM. These data indicate that chronic microbial translocation can be associated with increased activation and turnover of lymphocytes that results in decreased frequencies of naïve T cells, and an increased amount of memory and effector T cells and proliferating (Ki67+) T cells. Further support of these data is the increased frequencies of CCR5+ T cells in PTM. CCR5 is found only on the surface of memory and effector T cells (not naïve), as it is needed for lymphocytes to migrate to effector sites, such as the GI tract72, 73. Moreover, CCR5+CD4+ T cells are the primary targets for virus replication after SIV/HIV infection. Hence, it is possible that the increased frequency of these cells in PTM may contribute to the more rapid SIV disease progression we observe in these animals. It is of interest, however, that the increased target cells do not appear to affect transmission or infection, as previous studies using PTM have demonstrated that these animals get infected at the same rate as RM with similar peak and set-point viremia, despite inoculation size or infection route (i.e. mucosal infection vs. intravenous infection)56, 74. However, the increased immunological state and number of target cells in PTM provides a plausible mechanism for the more rapid progression to AIDS by destroying more activated CD4+ T cells via both viral mediated and bystander cell death with a reduced capacity to regenerate these important effector populations due to the reduced naïve T cell pool. Finally, given the differential frequencies of CCR5+CD4+ T cells in different sites, with the largest frequency of these targets for SIV replication in the small intestine, these data highlight the importance of examining multiple tissues when considering effector sites in the context of mucosal immunology.
Further evidence that microbial translocation is causing the immune activation and high frequencies of memory and activated T cells we observed is the functionality of T cells in PTM. Though PTM actually had lower frequencies of Th1 T cells that produce IFNγ compared to RM, these animals had very high frequencies of IL-17 producing T cells in all tissues. IL-17 production by T cells is typically in response to bacterial and fungal antigens66, 75, thus it is likely that the translocation of bacterial products we observe in PTM contributes to the increased IL-17 production by T cells. These data also have implications for viral pathogenesis, since even though IL-17 production is important for immunity against extracellular pathogens, these cells typically lack antiviral mediators, which may contribute to lack of control of viremia after SIV infection, while providing excellent targets for SIV infection, as these cells can be infected in vivo66, 75. Moreover, these cells have been shown to be preferentially lost from the GI tract in progressive HIV/SIV infections and in imbalance with Treg CD4+ T cells9, 66, 75. Of interest were the high frequencies of IL-17 producing CD8+ T cells. IL-17 production is generally associated with CD4+ T cells (Th17 cells)4-8, however more recently, several studies have addressed CD8+ T cells that produce IL-17 (Tc17 cells), particularly in the context of chronic inflammation76-79. Of relevance, Kader et al. recently demonstrated that SIV progression in RM is associated with dysregulation of both Th17 and Tc17 cells77. Here we show that not only do high frequencies of CD8+ T cells produce IL-17 in all tissues, but that there is a correlation between the frequencies of IL-17 producing CD4+ T cells and CD8+ T cells, which implies that these cells may be stimulated by the same types of antigens. IL-17 producing T cells can, themselves, contribute to mucosal health by providing cytokines such as IL-21, which is important for enterocyte homeostasis and induction of anti-bacterial defensins13, 15, 80. However, these T cells are also pro-inflammatory, suggesting that a delicate balance needs to be achieved to maintain mucosal integrity and health and prevent disease, which was demonstrated by Favre et al. in SIV infection of RM9. Indeed, there is an association between increased frequencies of Th17 cells and inflamed mucosa in inflammatory conditions such as Crohn's disease and ulcerative colitis13, 20, 22-24. Therefore, in the case of PTM, microbial translocation could likely be causing excessive skewing of T cells towards a Th17 phenotype, which may, in turn, contribute to immune activation and higher frequencies of memory T cells by enhancing an ongoing proinflammatory state in PTM.
This study highlights the importance of the consequences of damage to the GI tract and the resulting microbial translocation and consequent stimulation to the immune system, particularly since it was conducted in SIV naïve animals, in the absence of the effects of chronic virus infection. Microbial translocation has been well characterized in HIV and SIV infections, and clearly it is a factor contributing to the systemic, chronic immune activation that ultimately leads to disease progression and AIDS26, 37-42. However, to our knowledge, this is the first assessment of the extent to which damage to the GI tract and microbial translocation results in both local and systemic effects in the absence of chronic virus replication or chronic pro-inflammatory diseases. Not surprisingly, though this study demonstrates the extent to which microbial translocation results in immune activation, it also clearly demonstrates that microbial translocation and immune activation alone, in the absence of SIV or HIV replication is not sufficient to cause AIDS. Thus, in the case of SIV/HIV infection, though increased microbial translocation and immune activation likely play a large role in disease progression; virus replication, pro-inflammatory responses to the virus, and other contributing factors also are required for progression to AIDS. This study also emphasizes the degree to which dysfunction of the MALT locally results in systemic consequences. Moreover, the increased microbial translocation and consequent immune activation may likely be a mechanistic feature underpinning the increased disease progression rate observed in SIV-uninfected, captive PTM54, 55. This is likely due not only to the pre-existing damage to the GI tract, but rapid progression may also result as a consequence of increased activated CD4+ T cells as target cells for virus replication that exist due to the ongoing immune activation in PTM. Of note, these observations are not isolated to a single colony of PTM, as both diarrhea and rapid SIV progression has been observed in PTM worldwide (Shiu-Lok Hu, University of Washington, and Stephen J. Kent, University of Melbourne; personal communication). Given the increased intestinal permeability and microbial translocation we observed in uninfected PTM, this model should be well suited for assessing therapeutic interventions such as prebiotics and probiotics, as well as other dietary supplements in order to reduce microbial translocation and pathogenic consequences of damage to the GI tract. Taken together, these data highlight the importance of microbial translocation in disease progression and suggest that the PTM model is an ideal system to study therapeutic interventions that improve the structural and immunological functions of the GI tract.
Materials and Methods
Animals and Sample Collection
For this study, 13 SIV-uninfected pigtail macaques (Macaca nemestrina), and 10 SIV-uninfected rhesus macaques (Macaca mulatta) were sacrificed and the following tissues were collected: blood, spleen, axillary LN, mesenteric LN, inguinal LN, jejunum, duodenum, ileum, colon, and cecum. Lymphocytes were isolated from LNs and GI tract tissues by grinding tissues through a 0.22μm cell strainer. Lymphocytes were isolated from blood by ficoll gradient centrifugation. Lymphocytes were then viably cropreserved in fetal bovine serum supplemented with 10% dimethyl sulfoxide (Sigma), and stored in liquid nitrogen. Blood was collected from an additional 12 PTM and 12 RM for phenotypical analysis, and lymphocytes were collected as described above. Animals were housed and cared in accordance with American Association for Accreditation of Laboratory Animal Care standards in AAALAC accredited facilties, and all animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Flow Cytometry
Multicolor flow cytometric analysis was performed on isolated cells according to standard procedures using human mAbs that cross-react with PTM and RM. Predetermined optimal concentrations were used of the following antibodies: anti-CD3-Alexa700 (clone SP34-2, BDPharmigen), anti-CD8 PacBlue (clone RPA-T8, BDPharmigen), anti-CD4 PE-Cy5.5 (clone OKT4, eBioscience), anti-CD4 PE-Cy5.5 (clone L200, BDPharmigen), anti-Ki67 FITC (clone B26, BDPharmigen), anti-CCR5 PE (clone 3A9, BDPharmigen), Aqua Live/Dead amine dye (Invitrogen), anti-CD28 ECD (clone 28.2, Beckman Coulter), anti-CD95 PE-Cy5 (clone DX2, BDPharmigen), anti-CD14 APC (clone M5E2, BDPharmigen), anti-IFNγ PE-Cy7 (clone 4S.B3, BDPharmigen), and anti-IL-17 PE (clone eBio64CAP17, eBioscience). All samples were permeabilized and fixed using CytoFix/Perm Kit (BDPharmigen) and intracellularly stained to detect Ki67. For intracellular cytokine staining, cells were stimulated with phorbol myristate acetate (PMA, 5ng/mL) and ionomycin (1uM/mL) for 10-14 hours with brefeldin A (1ug/mL) added after 1 hour of stimulation. Flow cytometric acquisition was performed on at least 100,000 lymphocytes on a BD FACSAriaII cytometer driven by the FACS DiVa software (version 6.0; BD). Analysis of the acquired data was performed using FlowJo software (version 8.8.4; TreeStar). We used a threshold of 200 collected events for all analysis of specific cell subsets. Memory-effector T cells were determined by gating on both CD28+C95+ and CD28-subsets, while CD28+CD95- T cells were determined to be naïve cells.
Immunohistochemistry
Immunohistochemical staining was performed as previously described81. In brief, unselected specimens of tissues of interest were obtained at necropsy, fixed in 4% paraformaldehyde, and paraffin embedded. Immunohistochemistry was performed using a biotin-free polymer approach (MACH-3; Biocare Medical) on 5-μm tissue sections mounted on glass slides, which were dewaxed and rehydrated with double-distilled water. Antigen retrieval was performed by heating sections in 1× DIVA Decloaker reagent (Biocare Medical) in a pressure cooker (Biocare Medical) followed by cooling to room temperature. All slides were stained using the intelliPATH FLX autostaining system (Biocare Medical) according to experimentally determined optimal conditions. This included blocking tissues with Sniper Blocking Reagent (Biocare Medical) for 10 min followed by an additional blocking step with TNB containing 2% Blocking Reagent and 2% goat serum (Gibco) for 10 minutes at room temperature. Endogenous peroxidase was blocked with 1.5% (v/v) H2O2 in TBS (pH 7.4). Primary antibodies were diluted in TNB containing 2% Blocking Reagent and 2% goat serum for 1h at room temperature. Mouse or rabbit MACH-3 secondary polymer systems (Biocare Medical) were applied for 20 minutes each. Sections were developed with ImmPACT™ DAB chromogen (Vector Laboratories), counterstained with hematoxylin, and mounted in Permount (Fisher Scientific). All stained slides were scanned at high magnification (400×) using the ScanScope CS System (Aperio Technologies, Inc.) yielding high-resolution data from the entire tissue section. Primary antibodies used were mouse anti-LPS core (clone WN1 222-5; Hycult or provided by Dr. Robin Barclay), polyclonal rabbit anti-Claudin-3 (Labvision) and anti-MxA (clone M143; from Drs. Otto Haller and Georg Kochs, Department of Virology, Universitätsklinikum Freiburg, Germany).
Quantitative Image Analysis
To quantify the extent of epithelial barrier damage in the colon (claudin-3), microbial translocation into the lamina propria (LP) and lymph nodes (LPS) and immune activation in the colon and lymph nodes (MxA), 5-μm thick sections were cut from paraffin blocks of unselected colon tissue obtained at necropsy and stained with either monoclonal antibody against LPS or polyclonal antibody for claudin-3 and counter stained with heamotoxalin. High power (400×) whole tissue scans were obtained using an Aperio ScanScope as described above and imported into Photoshop CS3 (Adobe Systems Inc., Mountain View, California, USA). Images were manually trimmed to remove the submucosae, muscularis and residual luminal content, leaving only the LP mucosae to analyze. The percent area of the LP staining for LPS and MxA was determined in essence as previously described using Photoshop CS3 tools with plug-ins from Reindeer Graphics 81, 82. MxA was calculated as the area fraction of the cellular LP, while LPS was calculated as the area fraction of the entire LP. The percent area of LN staining for LPS was determined from whole LN scans as above but without the need to trim the image. The proportion of the epithelial barrier that was damaged during SIV infection was first determined by manually tracing (in green) the area of the lumen/GI epithelial tract interface that had no claudin-3 staining epithelial cells using the brush tool in Photoshop CS3. The remaining claudin-3 staining intact epithelial cell regions were then manually traced (in pink). The percent damage was calculated by determining the proportion of the image that was green (lack of claudin-3 stain) compared to the total epithelial surface area (green + pink) using plug-in tools from Reindeer Graphics.
Plasma LPS and sCD14 Levels
To determine plasma LPS levels we diluted plasma samples to 20% with endoxtoxin-free water and then heated to 70 °C for 10 min to inactivate plasma proteins. We then quantified plasma LPS with a commercially available Limulus Amebocyte assay (Cambrex) according to the manufacturer's protocol. Each sample was run in duplicate and background (water control) was subtracted.
To determine plasma sCD14 levels we diluted plasma samples 1:200 with dilution buffer and performed ELISA according to the manufacturer's protocol (R & D systems). Each sample was run in duplicate and background (water control) was subtracted.
Supplementary Material
Acknowledgments
We would like to acknowledge JoAnne Swerczek, Heather Cronise-Santis, Richard Herbert, and all the veterinary staff at the NIH animal center. We would like to thank the Bad Boys of Cleveland (BBC) for advice and helpful discussions. We would like to thank Jon Meddings, Marie Claire Arrieta, and Kyung Park at the University of Alberta. Judith A. Briant is funded by the Howard Hughes Medical Institute Research Scholars Program. These studies were supported by the Intramural National Institute of Allergy and Infectious Diseases, US National Institutes of Health program. This project has also been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does it mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Statistical analysis We performed Spearman's rank correlation, linear regression and Mann-Whitney U tests using Prism 5.0 software (Prism).
Disclosure: The authors declare no conflicts of interest.
References
- 1.Boirivant M, Amendola A, Butera A. Intestinal microflora and immunoregulation. Mucosal Immunol. 2008;1(1):S47–49. doi: 10.1038/mi.2008.52. [DOI] [PubMed] [Google Scholar]
- 2.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart DC, Dignass AU. Intestinal barrier function. Current Opinion in Clinical Nutrition & Metabolic Care. 2002;5:685–694. doi: 10.1097/00075197-200211000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 5.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 7.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 9.Favre D, et al. Critical Loss of the Balance between Th17 and T Regulatory Cell Populations in Pathogenic SIV Infection. PLoS Pathog. 2009;5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1:339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 12.Chung DR, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- 13.Brand S, et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827–838. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 14.Sugimoto K, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, et al. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 16.Wu Q, et al. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zelante T, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–2706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- 18.Kleinschek MA, et al. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J Immunol. 2006;176:1098–1106. doi: 10.4049/jimmunol.176.2.1098. [DOI] [PubMed] [Google Scholar]
- 19.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujino S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins SM, Vallance B, Barbara G, Borgaonkar M. Putative inflammatory and immunological mechanisms in functional bowel disorders. Baillieres Best Pract Res Clin Gastroenterol. 1999;13:429–436. doi: 10.1053/bega.1999.0037. [DOI] [PubMed] [Google Scholar]
- 22.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. 2008;57:1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 24.Monteleone G, Fina D, Caruso R, Pallone F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr Opin Gastroenterol. 2006;22:361–364. doi: 10.1097/01.mog.0000231808.10773.8e. [DOI] [PubMed] [Google Scholar]
- 25.Brenchley JM, et al. Preferential infection shortens the life span of human immunodeficiency virus-specific CD4+ T cells in vivo. J Virol. 2006;80:6801–6809. doi: 10.1128/JVI.00070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 27.Gordon SN, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol. 2007;179:3026–3034. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veazey RS, et al. Identifying the Target Cell in Primary Simian Immunodeficiency Virus (SIV) Infection: Highly Activated Memory CD4+ T Cells Are Rapidly Eliminated in Early SIV Infection In Vivo. J Virol. 2000;74:57–64. doi: 10.1128/jvi.74.1.57-64.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guadalupe M, et al. Severe CD4+ T-Cell Depletion in Gut Lymphoid Tissue during Primary Human Immunodeficiency Virus Type 1 Infection and Substantial Delay in Restoration following Highly Active Antiretroviral Therapy. J Virol. 2003;77:11708–11717. doi: 10.1128/JVI.77.21.11708-11717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandrea IV, et al. Acute Loss of Intestinal CD4+ T Cells Is Not Predictive of Simian Immunodeficiency Virus Virulence. J Immunol. 2007;179:3035–3046. doi: 10.4049/jimmunol.179.5.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sumpter B, et al. Correlates of preserved CD4(+) T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J Immunol. 2007;178:1680–1691. doi: 10.4049/jimmunol.178.3.1680. [DOI] [PubMed] [Google Scholar]
- 32.Klatt NR, et al. Availability of activated CD4+ T cells dictates the level of viremia in naturally SIV-infected sooty mangabeys. J Clin Invest. 2008;118:2039–2049. doi: 10.1172/JCI33814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giorgi JV, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 34.Paiardini M, et al. Cell-cycle dysregulation in the immunopathogenesis of AIDS. Immunol Res. 2004;29:253–268. doi: 10.1385/IR:29:1-3:253. [DOI] [PubMed] [Google Scholar]
- 35.Paiardini M, et al. Perturbations of cell cycle control in T cells contribute to the different outcomes of simian immunodeficiency virus infection in rhesus macaques and sooty mangabeys. J Virol. 2006;80:634–642. doi: 10.1128/JVI.80.2.634-642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurtrel B, et al. Apoptosis in SIV infection. Cell Death Differ. 2005;12(1):979–990. doi: 10.1038/sj.cdd.4401600. [DOI] [PubMed] [Google Scholar]
- 37.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 38.Jiang W, et al. Plasma Levels of Bacterial DNA Correlate with Immune Activation and the Magnitude of Immune Restoration in Persons with Antiretroviral, ÄêTreated HIV Infection. The Journal of Infectious Diseases. 2009;199:1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baroncelli S, et al. Microbial translocation is associated with residual viral replication in HAART-treated HIV+ subjects with <50copies/ml HIV-1 RNA. J Clin Virol. 2009 doi: 10.1016/j.jcv.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Marchetti G, et al. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS. 2008;22:2035–2038. doi: 10.1097/QAD.0b013e3283112d29. [DOI] [PubMed] [Google Scholar]
- 41.Ancuta P, et al. Microbial Translocation Is Associated with Increased Monocyte Activation and Dementia in AIDS Patients. PLoS ONE. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papasavvas E, et al. Delayed loss of control of plasma lipopolysaccharide levels after therapy interruption in chronically HIV-1-infected patients. AIDS. 2009;23:369–375. doi: 10.1097/QAD.0b013e32831e9c76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anselmi A, et al. Immune reconstitution in human immunodeficiency virus type 1-infected children with different virological responses to anti-retroviral therapy. Clinical & Experimental Immunology. 2007;150:442–450. doi: 10.1111/j.1365-2249.2007.03526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1:23–30. doi: 10.1038/mi.2007.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q, et al. Simian immunodeficiency virus-induced intestinal cell apoptosis is the underlying mechanism of the regenerative enteropathy of early infection. J Infect Dis. 2008;197:420–429. doi: 10.1086/525046. [DOI] [PubMed] [Google Scholar]
- 46.Sankaran S, et al. Rapid onset of intestinal epithelial barrier dysfunction in primary human immunodeficiency virus infection is driven by an imbalance between immune response and mucosal repair and regeneration. J Virol. 2008;82:538–545. doi: 10.1128/JVI.01449-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniel MD, et al. Simian models for AIDS. Cancer Detect Prev Suppl. 1987;1:501–507. [PubMed] [Google Scholar]
- 48.Letvin NL. Animal models for AIDS. Immunol Today. 1990;11:322–326. doi: 10.1016/0167-5699(90)90127-u. [DOI] [PubMed] [Google Scholar]
- 49.Silvestri G. AIDS pathogenesis: a tale of two monkeys. J Med Primatol. 2008;37(2):6–12. doi: 10.1111/j.1600-0684.2008.00328.x. [DOI] [PubMed] [Google Scholar]
- 50.Picker LJ. Immunopathogenesis of acute AIDS virus infection. Current Opinion in Immunology. 2006;18:399–405. doi: 10.1016/j.coi.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Veazey RS, et al. Gastrointestinal Tract as a Major Site of CD4+ T Cell Depletion and Viral Replication in SIV Infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 52.Alcantara S, et al. Thrombocytopenia is strongly associated with simian AIDS in pigtail macaques. J Acquir Immune Defic Syndr. 2009;51:374–379. doi: 10.1097/QAI.0b013e3181a9cbcf. [DOI] [PubMed] [Google Scholar]
- 53.Batten CJ, et al. Comparative evaluation of simian, simian-human, and human immunodeficiency virus infections in the pigtail macaque (Macaca nemestrina) model. AIDS Res Hum Retroviruses. 2006;22:580–588. doi: 10.1089/aid.2006.22.580. [DOI] [PubMed] [Google Scholar]
- 54.Stratov I, et al. Short communication: characteristics of effective immune control of simian/human immunodeficiency virus in pigtail macaques. AIDS Res Hum Retroviruses. 2006;22:27–32. doi: 10.1089/aid.2006.22.27. [DOI] [PubMed] [Google Scholar]
- 55.Hirsch VM, Johnson PR. Pathogenic diversity of simian immunodeficiency viruses. Virus Res. 1994;32:183–203. doi: 10.1016/0168-1702(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 56.Polacino P, et al. Differential pathogenicity of SHIV infection in pig-tailed and rhesus macaques. J Med Primatol. 2008;37(2):13–23. doi: 10.1111/j.1600-0684.2008.00325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brennan G, Kozyrev Y, Hu SL. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci U S A. 2008;105:3569–3574. doi: 10.1073/pnas.0709511105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brennan G, Kozyrev Y, Kodama T, Hu SL. Novel TRIM5 Isoforms Expressed by Macaca nemestrina. J Virol. 2007;81:12210–12217. doi: 10.1128/JVI.02499-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Russell RG, et al. Epidemiology and etiology of diarrhea in colony-born Macaca nemestrina. Lab Anim Sci. 1987;37:309–316. [PubMed] [Google Scholar]
- 60.Hukkanen RR, Liggitt HD, Anderson DM, Kelley ST. Detection of systemic amyloidosis in the pig-tailed macaque (Macaca nemestrina) Comp Med. 2006;56:119–127. [PubMed] [Google Scholar]
- 61.Rosenberg YJ, et al. Variation in T-lymphocyte activation and susceptibility to SIVPBj-14-induced acute death in macaques. J Med Primatol. 1991;20:206–210. [PubMed] [Google Scholar]
- 62.Brenchley JM, et al. CD4+ T Cell Depletion during all Stages of HIV Disease Occurs Predominantly in the Gastrointestinal Tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacDonald TT, Spencer J. Ontogeny of the gut-associated lymphoid system in man. Acta Paediatr Suppl. 1994;83:3–5. doi: 10.1111/j.1651-2227.1994.tb13219.x. [DOI] [PubMed] [Google Scholar]
- 64.Veazey RS, et al. Characterization of gut-associated lymphoid tissue (GALT) of normal rhesus macaques. Clin Immunol Immunopathol. 1997;82:230–242. doi: 10.1006/clin.1996.4318. [DOI] [PubMed] [Google Scholar]
- 65.Mattapallil JJ, et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 66.Brenchley JM, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cecchinato V, et al. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 2008;1:279–288. doi: 10.1038/mi.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laker MF, Bull HJ, Menzies IS. Evaluation of mannitol for use as a probe marker of gastrointestinal permeability in man. Eur J Clin Invest. 1982;12:485–491. doi: 10.1111/j.1365-2362.1982.tb02230.x. [DOI] [PubMed] [Google Scholar]
- 69.Laker MF, Menzies IS. Increase in human intestinal permeability following ingestion of hypertonic solutions. J Physiol. 1977;265:881–894. doi: 10.1113/jphysiol.1977.sp011750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronni T, Sareneva T, Pirhonen J, Julkunen I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J Immunol. 1995;154:2764–2774. [PubMed] [Google Scholar]
- 71.Haller O, Staeheli P, Kochs G. Interferon-induced Mx proteins in antiviral host defense. Biochimie. 2007;89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 72.Veazey RS, et al. Dynamics of CCR5 Expression by CD4+ T Cells in Lymphoid Tissues during Simian Immunodeficiency Virus Infection. J Virol. 2000;74:11001–11007. doi: 10.1128/jvi.74.23.11001-11007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Picker LJ, et al. IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J Clin Invest. 2006;116:1514–1524. doi: 10.1172/JCI27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho O, et al. Pathogenic infection of Macaca nemestrina with a CCR5-tropic subtype-C simian-human immunodeficiency virus. Retrovirology. 2009;6:65. doi: 10.1186/1742-4690-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sallusto F, Lanzavecchia A. Human Th17 cells in infection and autoimmunity. Microbes Infect. 2009;11:620–624. doi: 10.1016/j.micinf.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 76.Yen HR, et al. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol. 2009;183:7161–7168. doi: 10.4049/jimmunol.0900368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kader M, Bixler S, Piatak M, Lifson J, Mattapallil JJ. Anti-retroviral therapy fails to restore the severe Th-17: Tc-17 imbalance observed in peripheral blood during simian immunodeficiency virus infection. J Med Primatol. 2009;38(1):32–38. doi: 10.1111/j.1600-0684.2009.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huber M, et al. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol. 2009;39:1716–1725. doi: 10.1002/eji.200939412. [DOI] [PubMed] [Google Scholar]
- 79.Ortega C, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86:435–443. doi: 10.1189/JLB.0109046. [DOI] [PubMed] [Google Scholar]
- 80.Sugimoto K, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. The Journal of Clinical Investigation. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Estes JD, et al. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol. 2008;180:6798–6807. doi: 10.4049/jimmunol.180.10.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Estes J, et al. Collagen deposition limits immune reconstitution in the gut. J Infect Dis. 2008;198:456–464. doi: 10.1086/590112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.