

Abstract
Myosin VIIA mutations have been associated with non-syndromic hearing loss (DFNB2; DFNA11) and Usher syndrome type 1B (USH1B). We report clinical and genetic analyzes of a consanguineous Iranian family segregating autosomal recessive non-syndromic hearing loss (ARNSHL). The hearing impairment was mapped to the DFNB2 locus using Affymetrix 50K GeneChips; direct sequencing of the MYO7A gene was completed. The Iranian family (L-1419) was shown to segregate a novel homozygous missense mutation (c.1184G>A) that results in a p.R395H amino acid substitution in the motor domain of the myosin VIIA protein. Since one affected family member had significantly less severe hearing loss we used a candidate approach to search for a genetic modifier. This novel MYO7A mutation is the first reported to cause DFNB2 in the Iranian population and this DFNB2 family is the first to be associated with a potential modifier. The absence of vestibular and retinal defects, and less severe low frequency hearing loss, is consistent with the phenotype of a recently reported Pakistani DFNB2 family. Thus, we conclude this family has non-syndromic hearing loss (DFNB2) rather than Usher syndrome type 1B (USH1B), providing further evidence that these two diseases represent discrete disorders.
Keywords: DFNB2, genetic modifier, MYO7A gene, missense mutation, motor domain, myosin VIIA protein, USH1B
INTRODUCTION
Sensorineural hearing loss (SNHL) is the most prevalent human genetic sensory defect. It is estimated that globally 4 of every 10,000 children born have profound hearing loss 1. Non-syndromic SNHL accounts for ~70% of hereditary hearing loss and 80% of SNHL cases have an autosomal recessive mode of inheritance (ARNSHL). To date, 25 genes and 67 loci have been implicated in ARNSHL 2.
In 1995, mutations in MYO7A were associated with hearing impairment and vestibular dysfunction in Usher syndrome (type 1B; USH1B), the most frequent cause of deaf-blindness in humans 3. Since that time at least 126 different recessive mutations in MYO7A have been reported (Human Gene Mutation Database; www.hgmd.cf.ac.uk). Most of these mutations are associated with the USH1B phenotype, however, in 1997 Weil and colleagues and Liu and colleagues reported non-syndromic hearing loss at the DFNB2 locus in one Tunisian and two Chinese families, respectively 4, 5 (Table 1). A number of dominant alleles of MYO7A have also been implicated in progressive, non-syndromic hearing loss at the DFNA11 locus 6-10. shaker-1 mice that exhibit hearing impairment and vestibular dysfunction but not retinitis pigmentosa (RP) also carry mutations in the murine orthologue of MYO7A 11.
Table 1.
Reported 'DFNB2'-causing mutations in the MYO7A gene
| Family | Mutation | Domain | Type | Age of onset | Frequency loss | Vestibular dysfunction | RP | References |
|---|---|---|---|---|---|---|---|---|
| 1 Chinese (DFNB.01) | p.R244Pa | Motor | DFNB2? | Congenital | All frequencies | Some affected family members (CT) | No (ERG) | 5 |
| 1 Chinese (DFNB.05) | IVS3nt-2A>Ga,b p.V1199insT[FS]a,b |
Multiple domains | DFNB2? | Congenital | All frequencies | All affected family members (CT) | No (ERG) | 5 |
| 1 Tunisian | p.M599Ic | Motor | USH1B? | Birth up to 16 years | All frequencies | Some affected family members (CT) | Yes (FU) | 4 |
| 1 Pakistani (PKDF034) | p.E1716del | Unknown | DFNB2 | Congenital | All frequencies | No (CT) | No (ERG; FU) | 13 |
| 1 Iranian (L-1419) | p.R395H | Motor | DFNB2 | Seven months up to 7 years | All frequencies | No (CT) | No (FU) | This study |
CT, caloric tests; ERG, electroretinograms; FU, funduscopy; RP, retinitis pigmentosa.
These families were reported to have the DFNB2 phenotype (9). However, they both exhibit profound hearing loss across all frequencies and vestibular dysfunction is present in atleast some affected family members. Early onset RP was ruled out by ERG in affected family members (between 25 and 33 years of age) but late-onset RP has not been excluded. Independent studies have speculated that these families are more likely to have the USH1B phenotype (13, 27).
Compound heterozygous.
This mutation was originally associated with the DFNB2 phenotype. However, affected members of the Tunisian family were subsequently shown to develop RP as early as 25 years of age (USH1B) (25).
The existence of MYO7A mutations that are associated with a DFNB2 non-syndromic hearing loss phenotype distinct from the USH1B phenotypic spectrum is controversial. In this study, we characterize an Iranian ARNSHL family and identify a novel DFNB2-causing mutation in MYO7A. This missense mutation located in the motor domain represents one of only a few mutations that have been linked to the rare DFNB2 phenotype. The absence of vestibular and retinal phenotypes in this family, and the less severe low frequency hearing loss, is consistent with the clinical presentation of a recently reported Pakistani DFNB2 family 12 (Table 1). One affected individual was shown to have an atypically mild phenotype. Thus, we provide further evidence that an isolated DFNB2 phenotype exists and investigate a potential DFNB2 modifier in this family.
MATERIALS AND METHODS
Clinical Evaluation of Family
Family L-1419 is a five-generation consanguineous Iranian family segregating apparent ARNSHL (Fig.1). Hearing impaired persons appear in the 5th generation, consistent with autozygosity by descent. The reported onset of hearing loss ranges from seven months to 7 years of age (Table 2). To document the degree of hearing loss audiologic testing was completed on consenting family members (Fig.2A). Deficits in hearing were seen at all frequencies tested although low frequency hearing was less impaired. These audiograms were compared to those previously reported for the Pakistani DFNB2 family PKDF034 and they appear very similar (Fig.2B) 12. The less severe hearing impairment of individual V:2 in family L-1419 may be explained by a genetic modifier effect (Fig.2A; Table 2). No balance problems were reported by affected individuals and caloric tests performed on individuals V:1 and V:3 revealed normal vestibular function (Table 2). Sibling V:2 presented for caloric testing but decided not to complete the test. Funduscopic examination showed intact fundi and visual acuity tests revealed 20/20 vision in affected family members as late as 42 years of age. However, in the absence of electroretinograms (ERGs) in the affected family members, we cannot exclude a mild form of retinopathy. Ten milliliters of whole blood was obtained from family members by venipuncture and genomic DNA was extracted as described previously 13. Human research institutional review boards at the Welfare Science and Rehabilitation University and the Iran University of Medical Sciences, Tehran, Iran, and the University of Iowa, Iowa City, Iowa, USA approved all procedures. All family members involved in the study gave written informed consent.
Figure 1. Pedigree of the Iranian DFNB2 family L-1419.
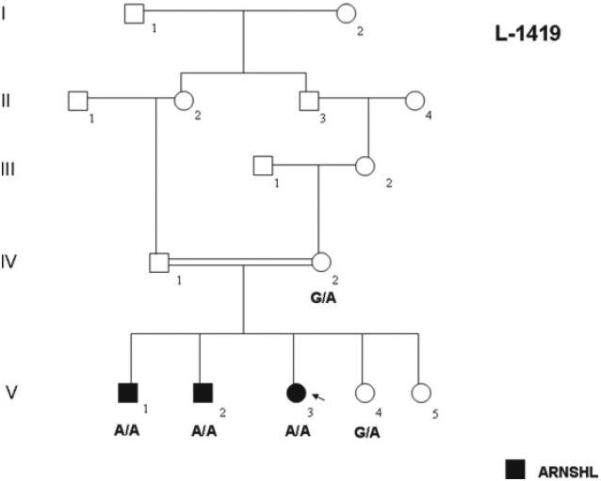
Genotypes for individuals carrying the c.1184G>A mutation are shown. Open symbols = unaffected; filled symbols = affected.
Table 2.
Clinical presentation of the Iranian DFNB2 family L-1419
| Patient | Age | Sex | Onset of hearing loss | Hearing threshold (dB) | Caloric test | Fundus examination | Visual acuity |
|---|---|---|---|---|---|---|---|
| V:1 | 39 | M | Seven months | ≥ 70 | Normal | Normal | 20/20 |
| V:2 | 31 | M | 7 years | ≥ 45 | Not tested | Normal | 20/20 |
| V:3 | 42 | F | 1.5 years | ≥ 70 | Normal | Normal | 20/20 |
Figure 2.
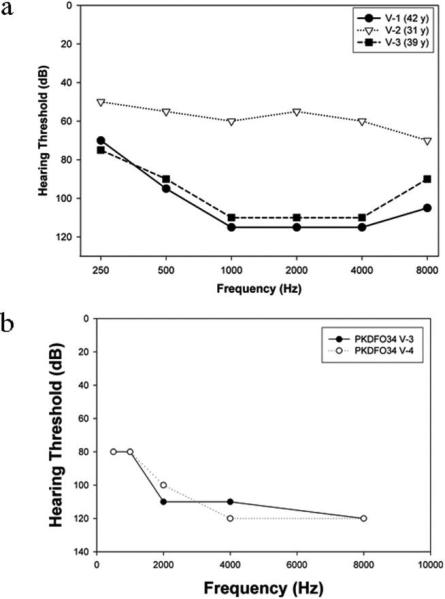
Audiograms of affected members of families L-1419 (A) and PKDF034 12 (B). The audiograms for family PKDF034 have been previously reported 12.
SNP Genotyping and Linkage Analysis of Family L-1419
Genomic DNA from individuals IV-1, IV-2, V-1, V-2, V-3, V-4 and V-5 (Fig.1) was genotyped for 50,000 SNPs using Affymetrix 50K Xba GeneChips at the Translational Genomics Research Institute (TGEN, Phoenix, Arizona). Genotypes were determined using the BRLMM genotyping algorithm 14. An autosomal, genome-wide parametric linkage analysis was performed since males and females appeared equally affected. All linkage analysis was performed with MERLIN 15. A subset of 9411 single nucleotide polymorphisms (SNPs) spaced at least 0.15 cM apart across the genome and with an average heterozygosity of 0.39 was chosen from the 50K Xba set to satisfy the linkage equilibrium requirements of the Lander-Green algorithm for linkage analysis 16. The selection and assembly of the data files were performed with linkdatagen 17. A parametric linkage analysis was run assuming a fully penetrant autosomal recessive model with a disease allele frequency of Pr(a)=0.0001 and penetrances of pr(disease|aa)=0.999 and pr(disease|aA)=Pr(disease|AA)=0.001. Haplotypes inferred with MERLIN were imported into Haplopainter 18. Linkage analysis allowing for the presence of hidden inbreeding loops was carried out with FEstim 19, 20.
PCR and Sequencing
The entire MYO7A [NM_000260] and PMCA2 [NM_001683] genes were amplified using gene-specific primers. For PMCA2, previously reported primers were used 21. Oligonucleotides used to amplify USH1C and SNPs in USH1G, PCDH15, WFS1, DFNM1 and DFNM2 are available upon request. Amplification reactions were cycled using a standard protocol on a GeneMate Genius thermocycler (ISC BioExpress, UT, USA). Sequencing was completed with a BigDye™ v3.1 Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. Sequencing products were read using an ABI 3730s sequencer (Perkin Elmer, Waltham, MA).
RESULTS
Linkage Mapping to the DFNB2 Locus
Genome-wide linkage analysis of family L-1419 detected 3761 Mendelian errors. The maximum parametric LOD score was 2.95 for a region on chromosome 11 and a region on chromosome 1 (Fig.3A,B). The region on chromosome 1 spanned approximately ~ 4 cM flanked by markers SNP_A-1739415 (rs10494078) and SNP_A-1682084 (rs478801) at chromosomal position 1p13.3(~3 Mb). The region on chromosome 11 was investigated further as it overlapped the known autosomal recessive deafness locus DFNB2. This region spanned ~ 5 cM flanked by markers SNP_A-1658804 (rs826056) and SNP_A-1710942 (rs949083) at chromosomal position 11q13.4-q14.1 (~5.5 Mb) (Fig.3A,B). Linkage analysis with FEstim also indicated that IV:1 was himself the offspring of a consanguineous marriage (estimated inbreeding coefficient F=0.031) with his parents estimated to be first cousins once removed. FEstim adjusted LOD scores for the two regions was LOD=2.4. Neither region could be eliminated due to the presence of the hidden inbreeding loop.
Figure 3. Linkage mapping and mutation analysis of family L-1419.
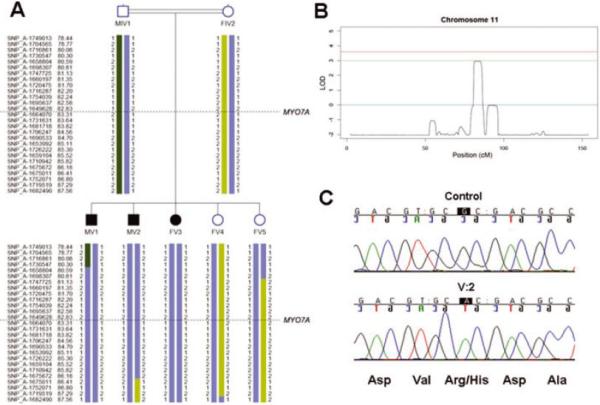
(A) Haplotype analysis for the chromosome 11 region. Genetic map positions in centimorgan (cM) are displayed on the left. The approximate location of the MYO7A gene is indicated. (B) Plot showing LOD score of 2.95 for the region on chromosome 11. LOD scores of 0 and 3 are indicated by solid lines. (C) Chromatogram showing the c.1184G>A mutation in family L-1419.
Mutation in MYO7A
The critical region on chromosome 11 contained the known ARNSHL locus DFNB2. Direct sequencing of the MYO7A gene revealed a novel homozygous missense mutation (c.1184G>A) in exon 11 that substitutes a histidine for an arginine (p.R395H) in the motor domain of the myosin VIIA protein (Fig.3C; Table 3). All affected individuals were homozygous for this mutation while their unaffected mother was heterozygous (Fig.1).
Table 3.
MYO7A mutation identified in the family L-1419
| Family | Ethnicity | Nucleotide alterationa | Amino acid alteration | Allele frequency | Domain |
|---|---|---|---|---|---|
| L-1419 | Iranian | c.1184G>Ab | p.R395H | 1/1 | Motor |
| Iranianc | c.1184G>Ab | p.R395H | 0/47 | Motor | |
| Europeanc | c.1184G>Ab | p.R395H | 0/129 | Motor |
Nucleotides numbered according to first coding ATG in exon 1.
Homozygous alteration.
Ethnically matched control subjects.
Centre d'Etude du Polymorphisme Humain (CEPH) control.
The p.R395 residue located in the motor domain of the myosin VIIA protein is exposed and highly conserved across species with a Conseq score of 9 ((http://conseq.tau.ac.il/). The arginine sidechain is strongly basic because its positive charge is stabilized by resonance whereas the nitrogens in the histidine sidechain have only a relatively weak affinity for protons and are only partially positive at neutral pH. This change in charge is likely to alter the local pH and compromise the structure and/or function of the motor domain. The c.1184G>A mutation was not observed in 47 (94 chromosomes) Iranian control or 129 (258 chromosomes) CEPH (Centre d'Etude du Polymorphisme Humain) control individuals (Table 3).
Investigation of a genetic modifier of the DFNB2 phenotype
The existence of a genetic modifier of MYO7A mutations has been suggested based on the variable phenotype of members of an American DFNA11 family 6, 22. Candidate modifier genes were selected based on their known interaction with MYO7A in hair cells or association with low frequency hearing, and most are members of the Usher type 1 and 2 protein network (Table 4) 23, 24. Since it has been suggested that the modifier is a common polymorphism not tightly linked to MYO7A mutations 22, the ATP2B2 (PMCA2) gene, a known deafness modifier 21, was screened for polymorphisms in individual V:3 with a typical DFNB2 presentation, and individual V:2 with atypically mild hearing impairment. Direct sequencing of the entire coding region and splice sites of the ATP2B2 gene did not reveal any polymorphic differences between these individuals, suggesting that the PMCA2 calcium pump does not modify the DFNB2 phenotype in this family (data not shown). The known ATP2B2 modifier allele (c.2075G>A) and a polymorphism (rs2289274) were also screened in individual V:1 (Table 4).
Table 4.
SNP genotyping in candidate modifier genes of the DFNB2 phenotype
| Gene | SNP | Genotype V:1 | Genotype V:2 | Genotype V:3 | References |
|---|---|---|---|---|---|
| ATP2B2 (PMCA2) | c.2075G>A (p.V586M) | G/G | G/G | G/G | 21 |
| rs2289274 (C>T) | C/T | C/T | C/T | ||
| USH1C (HARMONIN) | rs1055574 (C>T) | C/T | T/T | C/T | 31 |
| rs2240487 (A>G) | A/G | A/A | A/G | ||
| USH1G (SANS) | rs1013013 (G>A) | G/G | G/G | G/G | 32 |
| rs1558448 (G>C) | G/C | G/C | G/G | ||
| PCDH15 | rs10825114 (G>T) | G/G | G/G | G/G | 33 |
| rs1336190 (A>G) | A/G | A/G | A/G | ||
| WFS1 (WOLFRAMIN) | rs1801213 (C>G) | C/G | C/G | G/G | 34 |
| rs1801206 (C>T) | C/T | C/T | T/T | ||
| DFNM1 | rs2032555 (T>C) | T/C | T/C | T/C | 35 |
| rs12076134 (T>G) | T/G | T/T | G/G | ||
| DFNM2 | rs3735875 (G>A) | G/G | G/G | G/G | 36 |
| rs6991392 (A>G) | A/A | A/A | A/A |
We next selected two SNPs with high heterozygosity in each of 4 additional candidate modifier genes and 2 known deafness modifier loci (Table 4). Genotyping of V:1, V:2 and V:3 excluded all genes and loci except harmonin (USH1C). It should be noted, however, that there is a high probability these individuals are genotypically different at these SNPs by random chance.
To identify a modifier allele the entire coding region and splice sites of USH1C were sequenced in individual V:2. All variants were then genotyped in individual V:1 and V:3 (Table 5). Based on genotyping data coding SNP rs10832976 (c.2340C>T; p.V779V) and non-coding SNPs rs2240489 (C>G) and rs2041027 (C>T) could confer a protective effect on the hearing of individual V:2 and represent a modifier allele. Since none of these three SNPs alter the harmonin protein sequence predictive functional analysis was completed using FASTSNP (http://fastsnp.ibms.sinica.edu.tw/pages/input CandidateGeneSearch.jsp). However, this analysis demonstrated only a low probability of any of these variants being located within any exonic splicing enhancers (ESEs) or silencers (ESSs), or transcription factor binding sites.
Table 5.
SNP genotyping in harmonin (USH1C)
| SNP | Location | Genotype V:1 | Genotype V:2 | Genotype V:3 | Protein alteration |
|---|---|---|---|---|---|
| rs2240489 (C>G) | Intron 1 | C/G | G/G | C/G | None |
| rs2041027 (C>T) | Intron 2 | C/T | T/T | C/T | None |
| rs10832976 (C>T) | Exon 23 | C/T | T/T | C/T | p.V779V |
| rs1064074 (G>C) | Exon 24 | G/C | G/C | G/G | p.E819D |
| rs2072232 (G>C) | Intron 25 | G/C | G/C | G/G | None |
| rs10832795 (T>C) | Intron 26 | T/T | T/C | T/C | None |
DISCUSSION
It has been controversial whether the DFNB2 phenotype is a distinct entity or part of the USH1B disease spectrum. For example, the Tunisian family used to define the DFNB2 locus was diagnosed with hearing impairment and vestibular dysfunction in 1994 but when reassessed seven years later, affected persons were also found to have mild RP 25. This phenotypic progression in symptoms led the authors to conclude that DFNB2 and USH1B represent variable expression of the same disease. An independent group therefore re-examined clinical data from the Tunisian and Chinese 5 ‘DFNB2’ families (Table 1) and also concluded that these families most likely have USH1B 26. In contrast, however, a recent report described a Pakistani family with a MYO7A mutation and non-syndromic hearing loss (in the absence of vestibular dysfunction and RP), providing strong clinical data to support the recognition of a DFNB2 phenotype 12.
The clinical presentation of the Iranian family (L-1419) we describe in this report is noteworthy because of its similarity to the Pakistani DFNB2 family (PKDFO34; Table 1; Fig.2) 12. Late-onset RP was ruled out in the Pakistani family by electroretinography (ERG) and funduscopy 12. Likewise funduscopy and visual acuity tests up to 42 years of age were normal excluding severe but not mild RP in Iranian family L-1419 described here. The lack of signs of retinopathy at this age on funduscopy suggests the absence of RP but without measurement of ERGs a mild RP cannot be excluded. Flores-Guevara and colleagues recently concluded that ERG is required to diagnose retinopathy even in children with intact fundi 27. Of note, in the Tunisian and Chinese families, the ages of reported affected persons who had ophthalmologic examinations were 25-65 (Tunisian), 25-27 (Chinese DFNB.01) and 28-33 (Chinsese DFNB.05) years of age, respectively (Table 1) 5, 25. Since the three affected members of family L-1419 were shown to have normal vision by ophthalmological examination between 31 and 42 years of age, this suggests that RP is not a phenotype associated with their disease although a mild RP cannot be ruled out.
In both the Iranian and Pakistani DFNB2 families, vestibular function is reported to be normal, as confirmed by caloric testing (Table 2)12. The onset of hearing loss in the Pakistani family was reported to be congenital and in two of the Iranian family members the hearing impairment was recorded very early in life, suggesting a congenital defect. A mild-to-moderate hearing loss in Iranian family member V:2 is consistent with a genetic modifier effect. It is also noteworthy that the audiograms from both families show less severe low frequency hearing loss (Fig.2). These similarities suggest that the affected persons in these families share the DFNB2 phenotype.
Investigation of the molecular mechanisms underlying apparent genotype-phenotype correlations associated with MYO7A mutation has been limited. However, recent experiments by Riazuddin and colleagues have provided some important insights 12. Using GFP-tagged murine myosin VIIA constructs engineered with human DFNB2-causing mutations these researchers were able to show differences in expression of mutant protein in cultured mouse inner ear hair cells. They demonstrated that the mutant form of myosin VIIA protein (p.E1716del) responsible for the DFNB2 phenotype in their Pakistani family still localized to the hair cell stereocilia whereas the mutant proteins associated with the Chinese DFNB.01 family (p.R244P) 5 and two USH1B families (p.D437N and p.G1982R) 12 did not.
The authors investigated the nature of the p.R244P mutation further since patients in the Chinese family were reported to have normal ERGs 12. The human myosin VIIA p.R244 residue is equivalent to the p.R278 residue in chicken smooth muscle myosin II. From the crystal structure of chicken myosin II it has been demonstrated that p.R278 is located on the edge of a large cleft thought to close upon binding to actin; p.R278 makes a hydrogen bond with p.E428 (equivalent to p.D396 in human myosin VIIA) 28, 29. Introduction of a proline residue at amino acid position 278 is predicted to disrupt the opening and closing of this cleft. This impairment is supported by the absence of motor function in the murine myosin VIIA protein with the equivalent amino acid substitution (p.R233P) 12.
It is interesting that the p.R395H mutation described in Iranian DFNB2 family L-1419 in this study affects the p.R395 residue immediately adjacent to the p.D396 residue in human myosin VIIA. One hypothesis is that the local change in charge induced by substituting in a histidine residue interferes with opening and closing of the cleft. To test this hypothesis similar experiments to those described above by Riazuddin and colleagues would need to be completed using a mutant version of myosin VIIA carrying the p.R395H mutation. Since affected members of the Iranian family have no signs of RP at considerably older age (42 years old) than members of the Chinese family (27 years old), perhaps the relationship between MYO7A mutation and DFNB2/USH1B phenotypes is more complex than our current knowledge suggests.
Two interesting aspects to the linkage mapping of Iranian family L-1419 are the identification of a second locus with a high LOD score (2.95) on chromosome 1 and the likely presence of a hidden inbreeding loop (Fig.3). A hidden inbreeding loop can explain the appearance of two LOD scores very close to genome-wide significance as additional inbreeding will generate more homozygosity by descent than expected by chance alone. Based on our MYO7A screening results, the linked interval identified on chromosome 1 may be due to chance only and have no relevance to the phenotype.
The existence of a genetic modifier of MYO7A mutation has been proposed based on the variable phenotypic presentation of an American family with progressive hearing impairment and vestibular dysfunction at the DFNA11 locus 6, 22. It has been suggested that this modifier is a common polymorphism not tightly linked to the MYO7A mutation, although the modifier locus has not been identified 22. We considered the possibility of a genetic modifier effect on the phenotypic presentation of affected members of family L-1419 based on the better hearing of individual V:2. Since all affected individuals are homozygous for the locus on chromosome 1, this genomic region is not the site of a modifier gene, if one exists. Unfortunately, the size of this family precludes localizing the modifier gene by linkage mapping.
Based on this a candidate-gene strategy was adopted to identify the putative modifier gene. Screening of the ATP2B2 gene which encodes the PMCA2 calcium pump, a known deafness modifier gene 21, did not reveal any polymorphic differences between individual V:3 with a typical DFNB2 presentation and individual V:2 with atypically mild hearing impairment. Analysis of SNPs located close to other candidate modifier genes and loci (Table 4) in affected members family V:1, V:2 and V:3 suggested that USH1C might be the site of the genetic modifier. USH1C encodes harmonin, an F-actin-bundling protein that is thought to stabilize actin filaments in the stereocilia. The PDZ1 domain of harmonin has been shown to interact directly with the MYTH4 + FERM repeat at the C-terminal tail of MYO7A 30. Genotyping of USH1C SNPs in V:1, V:2 and V:3 indicated that rs10832976, rs2240489 or rs2041027 might represent the modifier allele. However, these variants do not alter the harmonin protein sequence and have only a low probability of residing within an ESE, ESS or transcription factor binding site, suggesting they are not modifying the hearing loss in family L-1419.
ACKNOWLEDGEMENTS
The authors sincerely thank the family for their participation in this study. R. Smith is the Sterba Hearing Research Professor, University of Iowa College of Medicine, who supported the project with National Institutes of Health (NIH)-NIDCD grant RO1 DCOO2842. K. Jalalvand and S. Arzhanginy are thanked for their contribution to this project. H. Najmabadi and K. Khahrizi supported this project with Iranian National Science Foundation Grants 85033/10 and 85073/23. M. Bahlo is supported by an Australian National Health and Medical Research Council (NHMRC) Career Development Award. M. Hildebrand is supported by an NHMRC Postdoctoral Training Fellowship. No researchers involved in this study report a conflict of interest.
Funding: NIH NIDCD grant RO1 DC002842 (RJHS). Iranian National Science Foundation Grant 85033/10 (KK). Australian National Health and Medical Research Council (NHMRC) Career Development Award (MB). NHMRC Postdoctoral Training Fellowship (MH).
Abbreviations
- ARNSHL
autosomal recessive non-syndromic hearing loss
- SNHL
sensorineural hearing loss
- MYO7A
myosin VIIA gene
- USH1B
Usher syndrome type 1B
- ERG
electroretinogram
- RP
retinitis pigmentosa
- RPE
retinal pigment epithelium
- ESE
exonic splicing enhancer
- ESS
exonic splicing silencer
REFERENCES
- 1.Smith RJ, Bale JF, Jr., White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879–90. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- 2.Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. 2008 http://webhost.ua.ac.be/hhh/
- 3.Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–1. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 4.Weil D, Kussel P, Blanchard S, et al. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16:191–3. doi: 10.1038/ng0697-191. [DOI] [PubMed] [Google Scholar]
- 5.Liu XZ, Walsh J, Mburu P, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16:188–90. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 6.Street VA, Kallman JC, Kiemele KL. Modifier controls severity of a novel dominant low-frequency MyosinVIIA (MYO7A) auditory mutation. J Med Genet. 2004;41:e62. doi: 10.1136/jmg.2003.013557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luijendijk MW, Van Wijk E, Bischoff AM, et al. Identification and molecular modelling of a mutation in the motor head domain of myosin VIIA in a family with autosomal dominant hearing impairment (DFNA11). Hum Genet. 2004;115:149–56. doi: 10.1007/s00439-004-1137-3. Epub 2004 Jun 2. [DOI] [PubMed] [Google Scholar]
- 8.Liu XZ, Walsh J, Tamagawa Y, et al. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet. 1997;17:268–9. doi: 10.1038/ng1197-268. [DOI] [PubMed] [Google Scholar]
- 9.Di Leva F, D'Adamo P, Cubellis MV, et al. Identification of a novel mutation in the myosin VIIA motor domain in a family with autosomal dominant hearing loss (DFNA11). Audiol Neurootol. 2006;11:157–64. doi: 10.1159/000091199. Epub 2006 Jan 9. [DOI] [PubMed] [Google Scholar]
- 10.Bolz H, Bolz SS, Schade G, et al. Impaired calmodulin binding of myosin-7A causes autosomal dominant hearing loss (DFNA11). Hum Mutat. 2004;24:274–5. doi: 10.1002/humu.9272. [DOI] [PubMed] [Google Scholar]
- 11.Gibson F, Walsh J, Mburu P, et al. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–4. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 12.Riazuddin S, Nazli S, Ahmed ZM, et al. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat. 2008;29:502–11. doi: 10.1002/humu.20677. [DOI] [PubMed] [Google Scholar]
- 13.Grimberg J, Nawoschik S, Belluscio L, et al. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di X, Matsuzaki H, Webster TA, et al. Dynamic model based algorithms for screening and genotyping over 100 K SNPs on oligonucleotide microarrays. Bioinformatics. 2005;21:1958–63. doi: 10.1093/bioinformatics/bti275. Epub 2005 Jan 18. [DOI] [PubMed] [Google Scholar]
- 15.Abecasis GR, Cherny SS, Cookson WO, et al. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. Epub 2001 Dec 3. [DOI] [PubMed] [Google Scholar]
- 16.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bahlo M, Bromhead CJ. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009 May 12; doi: 10.1093/bioinformatics/btp313. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Thiele H, Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–2. doi: 10.1093/bioinformatics/bth488. Epub 2004 Sep 17. [DOI] [PubMed] [Google Scholar]
- 19.Leutenegger AL, Labalme A, Genin E, et al. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am J Hum Genet. 2006;79:62–6. doi: 10.1086/504640. Epub 2006 Apr 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leutenegger AL, Prum B, Genin E, et al. Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet. 2003;73:516–23. doi: 10.1086/378207. Epub 2003 Jul 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz JM, Yang Y, Caride AJ, et al. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N Engl J Med. 2005;352:1557–64. doi: 10.1056/NEJMoa043899. [DOI] [PubMed] [Google Scholar]
- 22.Kallman JC, Phillips JO, Bramhall NF, et al. In search of the DFNA11 myosin VIIA low- and mid-frequency auditory genetic modifier. Otol Neurotol. 2008;29:860–7. doi: 10.1097/MAO.0b013e3181825651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adato A, Lefevre G, Delprat B, et al. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921–32. doi: 10.1093/hmg/ddi416. Epub 2005 Nov 21. [DOI] [PubMed] [Google Scholar]
- 24.Adato A, Michel V, Kikkawa Y, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–56. doi: 10.1093/hmg/ddi031. Epub 2004 Dec 8. [DOI] [PubMed] [Google Scholar]
- 25.Zina ZB, Masmoudi S, Ayadi H, et al. From DFNB2 to Usher syndrome: variable expressivity of the same disease. Am J Med Genet. 2001;101:181–3. doi: 10.1002/ajmg.1335. [DOI] [PubMed] [Google Scholar]
- 26.Astuto LM, Kelley PM, Askew JW, et al. Searching for evidence of DFNB2. Am J Med Genet. 2002;109:291–7. doi: 10.1002/ajmg.10384. [DOI] [PubMed] [Google Scholar]
- 27.Flores-Guevara R, Renault F, Loundon N, et al. Usher syndrome type 1: Early detection of electroretinographic changes. 2008. [DOI] [PubMed]
- 28.Holmes KC, Geeves MA. The structural basis of muscle contraction. Philos Trans R Soc Lond B Biol Sci. 2000;355:419–31. doi: 10.1098/rstb.2000.0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dominguez R, Freyzon Y, Trybus KM, et al. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 1998;94:559–71. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- 30.Boeda B, El-Amraoui A, Bahloul A, et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–99. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]