

Abstract
Mutations in the POLG gene have emerged as one of the most common causes of inherited mitochondrial disease in children and adults. They are responsible for a heterogeneous group of at least 6 major phenotypes of neurodegenerative disease that include: 1) childhood Myocerebrohepatopathy Spectrum disorders (MCHS), 2) Alpers syndrome, 3) Ataxia Neuropathy Spectrum (ANS) disorders, 4) Myoclonus Epilepsy Myopathy Sensory Ataxia (MEMSA), 5) autosomal recessive Progressive External Ophthalmoplegia (arPEO), and 6) autosomal dominant Progressive External Ophthalmoplegia (adPEO). Due to the clinical heterogeneity, time-dependent evolution of symptoms, overlapping phenotypes, and inconsistencies in muscle pathology findings, definitive diagnosis relies on the molecular finding of deleterious mutations. We sequenced the exons and flanking intron region from approximately 350 patients displaying a phenotype consistent with POLG related mitochondrial disease and found informative mutations in 61 (17%). Two mutant alleles were identified in 31 unrelated index patients with autosomal recessive POLG-related disorders. Among them, 20 (67%) had Alpers syndrome, 4 (13%) had arPEO, and 3 (10%) had ANS. In addition, 30 patients carrying one altered POLG allele were found. A total of 25 novel alterations were identified, including 6 null mutations. We describe the predicted structural/functional and clinical importance of the previously unreported missense variants and discuss their likelihood of being pathogenic. In conclusion, sequence analysis allows the identification of mutations responsible for POLG-related disorders and, in most of the autosomal recessive cases where two mutant alleles are found in trans, finding deleterious mutations can provide an unequivocal diagnosis of the disease.
Keywords: POLG, POLG1, Alpers syndrome, PEO, adPEO, arPEO, SANDO, SCAE, ANS, MEMSA, MCHS, mtDNA depletion, liver failure
INTRODUCTION
Mitochondrial DNA (mtDNA) biogenesis is highly dependent on the function of several nuclear-encoded proteins and the balance of deoxynucleotide pools within the mitochondrial matrix. MtDNA is replicated by DNA polymerase gamma (POLG) encoded by the nuclear POLG gene (MIM# 174763). Human POLG was identified in 1996 (Ropp and Copeland, 1996) and the first mutations within the gene identified as causing human disease were first described in 2001 (Van Goethem, et al., 2001). Since 2001, over 100 pathogenic mutations have been found to cause a vast array of both neurological and non-neurological disorders of variable age of onset and severity.
POLG mutations have been identified in severe mtDNA depletion syndromes such as childhood Myocerebrohepatopathy Spectrum disorders (MCHS) and Alpers-Huttenlocher syndrome [MIM# 203700] (Naviaux and Nguyen, 2004), as well as mtDNA deletion disorders in the Ataxia Neuropathy Spectrum (ANS) which includes spinocerebellar ataxia with epilepsy (SCAE) and mitochondrial recessive ataxia syndrome without ophthalmoplegia (MIRAS) (Winterthun, et al., 2005); Myoclonus Epilepsy Myopathy Sensory Ataxia (MEMSA); and autosomal dominant (ad) [adPEO; MIM# 157640] and recessive [arPEO; MIM# 258450] forms of progressive external ophthalmoplegia (PEO) (Lamantea, et al., 2002; Van Goethem, et al., 2001; Van Goethem, et al., 2003b), which may include sensory ataxic neuropathy with dysarthria and ophthalmoparesis (SANDO), arPEO+ (Milone, et al., submitted; Van Goethem, et al., 2003d) and cases of parkinsonism in PEO+ patients (Davidzon, et al., 2006; Hudson, et al., 2007; Luoma, et al., 2004). As a consequence of POLG failure, accumulation of multiple mtDNA deletions in post-mitotic tissues such as muscle and brain is noted in arPEO+ and adPEO+ (Milone, et al., submitted). Alpers syndrome appears to be the most common autosomal recessive disease caused by mutations in the POLG gene. This early-onset fatal disease is characterized by intractable seizures, hepatic failure, and global neurological deterioration (Naviaux and Nguyen, 2004).
The human POLG gene is located on chromosome 15q25, and its cDNA (GenBank database accession number NM_002693) comprises 4,465 bp including a 282 bp 5' untranslated region (UTR) and a 463 bp 3'UTR. The gene contains 23 exons, spanning approximately 18.5 kb. The translation-initiator methionine codon (Met1) is located in exon 2. The 23 exons range in size from 818 bp (exon 2) to 54 bp (exon 15). The 22 introns range in size from 1415 bp (intron 18) to 103 bp (intron 7). All of the 5' donor and 3' acceptor sites at each exon/intron junction conform to the GT-AG rule. Analysis of 1 kb of the 5'UTR region from the human POLG gene has not revealed any promoter consensus core (Ropp and Copeland, 1996).
The POLG protein (EC 2.7.7.7) is synthesized as a precursor containing an amino-terminal leader sequence (residues 1-25) that targets the protein to mitochondria and is cleaved off after import. The mature 140 kDa protein is divided into three functional domains: 1) a 3'→5' exonuclease (exo) domain (amino acid residues 26-417), 2) a linker domain (amino acid residues 418-755), and 3) a highly conserved carboxy-terminal polymerase (pol) domain (amino acid residues 756-1239) (Lecrenier, et al., 1997; Naviaux and Nguyen, 2004; Ropp and Copeland, 1996). The exo domain increases the fidelity of mtDNA replication by conferring a proofreading activity to the enzyme (Longley, et al., 2001).
A 55 kDa accessory subunit of POLG is encoded by the POLG2 gene, located on chromosome 17q21. This subunit confers high processivity on the protein complex by increasing its binding affinity for DNA (Lim, et al., 1999). Mutations in the POLG2 gene have been reported in a family with PEO and multiple mtDNA deletions (Longley, et al., 2006).
An increasing number of POLG mutations have been reported to be associated with a broad range of clinical phenotypes, some inherited in an autosomal recessive manner, others displaying autosomal dominant inheritance (DiMauro, et al., 2006; Horvath, et al., 2006; Tzoulis, et al., 2006). The POLG mutation database can be found at http://tools.niehs.nih.gov/polg/. Mutations were found to be equally distributed in the three domains of the protein. The p.A467T mutation in the linker region is the most common mutation; it leads to low intrinsic DNA polymerase activity and inefficient interaction with the accessory subunit (Chan, et al., 2005a). However, establishing the significance of amino acid variants in several instances is not straightforward and the clinical significance of many newly discovered missense mutations often remains undetermined. Multiple case studies with biochemical and molecular characterization are helpful in determining the clinical consequences of specific amino acid variations. These studies are particularly important in relation to Alpers syndrome because they provide crucial information needed for accurate genetic counseling, carrier testing and prenatal diagnosis of this severe disease.
In this paper we report the molecular genotypes and clinical consequences for a series of over 60 patients in whom we detected POLG sequence changes, out of approximately 350 patients referred to our laboratory for sequencing of the entire POLG coding region. We describe genotype-phenotype correlations involving 25 novel deleterious mutations or variants of possible clinical significance, interpret these sequence changes in the context of POLG protein structure, and discuss their implications for enzyme function.
MATERIALS AND METHODS
Patients and DNA
Tissue and blood samples of patients with clinical presentations suggestive of POLG deficiency were submitted to the Mitochondrial Diagnostics Laboratory at Baylor College of Medicine for biochemical and/or molecular evaluation. Clinical history of the patients was provided on a check list at the time of specimen submission and detailed clinical description was obtained in selected cases. Parental DNA analysis was performed for 27 of 33 (82%) cases with two mutated alleles to establish the configuration in the affected patient. Total (nuclear and mitochondrial) DNA was extracted from peripheral blood leukocytes or other tissues (muscle, liver, or skin fibroblast culture) using commercially available DNA isolation kits (Gentra Systems Inc., Minneapolis, MN) according to the manufacturer's protocols.
Molecular Analysis
Sequence-specific oligonucleotide primers linked to M13 universal primer sequences were designed to amplify the 22 coding exons and at least 50 nucleotides of each flanking intron (see Supplementary Table S1 for primer sequences). PCR products generated using Fast Start DNA polymerase (Roche, Indianapolis, IN) were purified on ExcelaPure 96-well UF PCR purification plates (Edge BioSystems, Gaithersburg, MD). Sequencing reactions were performed using the BigDye Terminator Cycle Sequencing kit (version 3.1) and analyzed on an ABI3730XL automated DNA sequencer with Sequencing Analysis Software v5.1.1 (Applied Biosystems, Foster City, CA, USA). DNA sequences were analyzed using Mutation Surveyor version 2.61 and the GenBank POLG sequence (ID NM_002693.1). Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence NM_002693.1, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
Phenotype Classifications
When sufficient clinical information was available, we assigned patients to one of 6 major diagnostic groups of POLG disease. These were: 1) Myocerebrohepatopathy Spectrum (MCHS), 2) Alpers Syndrome, 3) Ataxia Neuropathy Spectrum (ANS), 4) Myoclonus Epilepsy Myopathy Sensory Ataxia (MEMSA), 5) Autosomal Recessive Progressive External Ophthalmoplegia Plus (arPEO+), and 6) Autosomal Dominant Progressive Ophthalmoplegia Plus (adPEO+). If insufficient clinical information was available, or if symptoms were non-diagnostic, the patient diagnosis was “Unassigned” (U).
MCHS was defined by the clinical triad of: 1) myopathy or hypotonia, 2) developmental delay, or dementia, and 3) liver dysfunction. In addition, patients had either a liver biopsy that excluded classical Alpers hepatopathy (Nguyen, et al., 2006), or at least two of the following 8 findings: 1) neuropathy, 2) seizures, 3) elevated blood or cerebrospinal fluid lactic acid, 4) dicarboxylic aciduria, 5) renal tubular dysfunction with aminoaciduria, glucosuria, or bicarbonaturia, 6) hearing loss, 7) abnormal MRI with either cerebral volume loss, delayed myelination, or white matter disease, and 8) deficiency of either CIV (cytochrome c oxidase, COX) in isolation, or 2 or more electron transport complexes (CI, CII, CIII, or CIV) in skeletal muscle or liver biopsy. In some cases, patients came to diagnosis without, or before, the onset of liver dysfunction. In these cases, at least 3 of the 8 supportive diagnostic findings were required. Patients with POLG mutations meeting the diagnostic features for MCHS were first described by Ferrari, et al (their patient #9) (Ferrari, et al., 2005), and de Vries, et al (their patients #4-8) (de Vries, et al., 2007).
Alpers syndrome ((MIM# 203700); sometimes called Alpers-Huttenlocher syndrome, Alpers hepatopathic poliodystrophy, or hepatocerebral degeneration of childhood) was defined by the clinical triad of: 1) refractory, mixed-type seizures that often included a focal component, 2) psychomotor regression that was often episodic and triggered by intercurrent infection, and 3) hepatopathy with or without acute liver failure. In addition, either a liver biopsy was performed that showed characteristic histologic features (Nguyen, et al., 2006), or at least 2 of 11 additional clinical, laboratory, electrophysiologic, or neuroimaging features were present (Nguyen, et al., 2006).
Ataxia Neuropathy Spectrum (ANS) includes an overlapping clinical spectrum of disorders organized around ataxia and neuropathy in the absence of significant muscle weakness or myopathy. It embraces mitochondrial recessive ataxia syndrome (MIRAS) (Hakonen, et al., 2005), spinocerebellar ataxia and epilepsy (SCAE), and the ataxia neuropathy spectrum reported by Tzoulis, et al (Tzoulis, et al., 2006).
Myoclonus Epilepsy Myopathy Sensory Ataxia (MEMSA) includes an overlapping spectrum of disorders organized around the finding of myopathy, epilepsy, and ataxia in the absence of ophthalmoplegia. It may occur with or without ragged red fibers (Van Goethem, et al., 2003c).
ArPEO+ includes an overlapping spectrum of disorders organized around the finding of ophthalmoplegia demonstrating recessive inheritance (Van Goethem, et al., 2001). Most patients have additional symptoms. These may include sensory ataxia, neuropathy, dysarthria, and ophthalmoplegia (SANDO) (Van Goethem, et al., 2003b).
AdPEO+ includes a spectrum of disorders organized around the finding of ophthalmoplegia, but demonstrating autosomal dominant inheritance (Van Goethem, et al., 2001).
Molecular Modeling
PyMOL (DeLano Scientific, CA)(DeLano, 2002) was used to visualize and model amino acids in the three-dimensional model of the human POLG catalytic (pol) domain, based on the model pdb file (Graziewicz, et al., 2004).
RESULTS
Mutations of the POLG gene
POLG mutations (Table 1) were identified in a total of 61 unrelated families. These included 33 autosomal recessive cases with two defined pathogenic alleles identified in each index patient; 18 cases with one heterozygous pathogenic mutation identified; and 10 cases with one unclassified novel missense variant identified. Among the patients with two mutant alleles, 29 different pathogenic mutations were identified. These included 16 previously unreported alterations: p.Q68X, p.Q715X, c.2544_2545insC, c.2157+5G>A, c.2480+1G>A, p.G11D, p.L83P, p.R853Q, p.A862T, p.L886P, p.G888S, p.R943C, p.R1128H, p.R1138C, p.K1191R, and p.D1196N (Fig. 1). Among the novel mutations identified, 6 are null mutations caused by nonsense, frameshift, or splice site alterations and the remainder are missense mutations. In 28 unrelated patients who were found to carry one mutated allele, 16 different alterations were identified. Six of these were previously reported as pathogenic mutations. Six of the 10 unclassified missense variants are likely to be pathogenic considering the high degree of evolutionary conservation of the wild-type amino acids, their locations at sites of structural/functional importance, de novo origin, and the non-conservative nature of the amino acid substitution.
Table 1.
Phenotypes Associated with Compound Heterozygous or Homozygous POLG Mutations
| Nucleotide Change | Amino Acid Change | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Gender | Onset (yrs) |
DD or Dementia |
Sz | Liver | PEO | Lact | Dx | Other Symptoms | Allele 1 | Allele 2 | Allele 1 | Allele 2 | Domains |
| 1 | F | 23 | - | + | + | NA | NA | Alpers | Ataxia, neuropathy, hearing loss, valproate-associated liver failure |
c.1399G>A | c.1399G>A | p.A467T | p.A467T | L/L |
| 2 | M | 1 | + | + | + | - | + | Alpers | Stroke, hypotonia | c.1399G>A | c.2157+5_+ 6 gc>ag | p.A467T | c.2157+5 _+6 gc>ag splice? | L/L |
| 3 | F | 2 | + | + | + | - | - | Alpers | Ataxia | c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P |
| 4 | F | 2 | + | + | + | - | + | Alpers | Stroke | c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P |
| 5 | F | 1.5 | + | + | + | - | + | Alpers | Cortical atrophy, hypoglycemia |
c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P |
| 6 | F | 9 | + | + | + | - | - | Alpers | Stroke, ataxia, exercise intolerance |
c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P |
| 7 | M | 0.8 | + | + | - | - | - | Alpers | Hypotonia, failure to thrive |
c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P |
| 8 | M | 2 | + | + | + | - | - | Alpers | c.1399G>A | c.2542G>A | p.A467T | p.G848S | L/P | |
| 9 | M | 1 | + | + | - | - | - | Alpers | Family history of acute liver failure |
c.1399G>A | c.1270_1271del CT | p.A467T | p.L424G fsX28 | L/L-null |
| 10 | M | 1 | + | + | + | - | - | Alpers | c.1399G>A | c.2657T>C | p.A467T | p.L886P | L/P | |
| 11 | F | 3 | + | + | + | - | - | Alpers | Ptosis, ataxia, visual hallucinations |
c.1399G>A | c.202C>T | p.A467T | p.Q68X | L/Exo-null |
| 12 | M | 4 | + | + | - | - | - | Alpers | c.1399G>A | c.2143C>T | p.A467T | p.Q715X | L/L-null | |
| 13 | F | 1.5 | + | + | + | - | + | Alpers | Chorea, microcephaly, leukodystrophy |
c.1399G>A | c.2544_2545 insC c.3708G>T | p.A467T | p.T849H(i nsC) fs868X- p.Q1236H | L/P-null |
| 14 | M | 2 | + | + | - | - | - | Alpers | Chorea, Family history of Alpers syndrome |
c.1399G>A | c.2740A>C | p.A467T | p.T914P | L/P |
| 15 | F | 2 | + | + | + | - | - | Alpers | c.1399G>A | c.2740A>C | p.A467T | p.T914P | L/P | |
| 16** | F | 10 | + | + | + | - | - | Alpers | Cortical blindness, hearing loss |
c.248T>C | c.2662G>A | p.L83P | p.G888S | Exo/P |
| 17** | F | 1 | + | + | + | - | - | Alpers | Family history of Alpers syndrome |
c.1491G>C c.2243G>C c.3428A>G |
c.2542G>A | p.Q497H-p.W748S-p.E1143G | p.G848S | L/P |
| 18 | M | 1 | + | + | + | - | - | Alpers | Family history of Alpers syndrome |
c.3286C>T c.3708G>T |
c.3286C>T c.3708G>T | p.R1096C-p.Q1236H | p.R1096C-p.Q1236H | P/P |
| 19 | M | 0.5 | - | - | - | - | - | Alpers | Family history of Alpers syndrome. Diagnosed pre-symptomatically. |
c.752C>T c.1760C>T |
c.2542G>A | p.T251I-p.P587L | p.G848S | Exo-L/P |
| 20 | F | 4 | + | + | + | - | + | Alpers | Muscle weakness, respiratory failure |
c.2243G>C, c.3428A>G |
c.2480+1 G>A | p.W748S-p.E1143G | C.2480+1 G>A splice | L/P |
| 21 | F | 17 | + | + | - | - | - | ANS | Hearing loss | c.1399G>A | c.1491G>C c.2243G>C c.3428A>G |
p.A467T | p.Q497H-p.W748S-p.E1143G | L/L |
| 22 | M | 17 | + | + | - | - | - | ANS | Ataxia, exercise intolerance, cerebellar atrophy, SCAE |
c.2590C>T | c.2584G>A | p.R964C | p.A862T | P/P |
| 23 | F | 15 | - | + | + | + | + | ANS | Stroke, chorea, ataxia, ptosis, retinitis pigmentosa, liver transplant |
c.2554C>T |
c.32G>A c.1880G>C |
p.R852C | p.G11D-p.R627Q | P/MTS-L |
| 24** | F | 32 | - | - | - | + | - | arPEO+ | Neuropathy, myopathy, SANDO |
c.1399G>A | c.1399G>A | p.A467T | p.A467T | L/L |
| 25** | M | 60 | - | - | - | + | - | arPEO+ | Ataxia, neuropathy, myopathy, hearing loss, cerebellar atrophy, SANDO/SCAE |
c.1399G>A | c.2209G>C | p.A467T | p.G737R | L/L |
| 26** | F | 48 | - | - | - | + | + | arPEO+ | Neuropathy, myopathy, ptosis, SANDO |
c.1399G>A | c.3412C>T | p.A467T | p.R1138C | L/P |
| 27** | M | 33 | - | - | - | + | + | arPEO+ | Neuropathy, myopathy, SANDO |
c.2243G>C, c.3428A>G |
c.2243G>C, c.3428A>G | p.W748S-p.E1143G | p.W748S-p.E1143G | L/L |
| 28 | F | 3 | + | - | - | - | + | MCHS | Renal tubulopathy, dysmorphic featues, cataract, short stature, myopathy |
c.2209G>C | c.2827C>T, c.3428A>G | p.G737R | p.R943C-p.E1143G | L/P |
| 29 | F | 1 | + | - | + | - | + | MCHS | Fatigue, pancreatitis, cyclic vomiting |
c.752C>T, c.1760C>T |
c.3572A>G | p.T251I-p.P587L | p.K1191R | Exo-L/P |
| 30* | F | 0.2 | + | - | - | - | + | MCHS | Microcephaly, failure to thrive, hearing loss, abnormal MRI |
c.752C>T, c.1760C>T |
c.2558G>A | p.T251I-p.P587L | p.R853Q | Exo-L/P |
| 31 | M | 8 | NA | NA | NA | NA | NA | U | NA | c.2243G>C, c.3428A>G |
c.2542G>A | p.W748S-p.E1143G | p.G848S | L/P |
| 32 | M | 1 | + | - | - | - | - | U | Myopathy, RRF, elevated CK |
c.1550G>T | c.3586G>A | p.G517V | p.D1196N | L/P |
| 33 | M | 1 | + | - | + | - | - | U | microcephaly | c.1550G>T | c.3383G>A | p.G517V | p.R1128H | L/P |
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence NM_002693.1, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Novel mutations are shown in bold and neutral variants are in shown in blue.
Abbreviations: MCHS= Myocerebrohepatopathy Spectrum, ANS = Ataxia Neuropathy Spectrum, MEMSA= Myoclonus Epilepsy Myopathy Sensory Ataxia arPEO+ = autosomal recessive Progressive External Ophthalmoplegia, MTS = mitochondrial targeting sequence; Exo = exonuclease domain; L = linker region; P = Polymerase domain; DD = Developmental Delay; Sz = Seizures; Lact = Elevated lactate; Dx = Diagnosis.
Patient recognized early on the basis of family history positive for liver failure in a sibling.
These cases have been described in more details elsewhere (Milone et al., submitted; Bao et al., 2008; Brunetti-Pierri et al., 2008).
Figure 1.
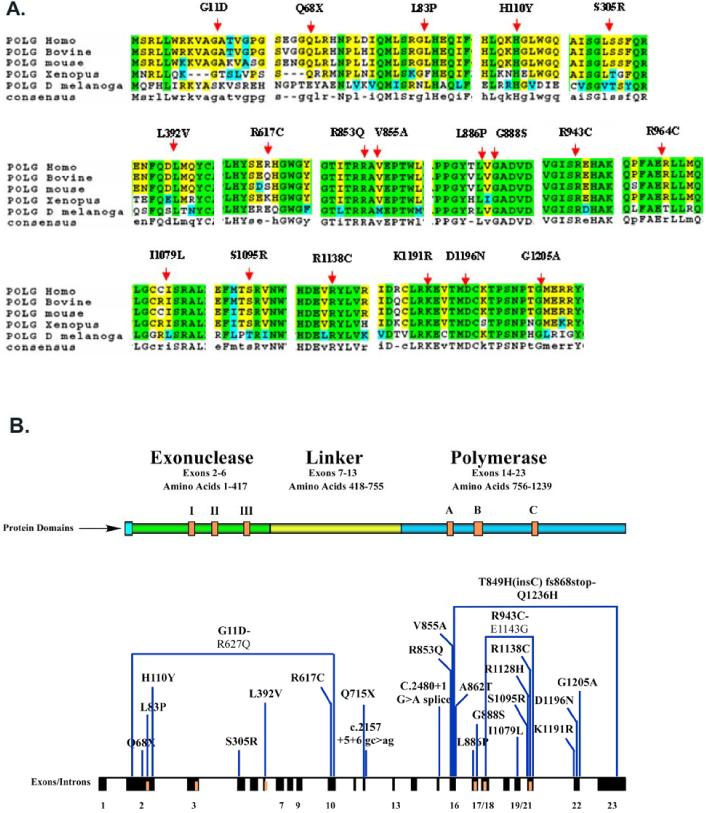
A. Amino acid alignment of the human POLG amino acid reference sequence versus POLG sequences of various species, with the locations of novel missense variants superimposed. B. New POLG Mutations. The position of the 25 novel mutations is indicated with reference to the exon-intron structure of the POLG gene locus, and the corresponding domains of the POLG protein.
Patients with two POLG mutant alleles identified
Among the 33 unrelated index patients with two pathogenic mutations (Table 1), 22 had clinical features of Alpers syndrome or had siblings with Alpers syndrome at the time of molecular diagnosis (Table 1). Four patients (#24 - 27) had arPEO+ and three patients (#21-23) had ANS. Three patients (#28-30) did not fulfill the clinical diagnostic triad for Alpers syndrome (triad of developmental delay, seizures and liver dysfunction) or the criteria for other autosomal recessive POLG-related diseases, but fit the criteria for MCHS. Patient #28 presented at the age of 3 years with developmental delay, renal tubulopathy, and myopathy in the absence of seizures and liver disease. Patient #1 showed seizures, neurosensory ataxia, and no liver disease until she was treated with valproic acid, which triggered a rapidly progressive and lethal liver failure. Patient #21 at 17 years of age had seizures and hearing loss, but no liver disease. No clinical information was provided for patient #31.
Six patients with Alpers syndrome were compound heterozygotes for the p.A467T and p.G848S mutations while two other patients were compound heterozygotes for p.A467T and p.T914P. The p.A467T mutation was the most common POLG mutation in our patient series. Previous studies have shown that the p.A467T bearing enzyme possesses only 4% of the wild-type DNA polymerase gamma activity and is compromised for its ability to interact with the POLG2 accessory subunit (Chan, et al., 2005a). In our cohort, patients heterozygous for p.A467T in trans with either p.G848S or p.T914P alleles presented with clinical features typical of Alpers syndrome, including seizures, liver disease and developmental delay.
We also found POLG mutations in two cases (patients #2 and #29) in whom the mutation in the paternal chromosome was not detected in the father's blood DNA. In these cases, analysis of 15 unlinked microsatellite markers was consistent with stated paternity. This suggests a de novo mutation origin on the paternal allele or paternal gonadal mosaicism.
In four cases (#17, #20, #21, and #31) the p.W748S mutation was found to be in cis with p.E1143G, as confirmed by parental studies. It was recently documented that one specific POLG SNP, p.E1143G, can modulate the deleterious effect of the p.W748S mutation (Chan, et al., 2006). This finding raises the possibility that other SNPs could potentially affect POLG enzymatic activity. Interestingly, p.E1143G was also found in cis with the novel p.R943C mutation in patient #28. Moreover, the SNP p.Q1236H, found in cis with p.R1096C in patient #18 and in cis with c.2544_2545insC in patient #13, could also have a modulatory effect, but this remains to be determined.
The Novel Mutations
We found five novel null mutations: p.Q68X, p.Q715X, c.2544_2545insC, c.2157+5G>A, and c.2480+1G>A; and one previously reported null mutation, c.1270_1271delCT (p.L424GfsX28); they are all predicted to result in premature translation termination and are therefore interpreted to be disease causing mutations. Messenger RNAs expressing these mutated alleles are expected to undergo nonsense mediated decay, resulting in mono-allelic expression similar to what has been described for the p.E873X allele of POLG (Chan, et al., 2005b).
The other eleven novel mutations identified in this study were all missense changes. Their pathogenicity was assessed based upon (a) co-segregation with disease phenotype, (b) de novo origin, (c) absence in 100 normal controls, (d) structurally non-conservative substitution of an evolutionary conserved amino acid, and (e) location in protein regions of structural/functional importance.
The p.G11D mutation was found in patient #23, who presented with features of Alpers syndrome but with a late onset at 15 years of age. This mutation is located within the mitochondrial targeting sequence, which is normally highly positively charged. Therefore, the change from the neutral small amino acid glycine into a negatively charged dicarboxylic amino acid, aspartate, is predicted to neutralize some of the positive charges in this region and potentially impair POLG transport into the mitochondria. The p.G11D mutation is the second reported mutation in the mitochondrial targeting sequence; the previously reported mutation, p.R3P, also results in a reduction of the positive charge in this region (Van Goethem, et al., 2001). The p.G11D mutation in patient #23 was found to be in cis with another reported mutation, p.R627Q, which resides in the linker region and was previously reported in compound heterozygosity with p.A467T in a family with an ataxia-myopathy syndrome (Luoma, et al., 2005). At the present time, it is unclear if either p.G11D or p.R627Q in isolation constitutes a pathogenic mutation. The patient who carried p.G11D and p.R627Q on one chromosome was compound heterozygous with another mutation, p.R852C.
In one of our patients (#30) presenting with MCHS, a novel allele p.R853Q, was found in a compound heterozygote state with p.T251I and p.P587L, which were both present in cis on the second allele [denoted as p.T251I+p.P587L (cis) from here on]. Although the p.R853Q allele is presently uncharacterized, another missense alteration at this amino acid position (p.R853W) has previously been reported to be pathogenic, and a mutation involving a neighboring amino acid change, p.R852C, was found in Alpers syndrome patients (Nguyen, et al., 2006). Furthermore, both the Arg852 and Arg853 residues are evolutionarily highly conserved from yeast to human. These data together support the classification of p.R853Q as a disease-causing mutation (Fig. 1A).
Two novel mutations, p.L83P and p.G888S, were identified in compound heterozygosity in a Chinese patient with Alpers syndrome (#16) who has been reported in more detail elsewhere (Bao, et al., 2007). Leu83, located between the polyglutamine stretch and the exo domain in an area of unknown function, appears to be highly conserved from yeast to man (http://tools.niehs.nih.gov/polg/). The invariant Gly888 resides in the pol domain and is only two residues away from a critical residue Asp890 within one of the three carboxylic amino acids (triad) involved in the chelation of Mg2+ at the active site (Fig. 2A). Mutation at the neighboring residue, p.A889T, has been associated with autosomal dominant progressive external ophthalmoplegia with an early age of presentation and ataxia, while a mutation on the other side, p.T885S, is associated with Alpers syndrome (Horvath, et al., 2006). Thus, the two novel missense variants p.L83P and p.G888S are most likely responsible for the clinical course of this patient.
Figure 2.
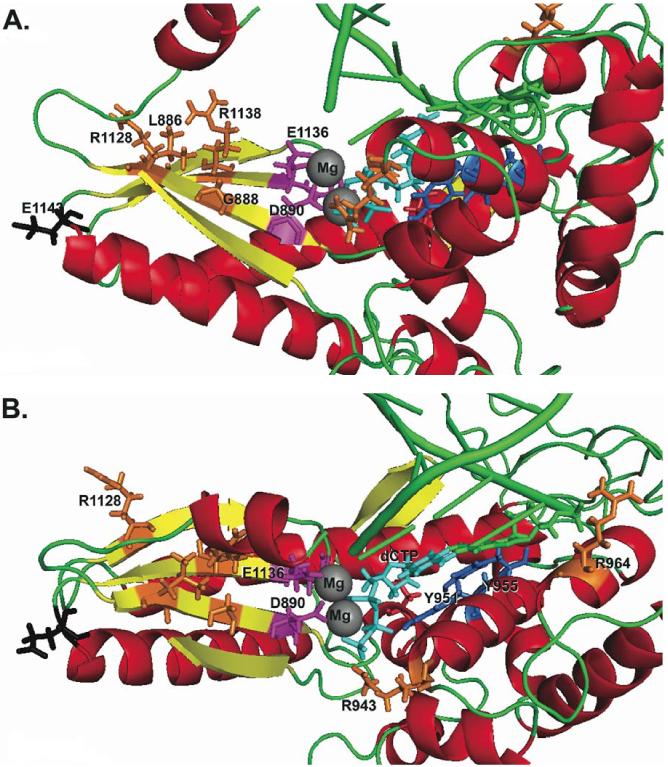
Molecular modeling of the human DNA polymerase gamma active site with amino acids of interest indicated. Critical amino acids important for catalysis are colored in. Those residues modified by POLG mutations are decorated: magenta for those carboxylic residues involved in chelating the active site Mg2+ ions (grey spheres); blue for those amino acids critical in dNTP recognition. Amino acid side chains colored in orange represent the sites of substitutions caused by novel disease mutations identified in POLG in this study. An incoming dNTP molecule is shown in turquoise. A. The polymerase active site viewed with the palm domain labeled. B. The polymerase active site rotated to view the fingers domain.
Patient #10, also presenting with Alpers syndrome, carried the novel p.L886P mutation in compound heterozygosity with the most common mutation, p.A467T. Leu886 is highly conserved from yeast to human and is located in the β-strand containing the enzyme active site (Fig. 2A). The change from a bulky hydrophobic amino acid leucine to a proline is predicted to cause a structural effect in this functionally important domain. The neighboring amino acids are evolutionarily conserved and, as described above, mutations at amino acid residues 885, 888, and 889 have all been identified in patients with Alpers syndrome (Fig. 1A).
The novel p.R943C mutation was identified, in compound heterozygosity with p.G737R, in patient #28, a girl who had a disease onset at 1 year of age with developmental delay, dysmorphic features, congenital cataract, and renal tubulopathy. Arg943 is evolutionarily highly conserved from yeast to human. While p.R943C has not previously been reported, another mutation affecting the same residue, p.R943H, has been found in patients with a severe form of autosomal dominant PEO (Lamantea, et al., 2002). Arg943 is critically important in binding the oxygen atoms of the gamma-phosphate of the incoming dNTP and helps to position the incoming dNTP for catalysis (Fig. 2B). The change from arginine to histidine at this position causes a substantial loss of catalytic activity (recombinant protein expressing p.R943H has only 0.2% of wild-type enzyme activity in vitro) (Graziewicz, et al., 2004). A heterozygous mutation of this residue to cysteine might therefore be expected to cause a similar dominant phenotype in vivo.
The novel mutation p.R1138C was identified in compound heterozygosity with p.A467T in patient #26, a 48-year-old woman who presented with dysphagia and features of arPEO+. Arg1138 is part of the evolutionarily conserved polymerase motif C found in all DNA polymerases and is invariant throughout evolution from yeast to human (Fig. 1A). In the 3-dimensional structural model reconstruction, this arginine residue is part of a beta-sheet within the palm domain (Fig. 2A) and substitution of this amino acid with cysteine most likely disrupts DNA polymerase catalysis.
Patient #29 had MCHS and pancreatitis. She carried the novel missense p.K1191R alteration and had p.T251I+p.P587L (cis) on the other chromosome. The p.K1191R mutation predicts a relatively conservative change of lysine to arginine at amino acid position 1191. While p.K1191R has not been previously reported, a p.K1191N mutation has been found in another patient with Alpers syndrome (Horvath, et al., 2006). Thus, the p.K1191R alteration is likely to be pathogenic. Testing of parental DNA revealed that the mother was heterozygous for the p.T251I+p.P587L (cis) mutations, confirming her carrier status. However, the father tested negative for both the p.T251I+p.P587L (cis) allele and for the p.K1191R allele, thus suggesting that either he is gonadal mosaic for the p.K1191R mutation or that the mutation occurred de novo on the paternal allele. Nonpaternity was ruled out in this case.
Patient #22, who has ANS, was found to be compound heterozygous for p.R964C and the novel mutation p.A862T. The p.R964C mutation was previously reported in the homozygous state, in an HIV infected patient who developed lactic acidosis following treatment with a nucleoside analog. Recombinant POLG with p.R964C showed only 14% activity in vitro compared to the wild-type control (Yamanaka, et al., 2007). The Arg964 is located in the pol domain of the POLG protein. The other alteration p.A862T is also located near the beginning of the pol domain preceding motif A. This residue is in a highly conserved region that is unique to the DNA polymerase γ subfamily, distinct from other family A DNA polymerases. Given the high degree of conservation of this residue from yeast to human (Fig. 1A), variants at this position are likely to be deleterious.
An uncharacterized novel variant, p.D1196N, was found in association with the known mutation p.G517V in patient #32, a 5-year-old male presenting with developmental delay, short stature, and myopathy. The p.G517V mutation, located within the linker region, has previously been reported in two three-generation families with autosomal dominant encephalopathy and variable disease onset (Horvath, et al., 2006). While Asp1196 is located in the C-terminal region of the pol domain and is conserved in mammals and yeast, its clinical significance is presently unclear since the corresponding substitution has been reported in chicken (Ropp and Copeland, 1996).
The p.G517V mutation was also found, in compound heterozygosity with p.R1128H, in a one-year old boy presenting with liver disease but no seizures (case #33). Arg1128 is part of the evolutionarily conserved motif C in the pol domain found in all DNA polymerases, which forms part of the palm domain. This residue is located in one of the palm β-sheets that correctly position two of the three carboxylic acids, Asp1135 and Glu1136 in the catalytic triad (Fig. 2B). This residue is conserved from yeast to human and a substitution to histidine at this position is predicted to be deleterious to the polymerase activity.
Emerging Differences in Age of Onset for POLG Disease
Table 2 illustrates the differences in age of onset for each of the 30 patients with homozygous or compound heterozygous genotypes for which a diagnosis could be assigned. The form of POLG disease that had the earliest age of onset was the Myocerebrohepatopathy Spectrum (MCHS). Among the 3 patients (#28-30) in our series with MCHS, the median age of onset was 1 year. Twenty of the 30 patients had Alpers syndrome. The median age of onset of Alpers syndrome was 2 years, and 80% of patients were between 0.9 and 9.5 years of age (Table 2). The median age of onset for patients with Ataxia Neuropathy Spectrum was 17 years, for autosomal recessive Progressive Ophthalmoplegia plus (arPEO+) it was 40, and for adPEO it was 46 (Table 2).
Table 2.
Age of Onset
| Age of Onset (yrs) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Diagnosis | N= | Median | 10th Pctl | 90th Pctl | Mean | SD | Min | Max |
| MCHS | 3 | 1.0 | * | * | 1.4 | 3.5 | 0.2 | 3 |
| ALPERS | 20 | 2.0 | 0.9 | 9.5 | 3.6 | 5.2 | 0.5 | 23 |
| ANS | 3 | 17 | * | * | 16 | 1.2 | 15 | 17 |
| arPEO+ | 4 | 40 | * | * | 43 | 13 | 32 | 60 |
| adPEO+ | 3 | 46 | * | * | 46 | 21 | 25 | 66 |
| Unassigned | 3 | |||||||
| Total | 36 | |||||||
Insufficient data for calculation
Patients with one identified POLG mutation and/or novel variant
Twenty eight patients were identified with one altered POLG allele and/or a single nucleotide polymorphism (Table 3). With the exception of the three mutations in the adPEO patients, the pathogenicity of these single POLG mutations are unclear and should be considered as uncharacterized variants until other supporting data is available. Twenty one of them carried one of the previously reported mutations (seven with p.G517V, three with p.G737R, six with p.Y831C, two with p.T251I+ p.P587L (cis), one with p.T914P, one with p.Y955C, and one with p.R964C). Each of the remaining 11 patients carried a novel missense variant. These novel variants occurred at evolutionarily conserved amino acid residues (Fig. 1A).
Table 3.
Phenotypes Associated with a Single POLG Variant
| Nucleotide Change | Amino Acid Change | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Gender | Onset (yrs) | DD or Dementia | Sz | Liver | PEO | Lact | Dx | Other Symptoms | Allele 1 | Allele 2 | Allele 1 | Allele 2 | Domains |
| 34 | M | 1 | + | + | + | - | + | ?Alpers | - | c.915C>G | - | p.S305R | - | Exo |
| 35 | M | 66 | - | - | - | + | - | adPEO | Hearing loss, ptosis | c.3235A>C | - | p.I1079L | P | |
| 36 | F | 46 | - | - | - | + | - | adPEO | Ptosis, hearing loss, muscle weakness, optic atrophy | c.3285C>G | - | p.S1095R | P | |
| 37 | F | 25 | - | - | - | + | + | adPEO | Ptosis, exercise intolerance, fatigue, ragged red fibers | c.2740A>C | - | p.T914P | P | |
| 38 | F | 16 | + | + | - | - | - | U | Short-stature, Leigh-like disease, neuropathy, stroke, dystonia, chorea, diabetes, myoclonus | c.1550G>T | - | p.G517V | L | |
| 39 | M | 2 | + | + | - | - | - | U | Half-sib with similar symptoms | c.1550G>T | - | p.G517V | L | |
| 40 | F | 2.5 | + | + | + | - | + | U | Chorea, myopathy, Leigh-like disease | c.1550G>T | - | p.G517V | L | |
| 41 | F | 6 | + | - | - | - | + | U | Sideroblastic anemia, splenomegaly | c.1550G>T | - | p.G517V | L | |
| 42 | M | 13 | + | - | - | + | - | U | Pearson syndrome to KSS | c.1550G>T | - | p.G517V | L | |
| 43 | F | 0.1 | NA | NA | NA | NA | NA | U | - | c.2492A>G | - | p.Y831C | P | |
| 44 | M | 0.1 | + | + | - | - | - | U | Hypotonia | c.2492A>G | - | p.Y831C | P | |
| 45 | M | 1.8 | - | - | - | - | + | U | Autism, hypotonia | c.2492A>G | - | p.Y831C | P | |
| 46 | F | 9 | + | + | - | - | - | U | - | c.2492A>G | - | p.Y831C | P | |
| 47 | F | 14 | NA | NA | NA | NA | NA | U | - | c.2492A>G | - | p.Y831C | P | |
| 48 | M | 19 | + | - | - | + | - | U | Neuropathy, ataxia, hearing loss, short stature, muscle weakness, fatigue | c.2492A>G | - | p.Y831C | P | |
| 49 | F | 5 | + | + | - | - | - | U | Failure to thrive | c.32G>A | - | p.G11D | MTS | |
| 50 | F | 3 | - | - | + | - | - | U | Hypotonia, failure to thrive, short stature, respiratory failure | c.328C>T | - | p.H110Y | Exo | |
| 51 | M | 0.5 | NA | NA | NA | NA | NA | U | NA | c.752C>T-c.1760C>T | - | p.T251I-p.P587L | Exo/L | |
| 52 | M | 4 | - | + | - | + | - | U | Neuropathy, ataxia, ptosis | c.752C>T-c.1760C>T | - | p.T251I-p.P587L | Exo/L | |
| 53 | M | 13 | + | - | + | - | - | U | Muscle weakness, optic atrophy | c.1174C>G | - | p.L392V | Exo | |
| 54 | F | 39 | - | - | - | - | + | U | Muscle weakness, exercise intolerance, hearing loss, arhytthmia | c.1849C>T | - | p.R617C | L | |
| 55 | M | 2 | + | - | - | - | - | U | - | c.2209G>C | - | p.G737R | L | |
| 56 | F | 3 | + | + | - | - | + | U | - | c.2209G>C | - | p.G737R | L | |
| 57 | M | 30 | NA | NA | NA | NA | U | - | c.2209G>C | - | p.G737R | L | ||
| 58 | M | 9 | - | - | - | + | - | U | Muscle weakness | c.2564T>C | - | p.V855A | P | |
| 59 | M | 2 | - | + | - | - | - | U | Ataxia, hypotonia | c.2590C>T | - | p.R964C | P | |
| 60 | F | 0.1 | + | - | + | - | + | U | Hearing loss, failure to thrive, Generalized ETC complex deficiency | c.2864A>G | - | p.Y955C | P | |
| 61 | F | 0.8 | + | + | - | - | - | U | Retinitis pigmentosa, hearing loss, failure to thrive | c.3614G>C | - | p.G1205 A | P | |
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence NM_002693.1, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Novel mutations are shown in bold and neutral variants are in shown in blue.
Abbreviations: adPEO = autosomal dominant Progressive External Ophthalmoplegia, U=unclassified Exo = exonuclease domain; L = linker region; P = Polymerase domain. DD = Developmental Delay; Sz = Seizures; Lact = Elevated lactate; Dx = Diagnosis
The p.H110Y mutation was found in a 3 year old female (patient #50) who had some features of Alpers syndrome but overall was classified as Undiagnosed. This mutation changes a highly conserved, positively charged histidine residue to tyrosine, a neutral aromatic amino acid.
The p.S305R variant found in a one year-old patient with Alpers syndrome (patient #34) causes a substitution within the exo domain. The clinical significance of this mutation is not known. However, a mutation affecting the adjacent amino acid, p.L304R, has been reported in patients with arPEO.
The p.I1079L variant was identified in a 66-year old man with adPEO (patient #35). Ile1079 is in the pol domain between motifs B and C and is conserved from yeast to man with the exception of the Drosophila POLG where a Leu resides. Thus, the pathogenic significance of this conservative substitution is unclear.
The p.S1095R variant was identified in a 46-year-old woman with PEO (patient #36). Ser1095 is surrounded by a block of highly conserved amino acids and is conserved from Drosophila to human. The adjacent amino acid Arg1096 is invariant from yeast to human. It is conceivable that substitution of a small amino acid serine to the bulky, highly charged basic amino acid arginine immediately adjacent to another arginine residue is not tolerated structurally and functionally.
The p.G1205A variant, seen in a patient with developmental delay, seizure disorder, and retinitis pigmentosa (patient #61), replaces a glycine residue which is highly conserved from Drosophila to human.
Similarly, the novel missense variant, p.V855A seen in a 9 year-old patient with muscle weakness and PEO (patient #58), is conserved from frog to human, and is surrounded by conserved amino acids.
The potential pathogenicity of the p.R617C allele (case #54) is more questionable. The p.Arg617 is located in the linker region and is not evolutionarily conserved. Therefore, there is not sufficient information to classify the p.R617C allele as either a pathogenic mutation or as a benign variant.
Summation of recurrent mutations
The frequencies of the most common mutations in the POLG gene, derived from a systematic survey of published articles reporting multiple cases, are summarized in Table 4. Only unrelated families with two identified mutant alleles are included in Table 4. Overall, the p.A467T mutation is the most common mutation, accounting for 36% of all mutant alleles, followed by p.G848S, p.T251I+p.P587L (in cis), p.W748S, and p.T914P with a frequency of 8%, 7.5%, 5%, 6.3%, and 3.4% respectively. Most of the remaining mutations (together accounting for 32% of all mutant alleles) are private mutations. The great majority (92.5% overall) of the mutated alleles in various populations are missense mutations, while frameshift and nonsense mutations account for a small fraction (7.5%) of the overall mutated alleles.
Table 4.
Frequency of the most common POLG mutations reported in the literature.
| Present study | Nguyen et al. 2006a | Horvath et al. 2006 | Ferrari et al. 2005 | de Vries et al. 2007 | Total | 95% CI | |
|---|---|---|---|---|---|---|---|
| Patients | 33 | 10 | 30 | 7 | 7 | 87 | |
| Alleles | 66 | 20 | 60 | 14 | 14 | 174 | |
| p.A467T | 22 (32%) | 10 (50%) | 19 (32%) | 5 (36%) | 7 (50%) | 63 (36%) | 29-43% |
| p.G848S | 9 (14%) | 2 (10%) | 1 (1.7%) | 1 (7%) | 1 (7%) | 14 (8%) | 4.4-13% |
| p.T251I-p.P587L (cis) | 3 (4.5%) | 0 | 9 (15%) | 1 (7%) | 0 | 13 (7.5%) | 4.0-12% |
| p.W748S | 6 (9%) | 1 (5%) | 2 (3%) | 2 (14%) | 0 | 11 (6.3%) | 3.2-11% |
| p.T914P | 2 (3%) | 1 (5%) | 3 (5%) | 0 | 0 | 6 (3.4%) | 1.3-7.4% |
| Splice, nonsense, frameshift mutations | 6 (9%) | 3 (15%) | 1 (1.7%) | 2 (14%) | 1 (7%) | 13 (7.5%) | 4.0-12% |
| Other missense mutationsb | 19 (29%) | 3 (15%) | 25 (42%) | 3 (21%) | 5 (36%) | 55 (32%) | 25-39% |
Cases previously reported by other groups were not included.
Missense mutations in cis were counted as one mutant allele.
DISCUSSION
In this study we report 25 novel mutations in 61 patients (Table 1, Table 3, Fig. 1B). These mutations add to the growing list of POLG gene mutations (http://tools.niehs.nih.gov/polg/). Missense and null mutations spread across regions encoding diverse structural/functional domains (mitochondrial targeting sequence, exo, linker, and pol domains) of the POLG protein help explain the wide spectrum of clinical phenotypes observed in patients with POLG mutations. In contrast, approximately 50% of mutations reported in deoxyguanosine kinase (DGUOK) are null mutations, and the majority of patients with mutations in DGUOK present with a phenotype of severe infantile hepatocerebral mtDNA depletion syndrome (Dimmock, et al., 2008).
Our study of a large cohort of 350 patients confirms earlier reports that A467T is the most common POLG disease mutation in patients displaying autosomal recessive inheritance, in Alpers, ANS and PEO syndromes (Table 4) (Davidzon, et al., 2005; Ferrari, et al., 2005; Naviaux and Nguyen, 2004; Nguyen, et al., 2005; Tzoulis, et al., 2006; Van Goethem, et al., 2003a; Van Goethem, et al., 2004; Winterthun, et al., 2005). We also noted that the five most common missense mutations, p.A467T, p.W748S, p.G848S, p.T914P and p.T251I+p.P587L (cis), together account for about 61% of disease alleles in cases with autosomal recessive diseases due to POLG mutations. Thus, testing of these 5 alleles would detect both alleles in over half of cases with recessive POLG-related disease, and would detect one mutant allele in about one third of cases. An efficient screening strategy for POLG disease may incorporate these five mutations in an initial screen, however rare and novel polymorphisms would be missed. Improvements in resequencing technologies and associated reductions in cost make diagnosis by resequencing feasible.
Our study found no gender bias among patients with childhood onset of disease (Tables 1 and 3). This finding is consistent with one study (Davidzon, et al., 2005), but not others (Ferrari, et al., 2005; Horvath, et al., 2006).
In our patient population, among patients with two mutant alleles, patients with disease onset before age 5 years displayed the more severe clinical phenotype of Alpers syndrome. Mutations in both alleles were also identified in three juvenile cases of ANS and four cases with adult onset arPEO+.
Compound heterozygotes for the p.A467T and p.W748S substitutions were reported to be common in other studies, and to have a significantly poorer survival and increased incidence of liver failure as compared with patients homozygous for either p.A467T or p.W748S (Tzoulis, et al., 2006). We identified only one case with compound heterozygosity for the p.A467T and p.W748S mutations. We found six compound heterozygotes for p.A467T/ p.G848S, two compound heterozygotes for p.A467T / p.T914P, and two compound heterozygotes for p.W748S/p.G848S Eight of these cases (80%) had a severe phenotype and died before reaching 2 years of age. Both the p.T914P and p.G848S mutations have been previously reported in compound heterozygosity with other mutations in patients with Alpers syndrome (de Vries, et al., 2007; Horvath, et al., 2006). The severe clinical effects of these mutations likely reflect the substitution of highly conserved amino acids located within the pol domain of the enzyme.
We found more than two mutations in six cases (Table 1). The p.T251I+p.P587L (cis) mutations were found in three cases, in trans with other putative pathogenic mutations. The p.Thr251 is situated in the exo region near the second of the three highly conserved exonuclease motifs (exo I, II, III) while Pro587 resides in the linker region. The degree of phylogenetic conservation of Pro587 is higher than that of Thr251, and there has been at least one case of PEO with the p.P587L mutation but without p.T251I. Thus, it is likely that the latter is a polymorphism (Gonzalez-Vioque, et al., 2006). However, pathogenicity of p.T251I or p.P587L, and the possibility of synergistic effects between them, await further experimental confirmation. The heterozygous p.T251I+p.P587L (cis) allele has previously been associated with a relatively mild phenotype (Di Fonzo, et al., 2003; Ferrari, et al., 2005; Horvath, et al., 2006), while all three of our compound heterozygous cases (patients #19, #29, and #30) presented with early onset (0.2, 1, and 0.5 year) Alpers syndrome or MCHS. These cases indicate that the presence of the p.T251I+P587L (cis) allele does not preclude a severe phenotype. This observation could indicate a dominant negative effect of the abnormal protein expressing the p.K1191R, p.R853Q, or p.G848S substitutions in combination with the p.T251I+P587L (cis) allele. Interestingly, all three of these mutations are located in the pol domain where other dominant mutations in PEO have been identified (Longley, et al., 2005).
Cases #17 and #21 each presented with three mutations; p.Q497H, p.W748S and p.E1143G, together in cis on a single allele, opposite a fourth mutation, p.G848S or p.A467T, respectively. The p.Q497H mutation has been reported in cis with p.W748S (Winterthun, et al., 2005), while p.W748S has never been found without p.E1143G (Winterthun, et al., 2005). The p.E1143G allele was recently shown to partially rescue deleterious effects of the p.W748S mutation upon DNA binding and polymerase activity; however, it is also shown to have a detrimental effect on protein stability (Chan, et al., 2006). In the yeast mtDNA polymerase, encoded by the MIP1 gene, the equivalent substitution of p.E1143G (E900G) causes a temperature sensitive phenotype that reduces polymerase protein levels (Baruffini, et al., 2007). Case #17, who carries the cis p.Q497H+p.W748S+p.E1143G allele opposite p.G848S, was diagnosed with severe Alpers syndrome and died at 13 months of age (Brunetti-Pierri, et al., 2008), whereas patient #21, carrying the cis p.Q497H+p.W748S+p.E1143G allele opposite p.A467T, did not experience disease onset until 17 years of age. This observation supports classification of p.G848S as a severe mutation.
Case #23 also carries two mutations in cis. The mutations: p.G11D, p.R627Q, and p.R852C were identified, with p.G11D and p.R627Q in cis and p.R852C on the second allele. Gly11, Arg627, and Arg852 reside in the mitochondrial targeting sequence; in the linker region, and in the pol domain, respectively. The clinical presentation was unusual, with onset of symptoms including myoclonic and focal seizures at 15 years of age. She was initially thought to have MELAS syndrome because of lactic acidosis and an occipital stroke at 19 years of age. It is noteworthy that following the stroke episode, when valproate treatment was initiated, the patient developed liver failure that required liver transplantation. This patient remains living nine years after liver transplant, but with slowly progressive neurological symptoms. This case indicates there are circumstances in non-Alpers POLG related mitochondrial disease where liver transplant would be appropriate, and emphasizes the susceptibility of hepatic failure from valproate treatment in patients with other phenotypes due to POLG mutations in addition to Alpers disease.
The clinical spectrum observed in patients with a single identified mutant allele is much broader than for autosomal recessive phenotypes. Patients with a single identified mutant POLG allele demonstrate no obvious correlation between genotype and phenotype, age of onset varies from newborn to late adulthood, and clinical symptoms range from severe Alpers-like phenotype, to mild late onset PEO (Table 3). Further study of genotype-phenotype correlation will be essential for accurate counseling.
Several cases in our cohort with early onset of severe disease were found to harbor only one definitive pathogenic mutation. However, the presence of a second mutant allele located in the promoter region or deep in an intron cannot be excluded. Since POLG is a large gene containing 23 exons, intragenic deletions and/or duplications may be present, which would escape detection by standard sequencing approaches. A high density exon-targeted oligonucleotide CGH (comparative genomic hybridization) array may be designed to address this possibility. Furthermore, the possibility of locus heterogeneity involving mutations in POLG2 (for example), or with molecular defects in other functionally-related genes, such as PEO1, DGUOK, TK2, and MPV17, should be considered. Digenic inheritance involving mutations in POLG and PEO1 has been documented in sporadic PEO (Van Goethem, et al., 2003a).
The pathogenicity of two mutations was unclear. Six heterozygous cases identified with p.Y831C demonstrated variable disease expression and age of onset. While p.Y831 has previously been reported as a pathogenic dominant mutation (Barthelemy, et al., 2002; Mancuso, et al., 2004), recent studies suggest that p.Y831C is a polymorphism (Stopinska, et al., 2006; Tiangyou, et al., 2006). The allele frequency of p.Y831C in our population was 0.9% (6 out of 700 alleles sequenced; 95% CI= 0.3-1.9%). Similarly, the p.G517V mutation has been reported as dominant in a three-generation family, but was also found in a German control group (Horvath, et al., 2006). We found 5 unrelated probands heterozygous for p.G517V. These patients displayed variable disease expression and age of onset (Table 3). We also identified p.G517V in compound heterozygosity with a second mutation in two presumptive autosomal recessive cases. However, neither patient demonstrated a classical presentation of Alpers syndrome. The pathogenic status of the p.G517V allele is less clear than the p.Y831C allele. The allele frequency of p.G517V in this selected population with suspected mitochondrial disease was 1.0% (7 out of 700 alleles sequenced; 95%CI= 0.4-2.0%). It is not clear whether this allele represents a benign polymorphism, or may be contributing to disease by poorly understood mechanisms, which allow it to function either as a dominant or a recessive allele. Further pedigree haplotype transmission and biochemical expression studies will be required to fully characterize function of the p.Y831C allele.
About half of our cases carrying one identified mutation had mutations affecting an amino acid in the pol domain (p.V855A, p.T914P, p.Y955C, p.R964C, p.I1079L, p.S1095R, and p.G1205A), similar to earlier findings (Luoma, et al., 2004; Van Goethem, et al., 2004). Other patients with a single identified mutation in our patient population had mutations/variants, including p.G517V, p.R617C, and p.G737R, resulting in amino acid substitutions in the linker region. While the possibility of autosomal recessive disease has not been excluded in these cases, these findings may indicate that mutations in either the linker or the pol domain may be causative lesions for autosomal dominant disease. In addition, we found two novel variants of unclear clinical significance: p.S305R and p.L392V, which cause substitutions in the exo domain of POLG.
Expression of the Alpers syndrome phenotype is associated with inheritance of two strongly pathogenic alleles of POLG, and DNA testing of POLG has become the standard diagnostic for confirmation of a clinical diagnosis of Alpers syndrome. Inheritance of one or two “weaker” POLG alleles gives rise to non-Alpers syndrome phenotypes that more likely demonstrate late-onset and more heterogeneous clinical manifestations. Predicting disease severity and prognosis from POLG genotype is currently challenging in many cases. However, examination of each variant in the context of functional domains, conservation and location in the 3-dimensional structure, along with information gained from in vitro studies contribute to the determination of pathogenicity of missense variants.
Already, POLG mutation analysis can provide a definitive diagnosis for many patients with Alpers syndrome. For this devastating disease, knowledge of causative lesions will facilitate meaningful and accurate counseling for family members. An increased understanding of the functional significance of POLG mutations may lead to carrier testing, prenatal diagnosis, and also postnatal pre-symptomatic diagnosis of affected siblings, allowing psychological preparation time for the parents as well as optimized clinical management from the early stages of the disease.
In this study, of 350 patient samples submitted for testing, 33 (9.4%) were found to carry two pathologic recessive mutations, and an additional 28 (8.0%) carried a single pathologic mutation. The higher identification of mutations in patients diagnosed with Alpers suggests that clinicians have become more proficient at diagnosing Alpers since the genetic basis for Alpers was initially reported (Naviaux and Nguyen, 2004) as compared to the adult-onset POLG-related diseases. Many adult-onset POLG-related diseases are poorly characterized, but with further characterization of phenotypes, it is possible that these patients would receive a more rapid, and accurate diagnosis. This study also suggests that there are likely additional novel lesions in POLG. Because of the contribution of observed allelic heterogeneity, the correlation to phenotypes in terms of severity, age of onset, and spectrum of organ system involvement, much further study will be required to fully understand the genetics underlying the POLG diseases.
Supplementary Table
ACKNOWLEDGMENTS
The authors would like to thank all referring physicians and patients who were included in this study and thank Drs. Sherine Chan and Lauranell Burch for critical review of this manuscript. This work was supported by DHHS, NIH, 5R01 CA100023 to LJW and by NIH intramural research funds to WCC. RKN was supported by grants from NIH (5R21-NS051815), the Lennox Foundation, Hailey's Wish Foundation, and the UCSD Christini Fund.
Contract grant sponsor: National Institutes of Health, Lennox Foundation, Hailey's Wish Foundation, and the UCSD Christini Fund. Contact grant number: 5R01 CA100023 to LJW and by NIH intramural research funds to WCC. RKN was supported by grants from NIH (5R21-NS051815), the Lennox Foundation, Hailey's Wish Foundation, and the UCSD Christini Fund.
REFERENCES
- Bao X, Wu Y, Wong LJ, Zhang Y, Xiong H, Chou PC, Truong CK, Jiang Y, Qin J, Yuan Y, Lin Q, Wu X. Alpers syndrome with prominent white matter changes. Brain Dev. 2008;30:395–300. doi: 10.1016/j.braindev.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Barthelemy C, de Baulny HO, Lombes A. D-loop mutations in mitochondrial DNA: link with mitochondrial DNA depletion? Hum Genet. 2002;110:479–487. doi: 10.1007/s00439-002-0708-4. [DOI] [PubMed] [Google Scholar]
- Baruffini E, Ferrero I, Foury F. Mitochondrial DNA defects in Saccharomyces cerevisiae caused by functional interactions between DNA polymerase gamma mutations associated with disease in human. Biochim Biophys Acta. 2007;1772:1225–1235. doi: 10.1016/j.bbadis.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Selby K, O'Sullivan M, Hendson G, Truong K, Waters PJ, Wong L-J. Rapidly progressive neurological deterioration in a child with Alpers syndrome exhibiting a previously normal brain MRI. Neuromuscul Disord. 2008 doi: 10.1055/s-0028-1093334. in press. [DOI] [PubMed] [Google Scholar]
- Chan SS, Longley MJ, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem. 2005a;280:31341–31346. doi: 10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- Chan SS, Longley MJ, Naviaux RK, Copeland WC. Mono-allelic POLG expression resulting from nonsense-mediated decay and alternative splicing in a patient with Alpers syndrome. DNA Repair (Amst) 2005b;4:1381–1389. doi: 10.1016/j.dnarep.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Chan SS, Longley MJ, Copeland WC. Modulation of the W748S mutation in DNA polymerase gamma by the E1143G polymorphismin mitochondrial disorders. Hum Mol Genet. 2006;15:3473–3483. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidzon G, Greene P, Mancuso M, Klos KJ, Ahlskog JE, Hirano M, DiMauro S. Early-onset familial parkinsonism due to POLG mutations. Ann Neurol. 2006;59:859–862. doi: 10.1002/ana.20831. [DOI] [PubMed] [Google Scholar]
- Davidzon G, Mancuso M, Ferraris S, Quinzii C, Hirano M, Peters HL, Kirby D, Thorburn DR, DiMauro S. POLG mutations and Alpers syndrome. Ann Neurol. 2005;57:921–923. doi: 10.1002/ana.20498. [DOI] [PubMed] [Google Scholar]
- de Vries MC, Rodenburg RJ, Morava E, van Kaauwen EP, Ter Laak H, Mullaart RA, Snoeck IN, van Hasselt PM, Harding P, van den Heuvel LP, Smeitink JA. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur J Pediatr. 2007;166:229–234. doi: 10.1007/s00431-006-0234-9. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. 2002 [Google Scholar]
- Di Fonzo A, Bordoni A, Crimi M, Sara G, Del Bo R, Bresolin N, Comi GP. POLG mutations in sporadic mitochondrial disorders with multiple mtDNA deletions. Hum Mutat. 2003;22:498–499. doi: 10.1002/humu.9203. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Davidzon G, Hirano M. A polymorphic polymerase. Brain. 2006;129:1637–1639. doi: 10.1093/brain/awl169. [DOI] [PubMed] [Google Scholar]
- Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, Tang LY, Truong C, Schmitt E, Sifry-Platt M, Lucioli S, Santorelli FM, Ficicioglu CH, Rodriguez M, Wierenga K, Enns GM, Longo N, Lipson MH, Vallance H, Craigen WJ, Scaglia F, Wong LJ. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008;29:330–331. doi: 10.1002/humu.9519. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, Parini R, Simonati A, Santer R, Zeviani M. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA. Brain. 2005;128:723–731. doi: 10.1093/brain/awh410. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Vioque E, Blazquez A, Fernandez-Moreira D, Bornstein B, Bautista J, Arpa J, Navarro C, Campos Y, Fernandez-Moreno MA, Garesse R, Arenas J, Martin MA. Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch Neurol. 2006;63:107–111. doi: 10.1001/archneur.63.1.107. [DOI] [PubMed] [Google Scholar]
- Graziewicz MA, Longley MJ, Bienstock RJ, Zeviani M, Copeland WC. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat Struct Mol Biol. 2004;11:770–776. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, Goethem GV, Lofgren A, Hackman P, Paetau A, Kaakkola S, Majamaa K, Varilo T, Udd B, Kaariainen H, Bindoff LA, Suomalainen A. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–441. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, Prokisch H, Lochmuller H, McFarland R, Ramesh V, Klopstock T, Freisinger P, Salvi F, Mayr JA, Santer R, Tesarova M, Zeman J, Udd B, Taylor RW, Turnbull D, Hanna M, Fialho D, Suomalainen A, Zeviani M, Chinnery PF. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- Hudson G, Schaefer AM, Taylor RW, Tiangyou W, Gibson A, Venables G, Griffiths P, Burn DJ, Turnbull DM, Chinnery PF. Mutation of the linker region of the polymerase gamma-1 (POLG1) gene associated with progressive external ophthalmoplegia and Parkinsonism. Arch Neurol. 2007;64:553–557. doi: 10.1001/archneur.64.4.553. [DOI] [PubMed] [Google Scholar]
- Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP, Zeviani M. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol. 2002;52:211–219. doi: 10.1002/ana.10278. [DOI] [PubMed] [Google Scholar]
- Lecrenier N, Van Der Bruggen P, Foury F. Mitochondrial DNA polymerases from yeast to man: a new family of polymerases. Gene. 1997;185:147–152. doi: 10.1016/s0378-1119(96)00663-4. [DOI] [PubMed] [Google Scholar]
- Lim SE, Longley MJ, Copeland WC. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J Biol Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276:38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley MJ, Graziewicz MA, Bienstock RJ, Copeland WC. Consequences of mutations in human DNA polymerase gamma. Gene. 2005;354:125–131. doi: 10.1016/j.gene.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Luoma PT, Luo N, Loscher WN, Farr CL, Horvath R, Wanschitz J, Kiechl S, Kaguni LS, Suomalainen A. Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum Mol Genet. 2005;14:1907–1920. doi: 10.1093/hmg/ddi196. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Filosto M, Oh SJ, DiMauro S. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol. 2004;61:1777–1779. doi: 10.1001/archneur.61.11.1777. [DOI] [PubMed] [Google Scholar]
- Milone M, Brunetti-Pierri N, Tang L-Y, Kumar N, Mezei MM, Josephs K, Powell S, Simpson E, Wong L-J. Sensory Ataxic Neuropathy with Ophthalmoparesis Caused by POLG1 Mutations. 2008 doi: 10.1016/j.nmd.2008.05.009. submitted. [DOI] [PubMed] [Google Scholar]
- Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- Nguyen KV, Ostergaard E, Ravn SH, Balslev T, Danielsen ER, Vardag A, McKiernan PJ, Gray G, Naviaux RK. POLG mutations in Alpers syndrome. Neurology. 2005;65:1493–1495. doi: 10.1212/01.wnl.0000182814.55361.70. [DOI] [PubMed] [Google Scholar]
- Nguyen KV, Sharief FS, Chan SS, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006;45:108–116. doi: 10.1016/j.jhep.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics. 1996;36:449–458. doi: 10.1006/geno.1996.0490. [DOI] [PubMed] [Google Scholar]
- Stopinska K, Grzybowski T, Malyarchuk BA, Derenko MV, Miscicka-Sliwka D. Optimization of the Y831C mutation detection in human DNA polymerase gamma by allelic discrimination assay. Acta Biochim Pol. 2006;53:591–595. [PubMed] [Google Scholar]
- Tiangyou W, Hudson G, Ghezzi D, Ferrari G, Zeviani M, Burn DJ, Chinnery PF. POLG1 in idiopathic Parkinson disease. Neurology. 2006;67:1698–1700. doi: 10.1212/01.wnl.0000238963.07425.d5. [DOI] [PubMed] [Google Scholar]
- Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G, Aarseth JH, Bindoff LA. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006;129:1685–1692. doi: 10.1093/brain/awl097. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Lofgren A, Dermaut B, Ceuterick C, Martin JJ, Van Broeckhoven C. Digenic progressive external ophthalmoplegia in a sporadic patient: recessive mutations in POLG and C10orf2/Twinkle. Hum Mutat. 2003a;22:175–176. doi: 10.1002/humu.10246. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Martin JJ, Dermaut B, Lofgren A, Wibail A, Ververken D, Tack P, Dehaene I, Van Zandijcke M, Moonen M, Ceuterick C, De Jonghe P, Van Broeckhoven C. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003b;13:133–142. doi: 10.1016/s0960-8966(02)00216-x. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Mercelis R, Lofgren A, Seneca S, Ceuterick C, Martin JJ, Van Broeckhoven C. Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology. 2003c;61:1811–1813. doi: 10.1212/01.wnl.0000098997.23471.65. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Schwartz M, Lofgren A, Dermaut B, Van Broeckhoven C, Vissing J. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet. 2003d;11:547–549. doi: 10.1038/sj.ejhg.5201002. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Luoma P, Rantamaki M, Al Memar A, Kaakkola S, Hackman P, Krahe R, Lofgren A, Martin JJ, De Jonghe P, Suomalainen A, Udd B, Van Broeckhoven C. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. doi: 10.1212/01.wnl.0000140494.58732.83. [DOI] [PubMed] [Google Scholar]
- Winterthun S, Ferrari G, He L, Taylor RW, Zeviani M, Turnbull DM, Engelsen BA, Moen G, Bindoff LA. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64:1204–1208. doi: 10.1212/01.WNL.0000156516.77696.5A. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Gatanaga H, Kosalaraksa P, Matsuoka-Aizawa S, Takahashi T, Kimura S, Oka S. Novel mutation of human DNA polymerase gamma associated with mitochondrial toxicity induced by anti-HIV treatment. J Infect Dis. 2007;195:1419–1425. doi: 10.1086/513872. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.