

Abstract
Extensive research has implicated the amyloid-β protein (Aβ) in the aetiology of Alzheimer’s disease (AD). This protein has been shown to produce memory deficits when injected into rodent brain and in mouse models of AD Aβ production is associated with impaired learning and/or recall. Here we examined the effects of cell-derived SDS-stable 7PA2-derived soluble Aβ oligomers on consolidation of avoidance learning. At 0, 3, 6, 9 or 12 h after training, animals received an intracerebroventricular injection of Aβ-containing or control media and recall was tested at 24 and 48 h. Immediately after 48 h recall animals were transcardially perfused and the brain removed for sectioning and EM analysis. Rats receiving injections of Aβ at 6 or 9 h post-training showed a significant impairment in memory consolidation at 48 h. Importantly, impaired animals injected at 9 h had significantly fewer synapses in the dentate gyrus. These data suggest that Aβ low-n oligomers target specific temporal facets of consolidation-associated synaptic remodelling whereby loss of functional synapses results in impaired consolidation.
Keywords: Amyloid β-protein, oligomers, memory consolidation, Alzheimer’s disease, synapse ultrastructure
1. Introduction
Alzheimer’s (AD) disease is the most common human dementia and as such places a huge emotional and economic burden on patients, carers and society (Ferri et al., 2005). Pathologically, the Alzheimer brain is characterized by atrophy and microscopically there are decreases in the numbers of neuronal cell bodies in the limbic and association cortices and certain subcortical nuclei (Gomez-Isla et al., 1997; Uylings and de Brabander, 2002) along with numerous amyloid plaques and neurofibrillary tangles (Terry, 1963; Kidd, 1964). Many studies have examined the relationship between cognitive impairment and plaque and tangle counts, and while in general, the number of neurofibrillary tangles correlates better with severity of dementia than the number of amyloid plaques, the most robust correlation in the staging of dementia and early AD is the magnitude of synapse loss (Davies et al., 1987; Masliah et al., 2001; Scheff et al., 2007). Quantification using electron microscopy or immunohistochemical staining for synaptic markers has documented significant decreases in synaptic density in the association cortices and hippocampus of AD brain (Davies et al., 1987; Bertoni-Freddari et al., 1989; DeKosky and Scheff, 1990; Masliah et al., 2001; Terry et al., 1991; Sze et al., 1997). Moreover, the decrease in synapse number and density seems disproportionate to the loss of neuronal cell bodies (Davies et al., 1987; Bertoni-Freddari et al., 1989; DeKosky and Scheff, 1990), suggesting that synapse loss may precede neuronal demise.
In AD the molecular pathways leading to synaptic pruning and dysfunction are not well understood, but substantial data indicate that the amyloid β-protein (Aβ) may be responsible for these affects (Klein, 2001; Walsh and Selkoe, 2004). Biochemical analysis of AD postmortem tissue has revealed a robust correlation between Aβ levels and the extent of synapse loss and severity of cognitive impairment (McLean et al., 1999). Aβ can exist in a variety of conformations and aggregation states and more recent studies have demonstrated that it is the non-fibrillar forms of Aβ, that preferentially alter neuroplasticity (Lambert et al., 1998; Wang et al., 2002; Walsh et al., 2002; Shreshta et al., 2006; LaCor et al., 2007; Calabrese et al., 2007; Shankar et al., 2007; 2008) and impair learned behaviours in rodents (Cleary et al., 2005; Lesné et al., 2006; Poling et al., 2008; Shankar et al., 2008). However, to date, no study has directly demonstrated the effect of soluble non-fibrillar Aβ on the activity-dependent alteration of synapse number and structure that is known to accompany memory consolidation (Marrone, 2007). Here we have examined the effect of cell-derived, SDS-stable low-n Aβ oligomers on the synapse remodelling that transiently increases in the molecular layer of the rat hippocampal dentate gyrus at the 6–9 h post-training time and returns to basal levels at 72 h following training in an avoidance conditioning paradigm (O’Malley et al., 1998). These SDS-stable low-n oligomers are detected with a variety of anti-Aβ antibodies, migrate on denaturing SDS-PAGE and elute from native size exclusion with sizes consistent for Aβ dimers and trimers (Walsh et al., 2005). Moreover, medium from cells containing such species prevent the learning-associated transient increase in synapse number and the effective consolidation of memory.
2. Methods
Reagents and Antibodies
All reagents were obtained form Sigma (Ireland) unless otherwise stated.
Preparation of Aβ-containing medium
7PA2 cells are a Chinese Hamster Ovary line that stably express human APP751containing the V717F mutation and have been used extensively to study the effects of SDS stable Aβ low-n oligomers (Walsh et al., 2002). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, penicillin/streptomycin, L-glutamine and G418. Once confluent, they were washed and then cultured overnight (15 h) in plain DMEM. Conditioned medium (CM) was collected, spun at 1000 g to remove cellular debris, aliquoted and stored at −80°C pending use.
Detection and immunodepletion of Aβ from 7PA2 CM
To confirm the presence and abundance of Aβ monomer and SDS-stable low-n oligomers, duplicate aliquots (3 ml) of CM were analysed using a sensitive immunoprecipitation (IP) western blot (Wblot) technique (Walsh et al., 2002). Samples were immunoprecipitated overnight with the novel polyclonal anti-Aβ antibody, AW8, used at a dilution of 1:80, AW8:CM, and Wblots were probed using the monoclonal antibody, 6E10 (1:1000, Signet, Dedham, MA). AW8 antiserum was raised against aggregated synthetic Aβ1–42 and contains antibodies to several different Aβ regions (McDonald et al., 2010) and is very effective at immunoprecipitating Aβ from 7PA2 CM, but only weakly recognizes APPsa. When used for immunohistochemistry AW8 readily detects amyloid deposits and congophilic amyloid angiopathy, but does not detect APP which is abundant in dystrophic neurites (McDonald et al., 2010). Aβ detection and quantification was carried out using IR800 secondary antibody (1:2500, Tebu-bio Ltd, UK) and a Li-Cor Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE).
Animal maintenance
Male Wistar rats, 375–425 g, were obtained from Charles River, UK and allowed to acclimate for 5 days prior to surgery. Animals were housed in groups of three with a 12 hr light-dark cycle at 22 ± 2 °C with food and water provided ad libitum. During this period the animals were introduced to the test room and handled for at least 3 days prior to surgery.
Surgical Procedures
All experiments were carried out in accordance to guide lines and under licence from the Department of Health, Ireland. Surgery was performed as previously described (Seymour et al., 2008). Briefly, rats were anaesthetised with a mixture of ketamine/medetomidine and a unilateral guide cannula (22 gauge, Plastics One, Roanoke, VA) was positioned in a small burr hole was drilled in the skull over the lateral ventricle (1.6mm lateral, 1mm posterior to bregma; Paxinos and Watson, 1986), secured with acrylic cement and a dummy cannula, cut to the same dimensions as the guide cannula, was used to maintain patency of the guide cannula. The animal was recovered with Antisedan and post-operative pain relief was provided by subcutaneous injection of Carprofen. In all cases animals were allowed to recover for at least one week prior to behavioural training.
Open-Field Analysis
To confirm that implantation of cannulae did not adversely affect overt behaviour or motor function, animals were placed in an open-field box for 5 min on each of two days prior to avoidance training. The total distance traveled and number of rears in the apparatus was determined using Ethovision™ software. Open-field behaviour was also assessed just prior to the 24 and 48 h recall times.
Passive avoidance learning and recall
At training, animals were placed in the light compartment and when they entered the dark compartment a light foot shock (0.75 mA scrambled every 0.5 sec) was delivered through the grid bar floor (Fox et al., 1995). Animals were judged to have acquired the task if they remained in the lighted chamber for a period of 30 s after the time of receiving the footshock. Following training animals were returned to their home cages and then tested for recall (10 min criterion) 24 h and 48 h post-training by placing them into the light compartment and noting their latency to enter the dark compartment. All behavioural experiments were carried out blind to the treated groups. Moreover, all animals that received an ICV injection were included in the behavioural analysis. Results were analysed using the Mann-Whitney U-test for non-parametric data, and p values of less than 0.05 were considered to be significant.
Intracerebroventricular (icv) injection of 7PA2 media
Following avoidance training, animals were injected with 5μl of either 7PA2 CM or 7PA2 CM immunodepleted of Aβ (ID 7PA2) at a rate of 1–2 μl/min delivered just following training (0 h), and at the 3, 6, 9 or 12 h post-training times (Figure 1A). The effect of the icv injections on memory recall was assessed at the 24 and 48 h post-training times.
Figure 1. Aβ oligomers induce amnesia of an avoidance conditioning paradigm when infused at a discrete post-training time.
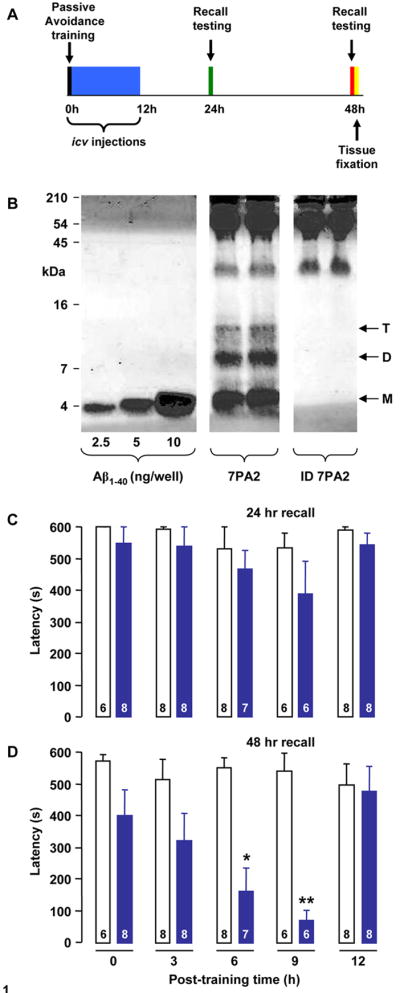
Animals were trained at time zero and injected with sample either immediately after training or at 3, 6, 9 or 12 h post-training. Thereafter animals were tested for recall first at 24 h post-training and then again at 48 h post-training (A). Immunoblotting with the anti-Aβ monoclonal antibody, 6E10, reveals the typical pattern of Aβ monomer (M), dimer (D) and trimer (T) in AW8 immunoprecipitates from 7PA2 CM (B). In contrast IP/wblot analysis of immunodepleted (ID) 7PA2 CM detected no Aβ. To allow estimation of Aβ concentration, known amounts of synthetic Aβ 1–40 were included on the same gel. Animals were injected once with 7PA2 CM or immunodepleted 7PA2 CM at 0, 3, 6, 9 or 12 h following training and tested for recall at 24 h and 48 h (C and D, respectively). 7PA2 CM-induced deficits in consolidation of avoidance training are dependent on timing of icv injection. At 24 hr recall there were no significant differences in escape latencies between the groups at any of the injection times studied. However, at the 48 h recall there was a strong time-dependent decrease in escape latency in the 7PA2 CM injected animals which was significant in the 6 and 9 hr injection groups (* p<0.05 and ** p<0.01, respectively; Mann-Whitney U test). Values are expressed as mean ± standard error and the group sizes are indicated in each column.
Electron microscopy
Following transcardial perfusion (4% paraformaldehyde/2.5% glutaraldehyde, pH 7.4) brains were dissected, coronal sections (100μm) collected at 3.3mm caudal to bregma, post-fixed (1% osmium tetroxide) and embedded with epoxy resin (Agar 100; Agar Scientific). The hippocampus was excised, re-embedded in resin and serial ultrathin sections (80nm) were collected in pairs on electron-lucent coated slot grids (2x1 mm; Agar Scientific, U.K.), counter-stained using uranyl acetate (5% w/v distilled water) and lead citrate (0.3% w/v in 0.1 M sodium hydroxide) and examined in a Tecnai G2 Spirit BioTWIN electron microscope.
Quantification of synapses employed double disector, unbiased stereology (Sterio, 1984). The number of synapses was counted at a magnification of 20,500x, with a Gunderson frame that covered an area of 13.3138μm2. The number of synapses present in the ‘look-up’ but not ‘reference’ section was estimated for each animal in up to 25 disector pairs according to previously described rules (Gundersen, 1977). A synapse was only counted if it contained 3 or more vesicles in the pre-synaptic element and a post-synaptic density. Parametric statistical comparisons were made using the Student’s t-test and p values <0.05 were taken to be significant.
3. Results
Intracerebroventricular infusions of medium containing SDS-stable 7PA2-derived soluble Aβ oligomers impair consolidation of avoidance conditioning in a time-dependent manner
Immunoprecipitation/western blotting confirmed the presence of Aβ monomer and SDS-stable Aβ dimer and trimer in 7PA2 CM, whereas there was no detectable Aβ in 7PA2 CM that had been previously immunodepleted of Aβ (Figure 1B). By comparison with synthetic Aβ 1–40 standards electrophoresed on the same gel, the total amount of Aβ monomer, dimer and trimer present in 7PA2 CM was estimated as 1.89 ng/ml, 0.82 ng/ml and 0.29 ng/ml, respectively. These concentrations are similar to those detected in 7PA2 CM by SEC/Wblot (Townsend et al., 2006) and by ELISA (Walsh et al., 2000).
All animals used in the avoidance conditioning behaved normally when tested in an open-field apparatus (Supplemental Figure 1) and were randomly assigned to receive a single injection of 7PA2 CM or ID-7PA2 CM at the 0, 3, 6, 9 or 12 h post-training times. At the 24 h recall time there was no significant difference in escape latency observed between the 7PA2 and ID-7PA2 treated groups at any of the post-training times examined (Figure 1C). In contrast, animals treated with 7PA2 CM exhibited an increasing reduction in their latencies to enter the darkened chamber and this trend became significant at the 6 and 9 h post-training times (Figure 1D). At all other time points examined the escape latencies of the 7PA2 were not significantly different from control counterparts (ID-7PA2). Moreover, all animals displayed normal open-field behaviour when assessed at the 24 and 48 h recall times (Supplemental Figure 2). The complete lack of effect of 7PA2 CM at the 12 h post-training time confirmed that Aβ exerts its amnesic action during a critical time period of memory consolidation.
Intracerebroventricular infusions of Aβ-containing 7PA2 CM prevents learning-associated synapse remodeling in the dentate gyrus of the hippocampal formation
In order to determine if the amnesic action of Aβ oligomers related to perturbations in the synapse remodeling that accompanies consolidation of the avoidance conditioning paradigm, we determined change in synapse density within the mid-molecular layer of the hippocampal dentate gyrus. The Aβ oligomers were infused at two post-training time points, 3 and 9 h, selected on the basis that the learning-associated increase in synapse density is known to occur at the 6 h post-training time in a variety of tasks, including the avoidance conditioning paradigm (O’Malley et al., 1998; Eyre et al., 2003). Ultrastructural analysis of the mid-molecular layer of the dentate gyrus revealed 7PA2 or ID-7PA2 infusions, at either time point, to be without effect on synapse form or integrity and all synapse profiles exhibited normal presynaptic vesicle content and intact postsynaptic densities at the 48 h post-training recall time (Figure 2A–D). Infusions of 7PA2 or ID-7PA2 at the 3 h post-training time were without effect on dentate synaptic complement (3.47 ± 0.06 and 3.38 ± 0.13 per μm3 respectively; Figure 2E). In contrast, synapse density was significantly reduced (~48%) in animals treated with 7PA2 media (2.32 ± 0.1 per μm3) at the 9 h post-training time, an effect not observed with infusion of ID-7PA2 media (4.44 ± 0.04 per μm3, Figure 2F). These observations suggest Aβ oligomers specifically inhibit synapse remodeling that accompanies learning and memory consolidation.
Figure 2. Aβ oligomers inhibit learning-associated synapse remodeling.
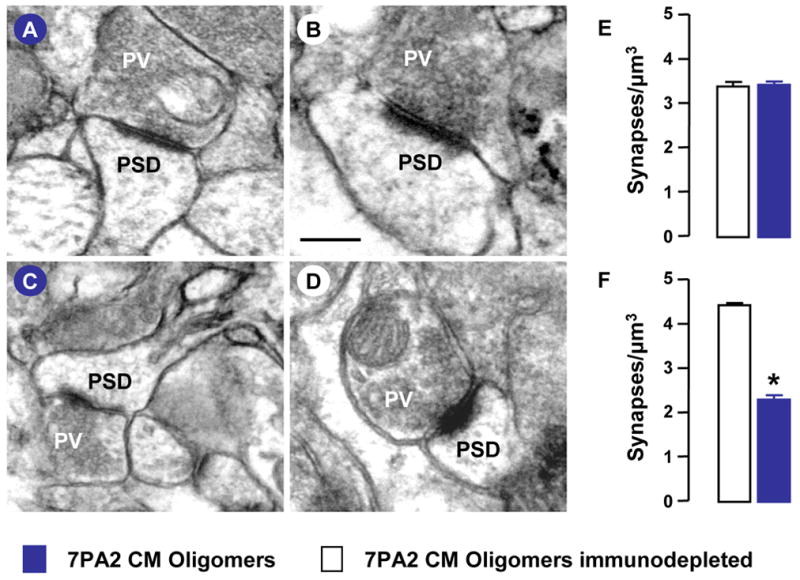
Animals received intracerebroventricular injections of 7PA2 CM, at 3 h and 9 h post-training, and were killed 48 h after training. Representative images of synapses in the dentate mid-molecular layer following 7PA2 (A, C) and immunodepleted 7PA2 (ID-7PA2) (B, D) treatment at 3 h post-training (A, B) and 9 h (D, E) post-training times are shown. Presynaptic vesicles (PV) and postsynaptic density (PSD) are indicated. The scale bar in Panel B applies to all images and corresponds to 1μm. Total synapse number in the dentate mid-molecular layer from 3 individual animals is shown for the 3 h (E) and 9 h (F) post-training injection times. Values are the mean ± SEM and significant differences (p<0.05; two-tailed Student t-test) are indicated by an asterisk.
4. Discussion
Considerable recent research has focused on small soluble forms of Aβ as a potential cause of the cognitive deficits that characterize AD (Klein et al., 2001; Walsh and Selkoe 2007) and in the present study we provide evidence that ventricular infusion of cell-derived low-n Aβ oligomers leads to reduced synapse density necessary for the synaptic remodellng that accompanies memory consolidation. Much of the previous work involving intracerebroventricular injection of either synthetic or cell-derived Aβ has focused on spatial reference memory or working memory (Nakamura et al., 2001; Cleary et. al., 2005; Lesné et al., 2006; Poling et al., 2008). In this study we employed a passive avoidance paradigm because its rapid acquisition ensures the synchronization of subsequent memory-associated neuroplastic events and afforded us the opportunity to examine long-term memory consolidation (Nakamura et al., 2001; Fox et al., 1995; Seymour et al., 2008).
Using this avoidance conditioning task we could demonstrate infusions of low-n Aβ oligomers to most effectively induce amnesia when administered in the 6 to 9 h post-training time, a period that coincides precisely with the transient increase in synapse number that accompanies consolidation of this task (O’Malley et al., 1998). Such time-dependent perturbations of memory consolidation are not unique to Aβ oligomers as, by way of example, infusions of antibodies to the neural cell adhesion molecule (NCAM) or peptide antagonists of its homophilic binding mechanism are effective only when administered at the 6 h post-training time when learning-associated synapse elaboration is initiated (Doyle et al., 1992a; Foley et al., 2000; Roullet et al., 1997).
Concentrating on the hippocampal dentate gyrus, a brain structure known to be involved in the task studied (Ambrogi Lorenzini et al., 1997; Trivedi and Coover, 2004; Czerniawski et al., 2009), we observed a significant reduction in synapse number in rats injected with Aβ-containing 7PA2 CM at 9 h post-training but not at the 3 h post-training time. These findings are in keeping with the observed behavioural effects. Specifically, oligomers injected between 0–9 h post-training exhibited impaired consolidation, but only injections at 6 and 9 h had a statistically significant effect and injections at 12 h had no effect. This suggests that the target of Aβ oligomers is involved in synapse formation and/or maintenance, is expressed in a highly time-dependent manner, and is most sensitive to Aβ between 9 and 12 h post-training. Assuming that the half-life of human Aβ, a parameter controlled by extracellular proteolysis, local uptake (by neurons and glia) and clearance from the brain, in the rat is similar to that determined in mice (~2 h; Cirrito et al., 2003) the trend for decreased latencies in animals injected between 0–6 h may simply reflect differences in the effective concentration of Aβ present when its cognate target is maximally expressed. For instance, assuming the target is most sensitive at 9 h, then the concentration of oligomers persisting at this time point from injections made at 0, 3 and 6 h would be ~5, 12.5 and 37.5%, respectively. Hence, the trend toward impairment evident in animals injected at 0 and 3 h post-training may be the consequence of the persistence of sub-toxic levels of oligomers.
Processing of information for long-term memory storage requires specific patterns of activity that lead to modification of synapse structure and eventually to change in neural connectivity (Lamprecht and LeDoux, 2004; Marrone, 2007). Increases in synaptic density following learning have been documented for a number of different paradigms including the avoidance learning paradigm used in this study (O’Malley et al., 1998). Indeed, it is well-established that synapses throughout the CNS are endowed with mechanisms to integrate synaptic activity levels over time and to convert these into signals that direct the maintenance and regulation of receptors and channels at synapses without which the ability to manipulate and store information becomes severely compromised (Malenka and Bear, 2004; Rao and Craig, 1997). The need to re-expose the animal to the training stimulus in order to observe the amnesia induced by the Aβ oligomers further suggests their action to be mediated at the level of the synapse. Task re-exposure, memory reconsolidation, reconstitutes the memory trace into an active form to allow enhancement of retrieval or new information to be incorporated (Nadel and Land, 2000; Nader, 2003). As precisely the same need for task re-exposure at 24h is necessary to observe the amnesia induced by intracerebroventricular infusions of antibodies to synapse-specific proteins, such as NCAM or glycosylated variant (Doyle et al., 1992a; Alexinsky et al., 1997; Foley et al., 2000; Seymour et al., 2008), it further suggests Aβ to compromise synapse structure and/or function and to exert an enduring effect on the malleability of the memory trace. It is therefore likely that perturbation of synapse formation and/or damage to existing synapses may underlie the memory impairments typical of AD. Indeed the early and extensive synaptic loss evident in AD brain supports this conclusion (Ferrer and Gullotta, 1990; Terry et al., 1991).
Our finding that Aβ oligomers can adversely alter synapses and disrupt the memory of learned behaviour is in keeping with previous studies that have demonstrated the deleterious effect of Aβ oligomers on memory (Cleary et al., 2005; Lesné et al., 2006; Poling et al., 2008) and synapse number (LaCor et al., 2007; Shankar et al., 2007; Calabrese et al., 2007). However, what sets this study apart is the fact that we have for the first time linked memory impairment, a specific form of Aβ and an in vivo structural change. Of course memory and synapse impairing effects for Aβ had been inferred from studies using transgenic animals (Jacobsen et al., 2006; Mucke et al., 2000), however, these studies are difficult to interpret due to potential complications or deleterious effects arising from the genetic manipulation itself and the lack of clarity about what form of Aβ is mediating the effect. In contrast, in the present study we are able to unambiguously assign the observed changes in synapse density and consolidation to sub-nanomolar concentrations of cell derived SDS-stable Aβ oligomers. The molecular mechanism by which these low-n oligomers mediate their effect remains to be established but this study suggests that a severe reduction in synapse number in the hippocampus may be responsible for the deficits in memory observed in AD.
Supplementary Material
Figure 1S. Open-field analysis reveals no behavioural differences in the experimental animal cohorts. Each animal was monitored in the open-field apparatus for two days prior to passive avoidance conditioning. There were no differences in total distance travelled (A) or vertical exploration (B) between the two groups. Data represents mean and 10–90 inter-quartile range of each group at either day 1 or day 2 prior to training.
Figure 2S. Open-field analysis reveals no behavioural differences between 7PA2-treated and ID-7PA2-treated animals. Each animal was monitored in the open-field apparatus at the 24 and 48 h recall times. There were no differences in total distance travelled (A) or vertical exploration (B) between the two groups. Data represents mean and 10–90 inter-quartile range of each group at each recall time.
Acknowledgments
We thank Alfred Welzel and Amaya Garcia-Munoz for technical assistance in preliminary experiments and Drs. Keith Murphy and Andrew Foley for useful discussion. This work was supported by funding from the European Community 7th Framework Programme (FP7/2007-2013) under grant agreement No. 200611 (DMW), by the NIH (1R01AG027443, DMW & DJS) and by Science Foundation Ireland Strategic Research Cluster Grant (07/IN.1/B1322, CMR).
Footnotes
The authors confirm that there are no actual or potential conflicts of interest relating to the work enclosed in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexinsky T, Przybyslawski J, Mileusnic R, Rose SP, Sara SJ. Antibody to day-old chick brain glycoprotein produces amnesia in adult rats. Neurobiol Learn Mem. 1997;67:14–20. doi: 10.1006/nlme.1996.3734. [DOI] [PubMed] [Google Scholar]
- Ambrogi Lorenzini CG, Baldi E, Bucherelli C, Sacchetti B, Tassoni G. Role of ventral hippocampus in acquisition, consolidation and retrieval of rat’s passive avoidance response memory trace. Brain Research. 1997;768:242–248. doi: 10.1016/s0006-8993(97)00651-3. [DOI] [PubMed] [Google Scholar]
- Bertoni-Freddari C, Fattoretti P, Meier-Ruge W, Ulrich J. Computer-assisted morphometry of synaptic plasticity during aging and dementia. Pathol Res Pract. 1989;185:799–802. [Google Scholar]
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S. Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol Cell Neurosci. 2007;35:183–193. doi: 10.1016/j.mcn.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Czerniawski J, Yoon T, Otto T. Dissociating space and trace in dorsal and ventral hippocampus. Hippocampus. 2009;19:20–32. doi: 10.1002/hipo.20469. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 1987;78:151–64. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Doyle E, Nolan PM, Bell R, Regan CM. Intraventricular infusions of anti-neural cell adhesion molecules in a discrete posttraining period impair consolidation of a passive avoidance response in the rat. J Neurochem. 1992;59:1570–1573. doi: 10.1111/j.1471-4159.1992.tb08477.x. [DOI] [PubMed] [Google Scholar]
- Eyre MD, Richter-Levin G, Avital A, Stewart MG. Morphological changes in hippocampal dentate gyrus synapses following spatial learning in rats are transient. European Journal of Neuroscience. 2003;17:1973–1980. doi: 10.1046/j.1460-9568.2003.02624.x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Gullotta F. Down’s syndrome and Alzheimer’s disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 1990;79:680–685. doi: 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- Foley AG, Hartz BP, Gallagher HC, Rønn LCB, Berezin V, Bock E, Regan CM. A synthetic peptide ligand of NCAM Ig1 domain prevents NCAM internalization and disrupts passive avoidance learning. J Neurochem. 2000;74:2607–2613. doi: 10.1046/j.1471-4159.2000.0742607.x. [DOI] [PubMed] [Google Scholar]
- Fox GB, O’Connell AW, Murphy KJ, Regan CM. Memory consolidation induces a transient and time-dependent increase in the frequency of neural cell adhesion molecule polysialylated cells in the adult rat hippocampus. J Neurochem. 1995;65:2796–2799. doi: 10.1046/j.1471-4159.1995.65062796.x. [DOI] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Alzheimer’s Disease International: Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gundersen HJG. Notes on estimation of the numerical density of arbitrary profiles: the edge effect. J Microsc. 1977;111:219–223. [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd M. Alzheimer’s disease- An electron microscopical study. Brain. 1964;87:307–320. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- LaCor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfribrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marrone DF. Ultrastructural plasticity associated with hippocampal-dependent learning: A meta-analysis. Neurobiology of Learning and Memory. 2007;87:361–371. doi: 10.1016/j.nlm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- McDonald JM, Savva GM, Brayne C, Welzel AT, Shankar GM, Selkoe DJ, Inceand PG, Walsh DM. The presence of SDS-stable Ab dimers is strongly associated with Alzheimer-type dementia. Brain. 2010 doi: 10.1093/brain/awq065. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel L, Land C. Memory traces revisited. Nat Rev Neurosci. 2000;1:209–212. doi: 10.1038/35044572. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces abound. Trends in Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1–42)-amyloid peptide. Brain Res. 2001;912:128–136. doi: 10.1016/s0006-8993(01)02704-4. [DOI] [PubMed] [Google Scholar]
- O’Malley A, O’Connell AW, Regan CM. Ultrastructrural analysis reveals avoidance conditioning to induce a transient increase in hippocampal dentate spine density in the 6 hour post-training period of consolidation. Neuroscience. 1998;87:607–613. doi: 10.1016/s0306-4522(98)00178-x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2. Academic Press; New York: 1986. [Google Scholar]
- Poling A, Morgan-Paisley K, Panos JJ, Kim EM, O’Hare E, Cleary JP, Lesné S, Ashe KH, Porritt M, Baker LE. Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav Brain Res. 2008;193:230–234. doi: 10.1016/j.bbr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Roullet P, Mileusnic R, Rose SP, Sara SJ. Neural cell adhesion molecules play a role in rat memory formation in appetitive as well as aversive tasks. Neuroreport. 1997;8:1907–1911. doi: 10.1097/00001756-199705260-00023. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Seymour CM, Foley AG, Murphy KJ, Regan CM. Intraventricular infusions of anti-NCAM PSA impair the process of consolidation of both avoidance conditioning and spatial learning paradigms in Wistar rats. Neuroscience. 2008;157:813–820. doi: 10.1016/j.neuroscience.2008.09.041. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha BR, Vitolo OV, Joshi P, Lordkipanidze T, Shelanski M, Dunaevsky A. Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol Cell Neurosci. 2006;33:274–282. doi: 10.1016/j.mcn.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Terry RD. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol. 1963;22:629–642. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi M, Coover G. Lesions of the ventral hippocampus, but not the dorsal hippocampus, impair conditioned fear expression and inhibitory avoidance on the elevated T-maze. Neurobiol Learn Mem. 2004;81:172–184. doi: 10.1016/j.nlm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Uylings HB, de Brabander JM. Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 2002;49:268–76. doi: 10.1006/brcg.2001.1500. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. Detection of intracellular oligomers of amyloid β-protein in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, Agnaf OE, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Abeta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of β-amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Research. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S. Open-field analysis reveals no behavioural differences in the experimental animal cohorts. Each animal was monitored in the open-field apparatus for two days prior to passive avoidance conditioning. There were no differences in total distance travelled (A) or vertical exploration (B) between the two groups. Data represents mean and 10–90 inter-quartile range of each group at either day 1 or day 2 prior to training.
Figure 2S. Open-field analysis reveals no behavioural differences between 7PA2-treated and ID-7PA2-treated animals. Each animal was monitored in the open-field apparatus at the 24 and 48 h recall times. There were no differences in total distance travelled (A) or vertical exploration (B) between the two groups. Data represents mean and 10–90 inter-quartile range of each group at each recall time.