

Abstract
The synthesis and structure–activity relationship analysis of a novel class of amide-based biaryl NR2B-selective NMDA receptor antagonists are presented. Some of the studied compounds are potent, selective, non-competitive, and voltage-independent antagonists of NR2B-containing NMDA receptors. Like the founding member of this class of antagonists (ifenprodil), several interesting compounds of the series bind to the amino terminal domain of the NR2B subunit to inhibit function. Analogue potency is modu-lated by linker length, flexibility, and hydrogen bonding opportunities. However, unlike previously described classes of NR2B-selective NMDA antagonists that exhibit off-target activity at a variety of monoamine receptors, the compounds described herein show much diminished effects against the hERG channel and α1-adrenergic receptors. Selections of the compounds discussed have acceptable half-lives in vivo and are predicted to permeate the blood–brain barrier. These data together suggest that masking charged atoms on the linker region of NR2B-selective antagonists can decrease undesirable side effects while still maintaining on-target potency.
Keywords: NMDA, GluN2B, NR2B-selective antagonists, Neuroprotection
1. Introduction
Ionotropic glutamate receptors mediate excitatory synaptic transmission in the central nervous system, and can be divided into three categories based on pharmacology and amino acid se-quence, which include N-methyl-d-aspartate (NMDA), kainate, and amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors.1–3 NMDA receptors are involved in neuronal development, learning, motor function, and pain transmission. Excessive activation of NMDA receptors can lead to neuronal death, and thus NMDA receptor over-activation has been proposed to occur in conditions in which extracellular glutamate is elevated, such as ischemic stroke and traumatic brain injury.4–8 In addition, it has also been proposed that NMDA receptors play a role in Alzheimer’s disease,9–12 Huntington’s disease,13–16 Parkinson’s disease,17–19 depression,20 and neuropathic pain.21–24
Structurally, NMDA receptors are heterooligomeric ligandgated cation channels containing two glycine-binding NR1 subunits, which exist as eight different RNA splice variants, and two glutamate-binding NR2 subunits. Simultaneous binding of the co-agonists glutamate and glycine leads to opening of the receptor channel, and subsequent influx of cations, including Ca2+. The resulting membrane depolarization contributes to the propagation of the excitatory signal, and the influx of Ca2+ can trigger intracellular signaling pathways.
There are numerous sites on the protein that could be pharmacologically exploited to modulate NMDA receptor function. For example, organic cations have been developed as selective blockers that bind within the ion conduction path in NMDA receptors (e.g., MK-801, 1, Fig. 1).25 In addition, competitive antagonists that bind to the co-agonist glycine site (Licostinel, 2)26 or glutamate site have been developed (Selfotel, 3).27,28 A number of subtype-selective non-competitive antagonists that bind to the amino terminal domain have been developed and potently inhibit receptors containing the NR2B subunit such as the negative allosteric modulators ifenprodil29,30 (4) and CP-101,606 (5).31–36 Initial clinical trials of competitive NMDA receptor blockers and channel blockers as neuroprotectants failed for several reasons including unfavorable side effects, which led to lowering of the dose below efficacious levels. In addition, the rapidly developing neurodegeneration after cerebral ischemia made it problematic to administer the compounds soon enough after the insult to prevent NMDA receptor-mediated cell death.37 To date only one NMDA receptor antagonist, the channel blocker memantine hydrochloride (6, Namenda ©), has been approved by the FDA for the treatment of Alzheimer’s disease.38,39
Figure 1.

The structures are shown for six classes of NMDA receptor antagonists.
A number of lines of evidence suggest that NR2B subtype-selective blockade of NMDA receptors can be beneficial in cerebral ischemia, epilepsy, Parkinson’s disease, depression, and perhaps Alzheimer’s disease.20,40–42 In addition, mice over-expressing NR2B-containing receptors show an increased sensitivity to pain, and NR2B-selective antagonists are antinociceptive in certain models of pain.24,43 While NR1 is ubiquitously expressed in the CNS, NR2B-containing receptors are localized in the forebrain, dorsal root ganglion, striatum, and the spinal cord.44–46 The differential distribution of NR2 receptor subtypes17 has supported the hypothesis that targeting specific NMDA receptor subunits, such as NR2B-containing receptors, will decrease untoward side-effects previously observed in clinical trials of non-selective NMDA receptor antagonists.
Comprehensive SAR studies of NR2B amino terminal domain (ATD) allosteric binding site antagonists suggest that one family of compounds (7, Fig. 2) contains a non-polar A ring connected via a basic amine linker of 9–11 Å to the B ring, which traditionally contains a hydrogen bond donor substituent.47–49 Previous studies in our lab involving enantiomeric propanolamines indicated the most potent compounds contain a 3,4-dichlorophenyl A ring and para-methanesulfonamide B ring.50 The optimal linker in the series was seven atoms, and a basic amine was required for activity. Compound 8 in that study showed excellent selectivity for NR2B-containing heterodimeric NR1/NR2 NMDA receptors and high potency in vitro and neuroprotective properties in an in vivo model of transient focal ischemia. Compound 8 did exhibit off-target monoamine receptor activity including the α1-adrenergic receptor and the human-ether-a-go-go (hERG) encoded cardiac potassium channel. The hERG channel is important in ventricular repolarization, and channel blockage can lead to an increased QT interval and potentially deadly ventricular arrhythmias and represents an off-target limitation addressed through SAR studies for a diverse set of chemical structures.51,52 Eliminating hERG activity, and interactions with other polyamine binding receptors including α1 and sigma receptors, has been a significant hurdle in the development of NR2B-selective NMDA receptor antagonists.
Figure 2.

Schematic of the NMDA receptor antagonist pharmacophore (7) compared to an NR2B-selective enantiomeric propanolamine (8) and an NR2B-selective screening hit (9).
Recently we explored the structural features of a novel NR2B-selective antagonist that was identified while screening a focused library of biaryl compounds (compound 9). This biaryl compound contains a thiosemicarbazide functionality within a seven-atom linker that would be non-ionizable under physiological conditions. We hypothesized that this structural feature would lead to decreased activity at off-target receptors compared to the propanolamines on which we previously worked. Here we describe the synthesis and SAR evaluation of compounds with various non-ionizable linker regions between the A and B rings of scaffold 7. Our goal in this study was to achieve selective, potent, and safe compounds through manipulation of the seven-atom linker and incorporation of various amides.
2. Chemistry
A series of hydrazide derivatives were prepared from the common starting material fragment propane hydrazide 12. Originating from para-aminophenylpropionic acid (10, Scheme 1), initial conversion of the carboxylic acid to methyl ester 11 was achieved using methanol and thionyl chloride. The ester was treated with methanesulfonyl chloride to yield the para-arylmethanesulfonamide, and finally converted to 13 via hydrazinolysis.53 Thiosemicarbazide analogue 14 was formed by reaction of 13 with 3,4-dichlorophenylisothiocyanate, while the use of the corresponding isocyanate gave semicarbazide 15. Bishydrazides 16 and 17 were synthesized using carbodiimide-mediated coupling between 13 and the corresponding carboxylic acid. Phthalamide derivative 18 was prepared via reaction of 13 with 3,4-dichlorophenylmalimide. Mesylate 21 was obtained from lithium aluminum hydride reduction of 3,4-dichlorophenylacetic acid (19) and treatment of the resultant alcohol 20 with methanesulfonyl chloride. The arylethyl propane hydrazide 22 was obtained by displacement of mesylate 21 by hydrazide 13 (Scheme 2).
Scheme 1.

Reagents and conditions: (a) SOCl2, MeOH, −10 °C to rt; (b) MsCl, DCM, pyridine, 0 °C to rt; (c) NHNH2·H2O, MeOH, reflux; (d) 3,4-dichlorophenylisothiocyanate or 3,4-dichlorophenylisocyanate, DMF, rt; (e) EDCI, 3,4-dichlorophenylacetic acid, DMAP, DMF 0 °C to rt; (f) 3,4-dichlorobenzoic acid, DCC, DMF, 0 °C to rt; (g) 5,6-dichloroisobenzofuran-1,3-dione, DMF, reflux.
Scheme 2.

Reagents and conditions: (a) LiAlH4, THF, reflux to rt; (b) pyridine, MsCl, 0 °C to rt; (c) 13, MeOH, reflux.
Amides 49–60 (Table 1) were prepared by EDCI-mediated coupling between acids of type 29–31 and amines 33, 34, 37, 40, 43, 48 and commercially available amines. The common carboxylic acids 29–31 were prepared by conversion to the aniline methyl ester (25, 26), formation of the para-arylmethanesulfonamide ester (27, 28), and saponification to give the desired carboxylic acids (Scheme 3).
Table 1.
Synthesis of amides 49–60a
| Amine | n = | Acid Z | Structure | ID |
|---|---|---|---|---|
| 33 | 1 | C |
|
49 |
| 48 | 1 | C |
|
50 |
| 1-(3,4-Dichlorophenyl) piperazine | 1 | C |
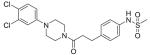
|
51 |
| 33 | 1 | O |
|
52 |
| 48 | 1 | O |
|
53 |
| 43 | 1 | O |
|
54 |
| 37 | 1 | O |
|
55 |
| 40 | 1 | O |
|
56 |
| 1-(3,4-Dichlorophenyl) piperazine | 1 | O |
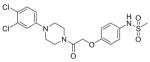
|
57 |
| 3,4-Dichlorophenethylamine | 2 | C |
|
58 |
| 34 | 1 | O |
|
59 |
| 34 | 1 | C |
|
60 |
All molecules were synthesized using EDCI, DMAP, DMF. Acid Z and n refer to atomic detail of compounds 29–31.
Scheme 3.

Reagents and conditions: (a) SOCl2, MeOH, −10 °C to rt; (b) H2, 5% Pd/C, MeOH, rt; (c) pyridine, MsCl, 0 °C to rt; (d) 1.0 M NaOH, MeOH, rt.
Various 3,4-dichlorophenyl alkyl amines were synthesized for subsequent amide bond formation (Scheme 4). The 3,4-dichlorophenyl-1,2-ethanediamine and the corresponding propyldiamine linkers 33 and 34 were prepared by refluxing excess 3,4-dichloroaniline (32) with the corresponding bromoalkyl hydrobromide.54 Mitsunobu conditions with 3,4-dichlorophenol (35) and N-Bocethanolamine followed by TFA deprotection gave amine 37.55 The sulfur-containing fragment, 3,4-dichlorophenylthioethanamine (40), was generated by ring opening of 2-oxazolidinone (39) with 3,4-dichlorothiophenol (38) followed by decarboxylation.56 Lithium aluminum hydride reduction of propionamide 42 gave 3,4-dichlorophenylpropionamine (43). Finally, the arylcinnamylamine derivative 48 was formed via Heck reaction of allyl phthalamide (45) and 3,4-dichloroiodobenzene (46) followed by cleavage with hydrazine.57
Scheme 4.

Reagents and conditions: (a) bromoethylamine hydrobromide or bromopropylamine HBr, toluene, reflux; (b) N-Boc-aminoethanol, PPh3, DIAD, THF, 0 °C to rt; (c) TFA, THF, rt; (d) 130 °C; (e) oxalyl choride, THF, DMF, rt; (f) Et2O, NH3 in 1,4-dioxane, rt; (g) LiAlH4, THF, 0 °C to rt; (h) allyl bromide, DMF, 50 °C; (i) 3,4-dichloroiodobenzene (46), Pd(OAc)2,Et3N, MeCN, 100 °C; (j) NH2NH2·H2O, EtOH, reflux.
Amide 67 was prepared as illustrated in Scheme 5. Mitsunobu conditions with para-nitrophenol (61) and N-Boc-hydroxyethylcarbamate gave 62. The nitro group was hydrogenolized to give aniline 63, followed by reaction with methanesulfonyl chloride to yield para-arylmethanesulfonamide 64. Deprotection with trifluoroacetic acid and formation of the hydrochloride salt afforded compound 65. Acylation of 3,4-dichloroaniline (32) with chloroacetyl chloride gave 66 which was combined with 65 to yield 67.
Scheme 5.

Reagents and conditions: (a) N-Boc-ethanolamine, PPh3, DIAD, THF, 0 °C to rt; (b) H2, 5% Pd/C, MeOH, rt; (c) MsCl, DIPEA, DCM, 0 °C to rt; (d) TFA, DCM, rt; (e) chloroacetyl chloride, Et3N, DCM, 0 °C to rt; (f) 65, Et3N THF, reflux.
Borane reduction of amide 49 resulted in ethanediamine derivative 68. Subsequent cyclization with N,N’-carbonyldiimidazole gave imidazolidinone 70. Analogue 71 was prepared in a similar fashion via amide 52 (Scheme 6).
Scheme 6.

Reagents and conditions: (a) LiAlH4, THF, 0 °C to rt; (b) CDI, THF, rt.
3. Results and discussion
3.1. Structure–activity-relationships
The potency and selectivity of all new compounds synthesized were evaluated using two electrode voltage clamp analysis of the effect of compounds on recombinant NMDA receptor function. We first screened the effect of 3 μM of each compound against current responses produced by maximally effective concentrations of the co-agonists glutamate and glycine at rat NR1/NR2A, NR1/NR2B, NR1/NR2C, NR1/NR2D as well as representative members of the AMPA-selective class of glutamate receptors (GluR1) and the kainate-selective glutamate receptor class (GluR6, Table 2). Compounds that produced response percentages less than 50% had marked inhibition of current responses. The data clearly show that most responses for all compounds in every receptor (NR1/NR2A, NR1/NR2C, NR1/NR2D, homomeric GluR1, homomeric GluR6) but the NR1/NR2B sub-type showed little to no effects with percentages greater than 90% in most cases. Furthermore, all but three compounds (16, 18, and 58) had less than 50% responses against the NR1/NR2B sub-type. This initial data clearly shows these compounds in general, had minimal effects (<10% change from control) at receptors comprised of NR1/NR2A, NR1/NR2C, NR1/NR2D, homomeric GluR1, homomeric GluR6, suggesting high NR2B-selectivity for this class of compounds.
Table 2.
In vitro analysis of subunit selectivity
| Compound | NR1/NR2A % response | NR1/NR2B % response | NR1/NR2C % response | NR1/NR2D % response | GluR1 % response | GluR6 % response |
|---|---|---|---|---|---|---|
| 14 | 104 ± 6.5 | 12 ± 1.2* | 94 ± 1.0 | 92 ± 0.7 | 100 ± 0.7 | 96 ± 0.6 |
| 16 | 104 ± 7.1 | 75 ± 2.0* | 91 ± 4.4 | 94 ± 1.8 | 101 ± 0.5 | 97 ± 1.0 |
| 15 | 101 ± 3.9 | 6 ± 1.4* | 96 ± 3.1 | 91 ± 1.3 | 100 ± 0.6 | 99 ± 1.2 |
| 22 | 106 ± 3.7 | 44 ± 1.8* | 97 ± 0.9 | 95 ± 2.9 | 100 ± 0.7 | 98 ± 1.0 |
| 17 | 102 ± 4.1 | 21 ± 2.6* | 97 ± 1.4 | 96 ± 0.7 | 100 ± 0.9 | 97 ± 1.0 |
| 18 | 101 ± 1.6 | 90 ± 3.2* | 102 ± 2.0 | 93 ± 4.8 | 98 ± 1.0 | 100 ± 0.8 |
| 49 | 99 ± 2.9 | 14 ± 2.8* | 101 ± 0.9 | 84 ± 3.4* | 103 ± 1.0 | 98 ± 2.3 |
| 51 | 103 ± 4.6 | 48 ± 5.6* | 100 ± 1.8 | 97 ± 1.9 | 104 ± 1.5 | 101 ± 2.4 |
| 50 | 99 ± 4.4 | 18 ± 5.0* | 95 ± 2.3 | 88 ± 2.7* | 104 ± 1.6 | 100 ± 0.8 |
| 58 | 99 ± 2.1 | 62 ± 5.0* | 102 ± 0.7 | 89 ± 2.4* | 102 ± 0.9 | 100 ± 0.5 |
| 68 | 89 ± 2.3* | 21 ± 0.9* | 104 ± 3.1 | 91 ± 2.0 | 102 ± 1.1 | 99 ± 1.1 |
| 52 | 92 ± 1.5* | 16 ± 1.2* | 98 ± 2.4 | 91 ± 1.2 | 101 ± 0.6 | 99 ± 3.3 |
| 70 | 96 ± 2.2 | 17 ± 1.3* | 97 ± 2.0 | 91 ± 0.9 | 100 ± 0.9 | 99 ± 0.5 |
| 69 | 91 ± 1.4* | 17 ± 1.2* | 100 ± 1.2 | 100 ± 2.3 | 101 ± 0.7 | 98 ± 0.7 |
| 57 | 89 ± 2.7 | 18 ± 4.0* | 94 ± 2.3* | 106 ± 6.6 | 101 ± 0.7 | 101 ± 0.4 |
| 54 | 99 ± 3.3 | 15 ± 3.3* | 96 ± 1.4 | 79 ± 2.0* | 100 ± 0.5 | 102 ± 1.6 |
| 55 | 97 ± 4.0 | 21 ± 2.3* | 96 ± 2.1 | 92 ± 2.2 | 100 ± 0.6 | 100 ± 1.3 |
| 56 | 96 ± 2.2 | 21 ± 7.7* | 95 ± 2.2 | 96 ± 3.4 | 101 ± 0.1 | 94 ± 2.4 |
| 71 | 105 ± 2.6 | 20 ± 2.9* | 98 ± 1.7 | 98 ± 1.2 | 98 ± 3.8 | 103 ± 0.89 |
| 67 | 108 ± 2.5* | 21 ± 3.2* | 94 ± 2.9 | 95 ± 1.8 | 97 ± 2.6 | 98 ± 1.2 |
| 53 | 100 ± 2.7 | 14 ± 2.3* | 95 ± 2.7 | 97 ± 2.3 | 101 ± 0.4 | 94 ± 4.1 |
| 59 | 106 ± 2.6 | 12 ± 2.0* | 97 ± 2.2 | 100 ± 2.2 | 103 ± 4.0 | 90 ± 2.8 |
| 60 | 96 ± 5.2 | 12 ± 1.4* | 90 ± 2.2 | 96 ± 2.2 | 100 ± 0.4 | 98 ± 7.1 |
Data shown are the current response in Xenopus oocytes expressing the indicated recombinant rat glutamate receptors recorded under two electrode voltage clamp (VHOLD −40 mV) in response to maximally effective concentration of glutamate and glycine (50 μM, 30 μM) in the presence of 3 μM of the indicated test compound, expressed as a percent of control response in the absence of test compound. Measurements are the mean ± SEM from 4 to 10 oocytes for each compound at each receptor subtype. For compounds showing a greater than 10% change from control
indicates p <0.05 (paired t-test).
We subsequently determined the concentration–effect curve for all compounds against recombinant human NR1/NR2B NMDA receptors. The concentrations that produced half-maximally effective inhibition (IC50) for all compounds are presented below in Table 3. While screening hit 9 (rat IC50 = 600 nM) contained a phenolic B ring, our previous studies50 focused on para-arylme-thanesulfonamide B ring substitution. The hybrid thiosemicarba-zide structure 14, which replaces the phenol functionality with a sulfonamide, exhibits increased potency (IC50 of 270 nM), consistent with our previous work with the propanolamine series. Changing the sulfur to an oxygen, as in semicarbazide 15, significantly enhanced potency (IC50 of 28 nM). By contrast, replacement of the aniline nitrogen with a carbon, as in bishydrazide 16 and monohydrazide 22, leads to significantly lowered IC50 values (IC50 5750, 4810 nM, respectively). Rigidifying the A ring into a larger ring system, such as 18, also decreased potency (IC50 41,800 nM). Compound 17, with a shortened linker (six atoms), was the only moderately active hydrazide analogue (IC50 521 nM). Hydrazides are known to lie perpendicular in space at the N-N bond,58 and further incorporation of the above analogues into the semicarbazide functionality insures limited bond rotation and flexibility which may be crucial for binding. Therefore, the key aniline nitrogen required for potency of semicarbazide 15 was incorporated into various amides. We postulated that reducing chain rigidity and increasing hydrophilicity would lead to potent compounds with a more diverse SAR profile. The ethanediamine amide 49 showed excellent potency (IC50 65 nM). Incorporation of a phenoxy oxygen proximal to the B ring, as in 52, showed similar potency (IC50 54 nM). Analogues of compounds 49 and 52 with eight-atom linkers (60 and 59), respectively) were found to be less effective than their seven-atom counterparts (IC50 values 750, 193 nM, respectively), suggesting a seven-atom linker is optimal in the amide series.
Table 3.
Comparison of in vitro on-target potency with selected off-target data
| Compound | NR2B IC50a (nM) | NR2B Kib (nM) | hERG (% displacement at 10 μM)c | hERG Kid (nM) | α1-Adrenergic (% displacement at 3 μM)e |
|---|---|---|---|---|---|
| 14 | 270 | 13 | <1 | ||
| 16 | 5750 | ||||
| 15 | 28 | 3 | 5 | ||
| 22 | 4810 | ||||
| 17 | 521 | ||||
| 18 | 41,800 | ||||
| 49 | 65 | 65 | 4620 | 1 | |
| 51 | 3600 | ||||
| 50 | 50 | 47* | 16 | ||
| 58 | 1860 | ||||
| 68 | 19 | >98 | 14 | 52 | |
| 52 | 54 | 119 | 43 | <1 | |
| 70 | 637 | 81 | <1 | ||
| 69 | 39 | 97 | 49 | ||
| 57 | 166 | 817 | 93 | 90 | |
| 54 | 64 | <1 | <1 | ||
| 55 | 380 | 42 | 11 | ||
| 56 | 362 | 75 | 5 | ||
| 71 | 730 | 300 | 71 | 23 | |
| 67 | 1110 | 67 | 21 | ||
| 53 | 72 | 74 | 22 | ||
| 59 | 193 | ||||
| 60 | 750 | 83 | 19 |
The IC50 value was determined from two electrode voltage clamp recordings of human NR1/NR2B receptor function, fitted as described in the Methods. For all experiments, data are the fitted IC50 value for the mean composite averaged from between 16 and 29 oocytes from 3 or more different frogs.
Ki values for NR2B receptors determined by displacement of 2 nM [3H] ifenprodil from Wistar rat cerebral cortex membranes.
Binding to the human ether-a-go-go potassium channel (hERG) expressed in HEK293 cells by displacement of 1.5 nM [3H]-astemizole. Each result represents the average of displacement binding experiments done in duplicate at 10 μM of the test compound, except where noted.
Ki values for human hERG channels determined by displacement of 1.5 nM [3H] astemizole from HEK-293 cell membranes transfected with human recombinant hERG channels (MDS Pharma). Data from multipoint displacement curves were fit by a non-linear, least squares regression analysis and the Ki calculated using the Cheng and Prusoff equation.
Percent displacement of 0.25 nM [3H] prazosin from Wistar rat brain membranes. Each result represents the average of displacement binding experiments done in duplicate at 3 μM of the test compound.
At 3 μM. All binding assays were performed by MDS Pharma, Bothell, WA.
A collection of amides were synthesized to probe the effect of the aniline nitrogen proximal to the A ring on activity. In contrast to the semicarbazide series where replacement of the aniline nitrogen with a carbon led to a 200-fold decrease in activity, such a replacement in the amide series, particularly propaneamide 54, had little effect (IC50 64 nM). Substitution of the aniline nitrogen with oxygen (55) or sulfur (56) however, did result in significant decreases in potency (IC50 380, 362 nM, respectively). Analogues with carbons containing a sp2 orientation, cinnamyl amides 50 and 53, both showed good activity (IC50 50, 72 nM, respectively), suggesting that conformation might be more important than hydrogen bonding interactions for binding to the allosteric site. The location of the amide in the linker region is also important for potency. For example, aniline acetamide 67 reduced NR2B potency to ~1000 nM IC50 value compared with 52, whose amide is centrally located in the linker.
The effect of rigidifying the ethanediamine linker by incorpora-tion into various heterocyclic structures was also probed. Both acyl piperazines 51 (Z = C) and 57 (Z = O) showed decreased potency from the parent ethanediamine amides. However 57 showed only a modest decrease in potency (IC50 166 nM) while 51 had an IC50 value over 3 μM, indicative of the varying effect of the phenoxy oxygen on activity depending on the particular scaffold. Imidazolidinone activity was also dependent on the presence of a phenoxy oxygen in the linker region of the molecule. Unlike the piperazine amides, cyclic ureas 70 (Z = C) and 71 (Z = O) had similar potency (IC50 637, 730 nM, respectively), although both were less active than their parent ethanediamine amides.
3.2. Off-target effects
Because previous classes of NR2B antagonists have shown off-target interactions with hERG potassium channels and α1-adrenergic receptors, we also evaluated the actions of selected new compounds with IC50 values less than 1 μM at NR1/NR2B recombinant NMDA receptors on these targets (Table 3). Thiosemicarbazide analogues exhibited minimal hERG and α1 activity. This confirmed our original hypothesis that charged nitrogen atoms are a strong structural determinant for off-target activity; elimination of the charge in the linker region attenuates affinity of the NR2B antagonists for the hERG channel.52
While amine 68 has a Ki value of 14 nM against hERG, the cor-responding amide 49 shows a greater than 300-fold decrease in the measured Ki value to 4620 nM. A similar case is seen with amine 69 (estimated Ki of 300 nM based on 97% displacement of [3H]astemizole at 10 μM) and corresponding amide 52 (estimated Ki ~12,000 nM). Also important is the 40-fold decrease in hERG binding observed with the incorporation of the phenoxy oxygen proximal to the B ring. Similar to the trend seen with hERG activity, neither amide 49 or 52 showed any α1-adrenergic binding (estimated as displacement of ligand at 3 μM) compared with the corresponding amines. Within the linear amide series, hERG activity was also influenced by the functionality proximal to the A ring. Sulfur-containing analogue 56 showed significant binding to the hERG channel (75% displacement at 10 lM) and ligand displace-ment consistent with low affinity for the α1-adrenergic receptor. The corresponding oxygen-containing analogue 55 reduced hERG channel binding (42% displacement at 10 μM) but exhibited little change in displacement of α1 ligands. This reinforces the value of introducing a phenoxy oxygen proximal to the B ring discussed above. It is clear that higher number of oxygen atoms in the linker leads to decreased off-target effects at hERG channels and α1-adrenergic receptors. While the sp3-carbon-containing amide ana-logue 54 exhibited negligible hERG and α1-adrenergic receptor binding, the sp2-carbon analogue 50 showed low potency binding to the hERG channel (estimated Ki ~3000 nM) without actions at the α1-adrenergic receptor.
Rigidifying the linker region also has a profound influence on off-target effects. While amide 52 exhibited minimal hERG and α1 binding, the corresponding acyl piperazine 57 showed more potent affinity for hERG (93% displacement) and the highest α1 activity of any compound tested (90% ligand displacement). Imidazolidinones 70 and 71 have hERG receptor activity at levels between linear amides and acyl piperazines but negligible α1 activity. This suggests that in addition to the presence of charge within the linker region, conformation and hydrogen bonding opportunities are important to off-target activity.
We also determined plasma half-life for a select group of compounds (50, 52, and 55) based on the overall in vitro data. Compound 52 showed a half-life of 0.92 ± 0.13 h (n = 3) when dosed at 3 mg/kg iv in rats. When the aniline nitrogen was replaced with an oxygen (55) or a carbon (50) the plasma half-life was shortened by half to 0.4 ± 0.16 (n = 3) h and 0.45 ± 0.07 (n = 3) h, respectively.
3.3. In vitro analysis of NMDA receptor antagonism
From these experiments we determined that compound 52 shows high potency and strong NR2B subunit selectivity. In addition, compound 52 was easily synthesized on large scale and exhibited the best half-life in rats (0.92 ± 0.13 h). We thus initiated a series of experiments to further probe the site and mechanism of action for this compound.
Compound 52 inhibits NR1/NR2B current responses with a half-maximally effective concentration of 36 nM (mouse subunits, n = 11 oocytes from 2 frogs), 57 nM (rat, n = 5–18 oocytes from 3 frogs), and 54 nM (human, n = 6–24 oocytes from 4 frogs). For all inhibition curves, the Hill slope ranged between 0.81 and 0.94. The amino acid identity between murine and human NR1 and NR2B subunits is ~98% with most changes occurring in a region of the receptor not expected to be part of the binding pocket for NR2B-selective inhibitors. Compound 52 did not inhibit receptor function fully, but rather showed a maximal inhibition of 88% in rat NR1/NR2B, 78% in mouse NR1/NR2B, and 90% inhibition in human (Fig. 3A and B). This result is consistent with other NR2B-selective antagonists, which act by a non-competitive mechanism to bring about incomplete inhibition.33,59–62 Compound 52 (3 μM) had minimal effects on recombinant heterodimeric NMDA receptors that contain other rat NR2 subunits (Fig. 3A). Determination of the IC50 value at rat NR1/NR2A (82 μM, n = 7), NR1/NR2C (58 μM, n = 6), NR1/NR2D (107 μM, n = 5) suggested that com-pound 52 was more than 1000-fold selective for rat NR1/NR2B over all other NR1/NR2 NMDA receptors. There were no detectable effects on recombinant kainate receptors (GluR6), recombinant AMPA (GluR1) receptors, or voltage-gated Na+ or K+ currents recorded from cultured cortical neurons (Fig. 3A). Furthermore, consistent with other NR2B-selective ligands, inhibition of NR1/NR2B receptor responses by compound 52 was not surmountable by increasing the concentrations of glycine or glutamate 10-fold (Fig. 3C), suggesting inhibition is non-competitive. The inhibition produced by 52 was voltage-independent (n = 5; Fig. 3D).
Figure 3.

Compound 52 is an NR2B-selective, non-competitive, voltage-independent NMDA receptor inhibitor. (A) Two electrode voltage clamp recordings from Xenopus oocytes were used to measure the mean ± SEM response to glutamate and glycine coapplied with 3 μM compound 52 on various glutamate receptors expressed as a percent of the maximal response evoked by saturating concentrations of agonists (50 μM glutamate plus 30 μM glycine) for the indicated NMDA receptors. Similar experiments were performed on recombinant GluR1 AMPA receptors (100 μM glutamate), and recombinant GluR6 kainate receptors pre-treated with 10 μM concanavalin-A (100 μM glutamate). The effect of 3 μM compound 52 was also evaluated on voltage-dependent sodium and potassium whole cell currents recorded from cultured cortical neurons and elicited by incremental 10 mV step depolarizations from −90 mV to +50 mV. The amplitudes of Na+ currents and K+ currents in the absence and presence of 3 μM compound 52 were measured at −10 mV and +50 mV, respectively. The numbers in parenthesis are the number of oocytes or neurons tested under each condition. *p <0.05, paired-t-test. (B) Concentration–effect curves for compound 52 were generated in oocytes expressing wild type and mutant NR1/NR2B receptors. The IC50 values of compound 52 were 0.057 μM on rat wild type receptors (n = 18), 52 μM on mouse amino terminal domain D101A point mutants (n = 4) and 563 μM on rat amino terminal domain deletion mutants (n = 3). (C) Increasing the concentration of glutamate does not surmount the inhibition by 0.054 μM of compound 52 (n = 4), suggesting compound 52 inhibits the receptor by a non-competitive mechanism. The mean ± SEM response as a percent control is summarized for experiments raising either glutamate or glycine concentration in the right panel. (D) Inhibition by compound 52 is independent of membrane potential. A representative recording from an oocyte expressing rat NR1/NR2B shows the current–voltage relationship for responses evoked by of 50 μM glutamate plus 30 μM glycine as closed circles and 0.054 μM compound 52 coapplied with 50 μM glutamate plus 30 μM glycine as open circles (representative of eight experiments).
These data are consistent with compound 52 exerting a nega-tive allosteric effect on NR2B receptor function through direct interaction with at least a portion of the ifenprodil binding site, which has been proposed to be contained within the amino terminal domain of the NR2B receptor.29,63,64 To test whether compound 52 and its analogues bind to the amino terminal domain, we evaluated a rat NR2B subunit in which residues within the amino terminal domain between S28 and H405 were deleted. We hypothesized that deletion of the amino terminal domain from the NR2B subunit, NR2B(ΔATD), would render the receptors insensitive to inhibition by compound 52 and its analogues. Functional properties of this deletion construct are similar to that of wild type receptors (e.g., glutamate and glycine EC50s are not altered, data not shown). As expected, compound 52 had virtually no effect on the current response of NR1/NR2B(ΔATD) when activated by maximal concentrations of glutamate and glycine (Fig. 3B). In addition, Asp101 (Δ101) within NR2B has been suggested to play an important role in ifenprodil binding. We find that mutation of this residue to alanine NR2B(Δ101A) increased the IC50 to ~30 μM(n =4 oocytes). This ~500-fold reduction in potency by NR2B(D101A) is consistent with compound 52 binding to the ifenprodil site on the NR2B subunit. Similar results were found for compounds 15, 49, and 68, which were 51−, 102−, 177-fold less potent when evaluated at NR2B(D101A) (n = 7, 13, 20 oocytes, respectively). A recently published homology model suggests the protonated piperidine of ifenprodil makes an electrostatic interaction with D101.65 Presumably, the amide compounds described here could make no such salt bridge. However, since D101A mutation reduces the potency (increases the IC50 value) for 15 and 52, we hypothesize that the amide compounds make H-bond(s) to D101 and are situated similarly to ifenprodil within the ligand binding site of the NR2B ATD.66
3.4. Brain penetration
To evaluate how this class of molecules might penetrate the brain, we estimated brain penetration for a number of compounds using the MDR1-MDCK in vitro assay (Table 4). It was determined that 52 was not expected to appreciably pass the blood–brain barrier (Papp(A–B) = 1.80) and is deemed a substrate for P-glycoprotein (Pgp) efflux (efflux ratio = 35). Acyl piperazine 57 shows improved brain penetration potential (Papp(A–B) = 4.8) but is also deemed a Pgp efflux substrate (efflux ratio = 8.9). The compound with the highest brain penetration potential tested in this in vitro model assay was imidazolidinone 71, which showed both the highest passive diffusion potential (papp(A–B) = 12.9), and lowest efflux ratio of 3.1. Thus, tying up the chain nitrogen atoms into a cyclic functionality, such as a piperazine or imidazolidinone, appears to increase the likelihood of crossing the blood brain barrier probably due to increased lipophilicity as well as decreasing the number of free H-bond donors. Furthermore, the ability of these compounds to act as a substrate for Pgp efflux is significantly decreased based on the Papp(B–A) results.
Table 4.
MDR1-MDCK permeability
Papp is the apparent permeability for the apical to basal (A–B) and the basal to apical (B–A) direction across MDR1-MDCK cell monolayers in Transwell® wells. Papp units are ×10−6 cm/s.
Efflux ratio = Papp (A–B)/Papp (B–A).
4. Conclusions
A novel series of amide-based NR2B-selective NMDA receptor antagonists was developed with high on-target potency and attractive off-target profiles. Compounds 52 and 71 in particular show an increased drug-like characteristics over the previous enantiomeric propanolamine series, including concomitant high potency with minimal hERG and α1 binding. In addition, 52 has an acceptable half-life in vivo while 71 is expected to cross the blood–brain barrier. These amide-based analogues provide new insight into ways by which off-target effects can be mitigated while retaining high potency and selectivity at NR2B containing receptors, and represent a starting point for further development of the potential therapeutic uses of NR2B-selective NMDA receptor antagonists.
5. Experimental procedures-biology
5.1. Expression of glutamate receptors in Xenopus laevis oocytes
All protocols involving the use of animals were approved by the Emory University IACUC. cRNA was synthesized from linearized template cDNA for rat glutamate receptor subunits according to manufacturer specifications (Ambion). Quality of synthesized cRNA was assessed by gel electrophoresis, and quantity was estimated by spectroscopy and gel electrophoresis. Stage V and VI oocytes were surgically removed from the ovaries of large, well-fed and healthy Xenopus laevis anesthetized with 3-amino-benzoic acid ethyl ester (1–3 gm/l) as previously described.67 Clusters of isolated oocytes were incubated with 292 U/mL Worthington (Freehold, NJ) type IV collagenase or 1.3 mg/mL collagenase (Life Technologies, Gaithersburg, MD; 17018–029) for 2 h in Ca2+-free solution comprised of (in mM) 89 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 10 HEPES, with slow agitation to remove the follicular cell layer. Oocytes were then washed extensively in the same solution and maintained in Barth’s solution comprised of (in mM) 88 NaCl, 1 KCl, 2.4 NaHCO3, 10 HEPES, 0.82 MgSO4, 0.33 Ca(NO3)2, and 0.91 CaCl2 and supplemented with 100 μg/mL gentamycin, 10 μg/mL streptomycin, and 10 μg/mL penicillin. Oocytes were manually defolliculated and injected within 24 h of isolation with 3–5 ng of NR1–1a (hereafter NR1) subunit and 7–10 ng of NR2 subunit in a 50 nl volume, or 5–10 ng in 50 nl of AMPA or kainate receptor cRNAs, and incubated in Barth’s solution at 15 °C for 1–7 d. Glass injection pipettes had tip sizes ranging from 10 to 20 μM, and were backfilled with mineral oil.
5.2. Two electrode voltage clamp recording from Xenopus laevis oocytes
Two electrode voltage clamp recordings were made 2–7 days post-injection as previously described.67 Oocytes were placed in a dual-track plexiglass recording chamber with a single perfusion line that splits in a Y-configuration to perfuse two oocytes. Dual recordings were made at room temperature (23 °C) using two Warner OC725B or OC725C two electrode voltage clamp amplifiers, arranged as recommended by the manufacturer. Glass micro-electrodes (1–10 Megaohms) were filled with 300 mM KCl (voltage electrode) or 3 M KCl (current electrode). The bath clamps communicated across silver chloride wires placed into each side of the recording chamber, both of which were assumed to be at a reference potential of 0 mV. Oocytes were perfused with a solution comprised of (in mM) 90 NaCl, 1 KCl, 10 HEPES, and 0.5 BaCl2; pH was adjusted to 7.4 or 7.6 by addition of 1 M NaOH. Oocytes expressing NR1/NR2A were pre-incubated before recording in recording solution supplemented with 50 μM BAPTA-AM at room temperature. Oocytes were recorded under voltage clamp at −40 mV. Final concentrations for control application of glutamate (50–100 μM) plus glycine (30 μM) to oocytes expressing NMDA receptors were achieved by dilution from 100 and 30–100 mM stock solutions, respectively. In addition, 10 μM final EDTA was obtained by adding a 1:1000 dilution of 10 mM EDTA, in order to chelate contaminant divalent ions such as extracellular Zn2+. Homomeric GluR1 AMPA receptors were activated by 100 μM glutamate. Homomeric GluR6 kainate receptors were incubated in concanavalin-A (10 μM) for 5 min, and activated by 100 μM glutamate. Concentration–response curves for experimental compounds acting on NMDA receptors were obtained by applying in successive fashion a maximally effective concentration of glutamate/glycine, followed by glutamate/glycine plus variable concentrations of experimental compounds. Concentration–response curves consisting of 5–8 concentrations were obtained in this manner. The baseline leak current at −40 mV was measured before and after recording, and the full recording linearly corrected for any change in leak current. Oocytes with glutamate-evoked responses smaller than 50 nA were not included in the analysis. The level of inhibition produced by experimental compounds was expressed as a percent of the initial glutamate response, and averaged together across oocytes from a single frog. Each experiment consisted of recordings from 3 to 10 oocytes obtained from a single frog. Results from ≥3 experiments using oocytes from three different frogs were pooled, and the percent responses at antagonist concentrations for each oocyte were fitted by the equation,
where minimum is the residual percent response in saturating concentration of the experimental compounds, IC50 is the concentration of antagonist that causes half of the achievable inhibition, and nH is a slope factor describing steepness of the inhibition curve. Minimum was constrained to be greater than or equal to 0.
5.3. Whole cell patch clamp recording of voltage-activated currents in neurons
Neuronal cultures were derived from E17 Sprague-Dawley rat pups. Briefly, cortical tissue was dissected, transferred into saline containing penicillin/streptomycin and 10 mM HEPES, and incu-bated in trypsin containing 0.02% DNase at 37 °C for 15 min. Tissue was then triturated and the supernatant resuspended in B27-supplemented Neurobasal medium (Gbco) containing 2 mM l-glutamine and 5% fetal bovine serum. Cells were plated onto poly-d-lysine-coated coverslips and after three days the media was replaced with serum-free medium. Cultures were maintained at 37 °C in a humidified 5% CO2-containing atmosphere. Whole cell patch clamp recordings (voltage clamp, holding potential −60 mV) from 5- to 10-day cultured cortical neurons were made with an Axopatch 200B amplifier (Axon Instruments, Union City, CA) at room temperature (23 °C). The recording chamber was continually perfused with recording solution composed of (in mM) 150 NaCl, 3 KCl, 2 CaCl2, 1.5 MgCl2, 5.5 glucose and 10 HEPES (pH 7.4 by NaOH; osmolality adjusted to 315 mOsm with sucrose). Thin wall glass pipettes were filled with (in mM) 110 d-gluconate (50% w/w), 110 CsOH (50% w/w), 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 NaATP, and 0.3 NaGTP (pH adjusted to 7.3 with CsOH and osmolality adjusted to 300 mOsm with sucrose). Recordings were made in the presence of 10 μM bicuculline, 10 μM CNQX, and 100 μM DL-APV to block both excitatory and inhibitory synaptic transmission. Drugs were applied by gravity and controlled by manual valves. Voltage-gated macroscopic whole cell currents were activated by 100 ms voltage steps from a holding potential of −60 mV to between −90 and +50 mV. The sensitivity of Na+ currents to 0.5 μM tetrodotoxin was confirmed at the end of each experiment; K+ channels were recorded in the presence of 0.5 μM tetrodotoxin to block Na+ channels. We evaluated the mean Na+ current from a number of whole cell recordings at −10 mV, and the mean K+ current at +50 mV using a paired t-test.
5.4. Estimation of brain penetration
Transwell® wells containing MDR1-MDCK cell monolayers were used for measuring the percent recovery of compound after dosing both sides of a cell monolayer with the test article (performed by Absorption Systems, Exton PA, USA). Briefly, monolayers were grown for 7–11 days at which time 5 μM of the test article was made by dilution from DMSO stocks into a Hank’s balanced salt solution (pH 7.4), final DMSO not greater than 1%, and added to: (a) the apical side for A–B permeability assessment, or separately (b) the basal side for the B–A permeability assessment, all at pH 7.4. After a 2 h incubation (37C) both the apical and the basal compartments were sampled and the amount of test article present determined by generic LC–MS/MS methods against a >4 point calibration curve. Apparent permeability (Papp) units are reported × 10–6 cm/s. Experiments were done in duplicate.
6. Experimental procedures: chemistry
6.1. General experimental procedures
All reagents were obtained from commercial suppliers and used without further purification. Reaction progress was monitored by thin layer chromatography (TLC) on precoated glass plates (Silica Gel 60 F254, 0.25 mm). Proton and carbon NMR spectra were recorded on an INOVA-400 (400 MHz), VNMRS 400 (400 MHz), INOVA-600 (600 MHz), or Mercury 300 Vx (300 MHz). The spectra obtained were referenced to the residual solvent peak. Mass spectra were performed by the Emory University Mass Spectroscopy Center on either a VG 70-S Nier Johnson or JEOL instrument. Elemental analyses were performed by Atlantic Microlab Inc. C, H, N agreed with proposed structures within ±0.4% of theoretical values. Flash chromatography was performed on a Teledyne Isco Combiflash Companion. HPLC (Varian) was used to determine the purity of some compounds. HPLC was performed on a C18 analytical column with UV detection using methanol and acetonitrile at 1 mL/min according to the following ratios: (a) 50% acetonitrile/50% methanol over 10 min, (b) 30% acetonitrile/70% methanol-70% acetonitrile/30% methanol gradient over 10 min and (c) 20% acetonitrile/80% methanol-80% acetonitrile/20% methanol gradient over 10 min.
6.2. Chemistry: experimental synthetic procedures
6.2.1. Methyl 3-(4-aminophenyl)propanoate (11)
Thionyl chloride (1.65 mL, 22.6 mmol, 3.3 equiv) was added dropwise to a solution of dry methanol (6.65 mL, 164 mmol, 24 equiv) at −10 °C. After stirring for 10 min, the propionic acid 10 (1.13 g, 6.84 mmol) was added to give a yellow suspension. The solution stirred for 1 h and warmed to room temperature. The resulting solution was concentrated in vacuo to give a yellow solid. The solid was suspended in EtOAc, and solid NaHCO3 was added until the salt dissolved fully. The layers were separated and the organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a yellow solid (1.06 g, 98%). 1H NMR (300 MHz, CDCl3) δ 7.00 (d, J = 8.3 Hz, 2H,) 6.60 (d, J = 8.3 Hz, 2H), 3.67 (s, 3H), 3.59 (br s, 2H), 2.85 (t, J = 7.6 Hz, 2H), 2.58 (t, J = 7.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 173.8, 144.9, 130.7, 129.3, 115.5, 51.8, 36.4, 30.4. m/z (EI+) calcd for C10H13NO2, 179.09; found, 180.30 [M+H]+.
6.2.2. Methyl 3-(4-(methylsulfonamido)phenyl)propanoate (12)
The ester 11 (7.38 g, 41.2 mmol) was dissolved in pyridine (17.0 mL, excess). After cooling to 0 °C, methanesulfonyl chloride (4.55 mL, 57.7 mmol, 1.4 equiv) was added dropwise. The reaction was warmed to room temperature and stirred for 20 h. The reaction was quenched with water and diluted with DCM. The layers were separated and the organics were washed with brine and concentrated in vacuo to give a red solid. The crude material was purified using silica gel chromatography (1 EtOAc/1 hexanes) to give a white solid (9.26 g, 87%). 1H NMR (400 MHz, CDCl3) δ 7.20 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 6.45 (br s, NH,1H), 3.68 (s, 3H), 3.00 (s, 3H), 2.94 (t, J = 7.6 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H).13C NMR (100 MHz, CDCl3) δ 173.4, 137.6, 135.2, 129.4, 121.4, 51.7, 38.5, 35.5, 30.1. m/z (APCI) calcd for C11H15NO4S, 257.07; found, 257.56 [M]+.
6.2.3. 3-(4-(Methylsulfonamido)phenyl)propanehydrazide (13)
Compound 12 (1.34 g, 5.21 mmol) was dissolved in MeOH (30 mL) and hydrazine monohydrate (0.49 mL, 15.6 mmol, 3.0 equiv) was added. The solution was refluxed for 12 h. The resulting solution was concentrated in vacuo to give a white solid. The crude material was purified using silica gel chromatography (10–20% MeOH/DCM gradient) to give a white solid. 1H NMR (DMSO-d6) δ 9.59 (s, 1H), 8.95 (s, 1H), 7.15 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 8.6 Hz, 2H), 4.16 (br s, 2H), 2.93 (s, 3H), 2.76 (t, J = 7.3 Hz, 2H), 2.28 (t, J = 7.3 Hz, 2H).
6.2.4. 4-(3,4-Dichlorophenyl)-1-(3-(4-(methylsulfonamido)-phenyl)propanoyl)thiosemicarbazide (14)
Hydrazide 13 (0.499 g, 1.94 mmol) was dissolved in DMF (15.0 mL). To this solution, 3,4-dichlorophenylisothiocyanate (0.278 mL, 1.94 mmol, 1.0 equiv) was added. The orange solution was stirred for 12 h at room temperature. The solution was concentrated in vacuo to give an orange residue which was purified using silica gel chromatography (3 EtOAc/1 hexanes) to give a white foam (0.670 g, 75%). 1H NMR (600 MHz, CD3OD) δ 7.96 (s, 1H), 7.76 (s, 1H), 7.44 (d, J = 9.1 Hz, 1H), 7.35 (dd, J1 = 9.1 Hz, J2 = 1.9 Hz, 1H), 7.22 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H), 2.95 (t, J = 7.6 Hz, 2H), 2.91 (s, 3H), 2.61 (t, J = 8.1 Hz, 2H).13C NMR (150 MHz, CD3OD) δ 183.7, 174.9, 164.9, 140.1, 138.7, 137.6, 132.8, 131.3, 131.1, 130.6, 130.5, 129.9, 122.3, 122.2, 39.2, 36.6, 31.4. HRMS calcd for C17H18Cl2N4O3S2, 460.0197; found, 459.01074 [M−H]+. Anal. (C17H18Cl2N4O3S2) C, H, N.
6.2.5. 4-(3,4-Dichlorophenyl)-1-(3-(4-(methylsulfonamido)-phenyl)propanoyl)semicarbazide (15)
Hydrazide 13 (0.100 g, 0.39 mmol) was dissolved in DMF (3.0 mL). To this solution, 3,4-dichlorophenyisocyanate (0.073 g, 0.39 mmol, 1.0 equiv) was added and the mixture was stirred at room temperature for 12 h. The resulting solution was concentrated in vacuo to give a white solid. The crude material was purified using silica gel chromatography (10–50% MeOH/DCM gradient) to give a white solid (0.114 g, 66%). 1H NMR (400 MHz, DMSO-d6) δ 9.69 (br s, 1H), 9.60 (br s, 1H), 9.12 (br s, 1H), 8.27 (br s, 1H), 7.85 (d, J = 2.4 Hz, 1H), 7.51 (d, J = 8.8 Hz), 7.41 (m, 1H), 7.20 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 2.94 (s, 3H), 2.80 (t, J = 8.0 Hz, 2H), 2.43 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.2, 155.8, 140.7, 137.4, 136.9, 131.5, 131.1, 129.7, 123.8, 120.9, 120.6, 119.3, 35.5, 30.6. HRMS calcd for C17H18Cl2N4O4S, 444.0426; found 444.04663 [M]+. Anal.(C17H18Cl2N4O4S) C, H, N.
6.2.6. N’-(2-(3,4-Dichlorophenyl)acetyl)-3-(4-(methylsulfonamido)phenyl)propanehydrazide (16)
3,4-Dichlorophenylacetic acid (0.800 g, 3.9 mmol), EDCI (0.750 g, 3.9 mmol, 1.0 equiv) and DMAP (0.470 g, 3.9 mmol, 1.0 equiv) were dissolved in DMF (10.0 mL) at 0 °C and stirred for 45 min. Hydrazide 13 (1.0 g, 3.9 mmol, 1.0 equiv) was added and the mixture was warmed to room temperature and stirred for 12 h. The reaction was quenched with 1.0 N HCl and extracted with EtOAc. The organics were dried over MgSO4 and concentrated in vacuo to give an off-white powder. The crude material was purified using silica gel chromatography (5% MeOH/DCM) to give a white solid which was further purified by recrystallization with EtOAc (0.103 g, 6.0%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.88 (s, 1H), 9.60 (s, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 1.9 Hz, 1H), 7.28 (dd, J1 = 8.6 Hz, J2 = 1.9 Hz, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.6 Hz, 2H), 2.94 (s, 3H), 2.78 (t, J = 7.3 Hz, 2H), 2.40 (t, J = 7.3 Hz). 13C NMR (150 MHz, DMSO-d6) δ 170.2, 168.1, 136.8, 136.3, 131.1, 130.8, 130.4, 129.6, 129.4, 129.3, 129.0, 120.2, 34.7, 30.0, 24.4. HRMS calcd for C18H19Cl2N3O4S, 444.0473; found, 444.04802 [M]+. Anal. (C18H19Cl2N3O4S) C, H, N.
6.2.7. 3,4-Dichloro-N’-(3-(4-(methylsulfonamido)phenyl)propanoyl)benzohydrazide (17)
The hydrazide 13 (0.500 g, 1.9 mmol) was dissolved in DMF (10.0 mL). To this solution, 3,4-dichlorobenzoic acid (0.370 g, 1.9 mmol, 1.0 equiv) was added. Finally, DCC (1.0 M in DCM, 1.9 mL, 1.9 mmol, 1.0 equiv) was added. The mixture was stirred at room temperature for 12 h. The white precipitate was filtered off, and the resulting solution was concentrated in vacuo to give a brown residue. The crude material was purified using silica gel chromatography (10% MeOH/DCM) to give a white solid (0.648 g, 77%). 1H NMR: (600 MHz, CD3OD) δ 8.03 (d, J = 1.9 Hz,1H), 7.86 (dd, J1 = 8.1 Hz, J2 = 1.0 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.1 Hz, 2H), 2.97 (t, J = 7.6 Hz, 2H), 2.92 (s, 3H), 2.61 (t, J = 8.1 Hz, 2H). 13C NMR: (150 MHz, CD3OD) δ 174.4, 166.8, 138.8, 137.8, 137.5, 134.1, 134.1, 132.1, 131.0, 130.6, 128.6, 122.4, 39.1, 36.7, 31.8. HRMS calcd for C17H17Cl2N3O4S, 429.0317; found, 430.03129 [M+H]+. Anal. (C17H17Cl2N3O4S) C, H, N.
6.2.8. N-(5,6-Dichloro-1,3-dioxoisoindolin-2-yl)-3-(4-(methylsulfonamido)phenyl)propanamide (18)
The hydrazide 13 (0.250 g, 0.972 mmol) was dissolved in DMF (5.0 mL). To this solution, 5,6-dichloroisobenzofuran-1,3-dione (0.211 g, 0.972 mmol, 1.0 equiv) was added. The mixture was refluxed for 12 h. The resulting solution was concentrated in vacuo to give a yellow residue. The crude material was purified using silica gel chromatography (10% MeOH/DCM) to give a yellow solid (0.220 g, 50%).1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 8.07 (s, 1H), 7.89 (s, 1H), 7.23–7.24 (mult, 2H), 7.19–7.17 (mult, 2H), 3.35 (br s, 2H), 3.00–2.95 (mult, 2H), 2.91 (s, 3H), 2.70 (t, J = 7.6 Hz, 1H), 2.60 (t, J = 7.6 Hz, 1H).13C NMR (150 MHz, CDCl3) δ 189.0, 174.2, 138.7, 137.9, 137.7, 137.4, 135.6, 133.2, 130.6, 122.3, 39.1, 36.7, 31.9. HRMS calcd for C18H15Cl2N3O5S, 455.0109; found, 456.01001 [M+H]+. Anal. (C18H15Cl2N3O5S·0.25 CH3OH) C, H, N.
6.2.9. 2-(3,4-Dichlorophenyl)ethanol (20)
Lithium aluminum hydride (0.90 g, 24 mmol, 1.0 equiv) was dissolved in THF (50.0 mL) at 0 °C, and then warmed to room temperature for 30 min. To this suspension, a solution of 3,4-dichlorophenyl acetic acid (19, 5.0 g, 24 mmol) and triethyl amine (3.0 mL, 24 mmol, 1.0 equiv) in THF (37.5 mL) was added dropwise. The reaction mixture was refluxed for 1 h. The reaction was cooled to room temperature and poured into water (125 mL) to give a bright yellow solution. The mixture was extracted with EtOAc (2×). organics were dried over Na2SO4 and concentrated in vacuo to give a yellow oil (3.76 g, 81%). The crude material was carried on without further purification. 1H NMR (300 MHz, CDCl3) δ 7.38 (d, J = 8.3 Hz, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.08 (dd, J1 = 8.3 Hz, J2 = 1.9 Hz), 3.87 (quart, J = 6.6 Hz, 2H), 2.83 (t, J = 6.6 Hz, 2H), 1.45 (br s, 1H).
6.2.10. 3,4-Dichlorophenethyl methanesulfonate (21)
Crude alcohol 20 (3.76 g, 2.79 mmol) was dissolved in pyridine (30.0 mL). Methanesulfonyl chloride (1.7 mL, 30 mmol, 1.1 equiv) was added dropwise at room temperature and the reaction was stirred for 1 h. The mixture was poured into ice water and warmed to room temperature. The organics were extracted with EtOAc (3×), dried over Na2SO4, and concentrated in vacuo to give a yellow liquid. The crude material was purified using silica gel chromatography (1 EtOAc/1 hexanes) to give a yellow oil (4.25 g, 80%). 1H NMR (300 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.10 (dd, J1 = 8.1 Hz, J2 = 1.9 Hz, 1H), 4.41 (t, J = 6.6 Hz, 2H), 3.03 (t, J = 6.6 Hz, 2H), 2.95 (s, 3H).
6.2.11. N’-(3,4-Dichlorophenethyl)-3-(4-(methylsulfonamido)phenyl)propanehydrazide (22)
Hydrazide 13 (0.300 g, 1.11 mmol) was dissolved in methanol (11.0 mL). To this solution, mesylate 21 was added dropwise (0.287 g, 1.11 mmol, 1.0 equiv). The solution was refluxed for 48 h, cooled to room temperature, and concentrated in vacuo to give a white solid. The crude material was purified using silica gel chromatography (10% MeOH/DCM) to give a white foam (0.065 g, 14%). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.3 Hz, 1H), 7.29 (d, J = 1.90 Hz, 1H), 7.20 (d, J = 8.6 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 7.04 (dd, J1 = 8.1 Hz, J2 = 2.2 Hz, 2H), 6.73 (br s, 1H), 6.25 (br s, 1H), 4.60 (br s, 1H), 3.04 (mult, 2H), 2.99 (s, 3H), 2.96 (t, J = 7.3 Hz, 2H), 2.69 (t, J = 7.3 Hz, 2H), 2.41 (t, J = 7.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 172.0, 139.8, 137.7, 135.4, 132.4, 130.8, 130.6, 130.4, 129.7, 128.3, 121.4, 52.6, 39.4, 36.2, 33.6, 30.9. HRMS calcd for C18H21Cl2N3O3S, 429.0681; found, 430.07001 [M+H]+ Anal. (C18H21Cl2N3O3S) C, H, N.
General method for preparation of esters 25 and 26, exemplified by 25.
6.2.12. Methyl 2-(4-nitrophenoxy)acetate (25a)
Thionyl chloride (17 mL, 234 mmol, 3.3 equiv) was added dropwise to a solution of dry methanol (70 mL, 1704 mmol, 24 equiv) at −10 °C. After stirring for 10 min, 4-nitrophenoxyacetic acid (14.0 g, 71 mmol) was added to give a white precipitate in an orange suspension. The solution stirred for 1 h and slowly warmed to room temperature. The resulting solution was concentrated in vacuo to give an off-white solid. The solid was dissolved in EtOAc, and NaHCO3 (satd) was added. The layers were separated and the organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a white solid (14.3 g, 95%). 1H NMR (300 MHz, CDCl3) δ 8.23 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 9.2 Hz), 4.75 (s, 2H), 3.84 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 168.4, 162.7, 142.5, 126.2, 114.9, 65.5, 52.8.
6.2.13. Methyl 2-(4-aminophenoxy)acetate (25)
Ester 25a (14.3 g, 68 mmol) was dissolved in dry methanol (100 mL) and 5% palladium on carbon catalyst (1.43 g, 10 wt %) was added. The suspension was hydrogenolyzed using a balloon for 12 h. The mixture was filtered over a pad of Celite, and the resulting solution was concentrated in vacuo to give a pink oil which turned to a solid upon trituration with chloroform. The crude material was purified using silica gel chromatography (1 EtOAc/2 hexanes) to give a pink solid (11.7 g, 95%). 1H NMR (400 MHz, CDCl3) δ 6.74 (d, J = 8.6 Hz, 2H), 6.60 (d, J = 8.6 Hz), 4.54 (s, 2H), 3.77 (s, 3H), 3.50 (br s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.9, 150.9, 141.2, 116.3, 116.0, 66.4, 51.2. m/z calcd for C9H11NO3, 182.18; found, 182.081 [M]+.
6.2.14. Methyl 4-(4-nitrophenyl)butanoate (26a)
Compound 26a was obtained from 4-(4-nitrophenyl)butanoic acid (off-white solid, 5.01 g, 94%). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 9.1 Hz, 2H), 7.22 (d, J = 9.1 Hz, 2H), 3.53 (s, 3H), 2.63 (t, J = 7.6 Hz, 2H), 2.22 (t, J = 7.1 Hz, 2H), 1.85 (quint, J = 7.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 173.3, 149.4, 146.3, 129.2, 123.5, 51.4, 34.7, 32.9, 25.4.
6.2.15. Methyl 4-(4-aminophenyl)butanoate (26)
Compound 26 was obtained from compound 26a (pink solid, 4.34 g, 100%). 1H NMR (400 MHz, CDCl3) δ 6.93 (d, J = 7.0 Hz, 2H), 6.57 (d, J = 6.7 Hz, 2H), 3.68 (br s, 2H), 3.63 (s, 3H), 2.51 (t, J = 7.3 Hz, 2H), 2.30 (t, J = 7.3 Hz, 2H), 1.88 (quint, J = 7.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 173.9, 114.5, 130.8, 129.0, 115.0, 51.2, 34.0, 33.1, 26.6. HRMS calcd for C11H15NO2, 193.1103; found, 194.11743 [M+H]+.
Sulfonamides 27 and 28 were obtained in the same manner as compound 12.
6.2.16. Methyl 2-(4-(methylsulfonamido)phenoxy)acetate (27)
Compound 27 was obtained from compound 25 (orange solid, 15.0 g, 90%). 1H NMR (300 MHz, CDCl3) δ 7.22 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 4.65 (s, 2H), 3.83 (s, 3H), 2.96 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 169.5, 155.8, 130.6, 124.2, 115.6, 65.5, 52.5, 38.7. HRMS calcd for C10H13NO5S, 259.0514; found, 264.00073 [M+Li]+.
6.2.17. Methyl 4-(4-(methylsulfonamido)phenyl)butanoate (28)
Compound 28 was obtained from compound 26 (pink solid, 6.5 g, 107%). The crude material was used without further purification. 1H NMR (300 MHz, CDCl3) δ 7.43 (br s, 1H), 7.16 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 3.62 (s, 3H), 2.94 (s, 3H), 2.57 (t, J = 7.3 Hz, 2H), 2.29 (t, J = 7.3 Hz, 2H), 1.88 (quint, J = 7.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 174.1, 138.7, 134.9, 129.6, 121.5, 51.6, 39.3, 38.5, 33.3, 26.4.
General method for preparation of carboxylic acids 29–31, exemplified by 30.
6.2.18. 3-(4-(Methylsulfonamido)phenyl)propanoic acid (30)
The sulfonamide ester 12 (1.16 g, 4.5 mmol) was dissolved in methanol (50 mL). To this solution, 1.0 N NaOH (17.0 mL, 17.0 mmol, 3.8 equiv) was added. The mixture was stirred at room temperature for 12 h. The pH of the solution was adjusted to ~3 with a solution of aqueous HCl. The volume of methanol was reduced by rotary evaporation (40 mbar), upon which the product crashed out of solution. The yellow crystals were collected by filtration and dried in vacuo (0.900 g, 82%). 1H NMR (400 MHz, CD3OD) δ 7.21 (d, J = 8.6 Hz, 2H), 7.17 (d, J = 8.6 Hz, 2H), 2.91 (s, 3H), 2.89 (t, J = 7.6 Hz, 2H), 2.59 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 176.7, 139.0, 137.7, 130.5, 122.3, 39.1, 36.8, 31.4. m/z (APCI) calcd for C10H13NO4S, 243.06; found, 242.05 [M−H]+.
6.2.19. 2-(4-(Methylsulfonamido)phenoxy)acetic acid (29)
Compound 29 was obtained from compound 27 (pink crystals, 5.6 g, 97%). 1H NMR (300 MHz, CD3OD) δ 7.20 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 9.0 Hz, 2H), 4.65 (s, 2H), 2.88 (s, 3H). 13C NMR (75 MHz, CD3OD) δ 172.7, 157.3, 132.8, 125.0, 116.5, 66.2, 38.8. m/z (APCI) calcd for C9H11NO5S, 245.0358; found, 489.064 [M×2]+.
6.2.20. 4-(4-(Methylsulfonamido)phenyl)butanoic acid (31)
Compound 31 was obtained from compound 28 (off-white solid, 3.24 g, 53%). 1H NMR (300 MHz, DMSO-d6) δ 12.04 (br s, 1H), 9.58 (br s, 1H), 7.12 (mult, 4H), 2.91 (s, 3H), 2.51 (t, J = 7.3 Hz, 2H), 2.18 (t, J = 7.3 Hz, 2H), 1.74 (quint, J = 7.3 Hz, 2H). 13CNMR (75 MHz, DMSO-d6) δ 175.0, 138.0, 136.8, 129.8, 121.0, 34.4, 33.7, 28.3, 27.0. HRMS calcd for C11H15NO4S, 257.0722; found, 256.06500 [M+H]+.
General method for preparation of amines 33 and 34, exemplified by 33.
6.2.21. N1-(3,4-Dichlorophenyl)ethane-1,2-diamine (33)
Bromoethylamine hydrobromide salt (5.0 g, 25 mmol) was suspended in toluene (100 mL) and 3,4-dichloroaniline (32, 12g, 74 mmol, 3.0 equiv) was added. The brown solution was refluxed for 1 h. The resulting mixture was cooled to room temperature to give a brown precipitate which was filtered off and washed with toluene. The solid was then treated with 20% w/v NaOH (100 mL) and extracted with DCM (2×). The organics were extracted with an acetic acid/sodium acetate buffer (300 mL of 0.1 M pH 5.5). The resulting aqueous layer was then basified with 20% w/v NaOH (100 mL) and extracted with DCM (2×). The combined organics were collected and washed with water, dried over MgSO4, and concentrated in vacuo to give a brown oil. The crude material was purified using silica gel chromatography (100% DCM to 10% MeOH/DCM + 1% Et3N gradient) to give a brown residue (3.60 g, 72%). 1H NMR (400 MHz, CDCl3) δ 7.17 (d, J = 8.6 Hz, 1H), 6.69 (s, 1H), 6.46 (d, J = 8.6 Hz, 1H), 4.25 (br s, 1H), 3.13 (t, J = 5.1 Hz, 2H), 2.96 (t, J = 5.1 Hz, 2H), 1.41 (br s, 2H). 13C NMR (150 MHz, CDCl3) δ 148.1, 133.0, 131.8, 114.0, 113.0, 46.2, 40.9. HRMS calcd for C8H10Cl2N2, 204.0221; found, 205.02914 [M+H]+.
6.2.22. N1-(3,4-Dichlorophenyl)propane-1,3-diamine (34)
Compound 34 was obtained from compound 32 and bromopro-pylamine hydrobromide (brown residue, 45%). 1H NMR (400 MHz, CDCl3) δ 7.06 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 2.9 Hz, 1H), 6.32 (dd, J1 = 8.0 Hz, J2 = 2.9 Hz, 1H), 4.76–4.27 (br s, 1H), 3.01 (t, J = 6.7 Hz, 2H), 2.73 (t, J = 6.7 Hz, 2H), 2.25–1.84 (br s, 2H), 1.64 (quint, J = 6.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 148.0, 132.3, 130.3, 118.6, 113.2, 112.3, 41.9, 39.9, 31.7. HRMS calcd for C9H12Cl2N2, 218.0378; found, 219.04479 [M+H]+.
6.2.23. tert-Butyl 2-(3,4-dichlorophenoxy)ethylcarbamate (36)
To a solution of dry THF (40.0 mL), 3,4-dichlorophenol (35, 1.11 g, 6.82 mmol, 1.1 equiv), N-Boc-aminoethanol (0.962 mL, 6.20 mmol) and triphenyl phosphine (2.28 g, 8.68 mmol, 1.40 equiv) were added. Diisopropylazodicarboxylate (1.71 mL, 8.68 mmol, 1.4 equiv) was added dropwise over 5 min at 0 °C. The mixture was warmed to room temperature and stirred for 12 h. The resulting solution was washed with 1.0 N NaOH (20 mL), water (20 mL), and brine (20 mL). The organics were dried over MgSO4 and concentrated in vacuo to give a yellow oil. The crude material was purified using silica gel chromatography (1 EtOAc/1 hexanes) to give a colorless oil (1.30 g, 68%). 1H NMR (400 MHz, CDCl3,) δ 7.33 (d, J = 8.9 Hz, 1H), 7.00 (d, J = 2.9 Hz, 1H), 6.76 (dd, J1 = 8.9 Hz, J2 = 2.9 Hz, 1H), 4.96 (br s, 1H), 3.99 (t, J = 5.1 Hz, 2H), 3.55–3.50 (mult, 2H), 1.46 (s, 9H).
6.2.24. 2-(3,4-Dichlorophenoxy)ethanamine (37)
Carbamate 36 (1.30 g, 4.25 mmol) was dissolved in THF (30.0 mL) and cooled to 0 °C. To this solution trifluoroacetic acid (10.0 mL, 130 mol, 31 equiv) was added dropwise to give a brown solution, which was warmed to room temperature and stirred for 2 days. The mixture was basified with solid sodium bicarbonate and partitioned between water and ethyl acetate. The layers were separated and the organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a yellow residue. The crude material was purified using silica gel chromatography (10% MeOH/DCM) to give a yellow oil (0.826 g, 94%). 1H NMR (300 MHz, CDCl3) δ 7.28 (d, J = 9.0 Hz, 1H), 6.92 (d, J = 2.8 Hz, 1H), 6.67 (dd, J1 = 9.0 Hz, J2 = 2.8 Hz), 4.05 (t, J = 4.7 Hz, 2H), 3.52 (t, J = 4.3 Hz, 2H). 13CNMR (75 MHz, CDCl3) δ 156.6, 133.2, 125.5, 116.7, 114.4, 64.3, 39.5.
6.2.25. 2-(3,4-Dichlorophenylthio)ethanamine (40)
Thiophenol (38, 1.00 g, 5.58 mmol) and oxazolidinone (38, 0.486 g, 5.58 mmol, 1.0 equiv) were heated to 130 °C, and stirred for 2 h. After cooling to room temperature, the resulting green solid was taken up in 10% HCl and heated for several minutes. The mixture was filtered and the filtrate was made basic (pH 10) with solid NaOH and extracted with DCM. The combined extracts were washed with water and brine, dried over MgSO4, and concentrated in vacuo to give a colorless residue. The crude material was purified using silica gel chromatography (10% MeOH/DCM) to give a colorless residue (0.338 g, 27%). 1H NMR (600 MHz, CDCl3) δ 7.43 (d, J = 1.9 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.18 (dd, J1 = 8.6 Hz, J2 = 1.9 Hz, 1H), 3.02 (t, J = 6.7 Hz, 2H), 2.94 (t, J = 6.2 Hz, 2H), 1.59 (br s, 2H). HRMS calcd for C8H9Cl2NS, 220.9833; found, 221.99072 [M+H]+.
6.2.26. 3-(3,4-Dichlorophenyl)propanamide (42)
3-(3,4-Dichlorophenyl)propionic acid (41, 3.00 g, 14 mmol) was dissolved in benzene (30.0 mL) and treated with oxalyl choride (5.8 mL, 68 mmol, 5.0 equiv) and DMF (catalytic). The mixture was stirred for 12 h at room temperature. The resulting solution was concentrated in vacuo to give an opaque liquid and dried in vacuo overnight. 1H, 13C NMR confirms the presence of the acid chloride. The acid chloride was dissolved in ether (24.0 mL), and ammonia (0.5 M solution in 1,4-dioxane, 82 mL, 41 mmol, 3.0 equiv) was added dropwise to give a white precipitate. The mixture was stirred at room temperature for 2 h. The reaction was quenched with water and diluted with ether, and the layers were separated. The organics were washed with 2.0 N HCl, 10% NaHCO3 solution, and brine. They were then dried over MgSO4 and concentrated in vacuo to give a colorless oil which solidified upon trituration with DCM and hexanes (1.40 g, 47%). 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.3 Hz, 1H), 7.29 (d, J = 1.9 Hz, 1H), 7.04 (dd, J1 = 8.3 Hz, J2 = 1.9 Hz, 1H), 6.04 (br s, 1H), 5.61 (br s, 1H), 2.90 (t, J = 7.6 Hz, 2H), 2.49 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 174.3. 141.1, 132.5, 130.1, 128.1, 37.0, 30.4.
6.2.27. 3-(3,4-Dichlorophenyl)propan-1-amine (43)
Amide 42 (0.446 g, 2.0 mmol) was dissolved in THF (15.0 mL). After cooling to 0 °C, lithium aluminum hydride (2.0 M sol’n in THF, 3.1 mL, 6.1 mmol, 3.0 equiv) was added dropwise. The mixture was stirred at room temperature for 12 h. The mixture was diluted with EtOAc and water, filtered over a pad of Celite, and the resulting layers were separated. The organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a yellow residue (0.400 g, 96%). 1H NMR (400 MHz, CD3O) δ 7.40–7.36 (mult, 2H), 7.13 (dd, J1 = 8.9 Hz, J2 = 2.0 Hz, 1H), 2.68–2.61 (mult, 4H), 1.80–1.73 (mult, 2H). 13C NMR (100 MHz, CD3OD) δ 143.0, 131.9, 130.3, 130.2, 129.4, 128.2, 40.5, 33.5, 32.0. HRMS calcd for C9H11Cl2N, 203.0269; found, 204.03458 [M+H]+.
6.2.28. 2-Allylisoindoline-1,3-dione (45)
Potassium phthalimide (44, 2.59 g, 14 mmol, 1.4 equiv) was added to anhydrous DMF (100 mL). To this solution allyl bromide (0.86 mL, 10.0 mmol) was added dropwise. The reaction was heated to 50 °C and stirred for 12 h. The DMF was removed in vacuo and the remaining yellow liquid was dissolved in DCM. The solution was washed with water (2×) and brine (2×). The organics were dried over MgSO4 and concentrated in vacuo to give an off-white solid. The crude material was purified using silica gel chromatography (50% DCM/50% hexanes) to give a white solid (1.80 g, 96%). 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J1 = 5.7 Hz, J2 = 2.9 Hz, 2H), 7.73 (dd, J1 = 5.7 Hz, J2 = 2.9 Hz, 2H), 5.90 (mult, 1H), 5.28–5.29 (mult, 2H), 4.31 (d, J = 5.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 168.2, 134.2, 132.3, 131.7, 123.5, 118.0, 40.2.
6.2.29. 2-(3,4-Dichlorocinnamyl)isoindoline-1,3-dione (47)
N-Allylphthalimide 45 (0.660 g, 3.53 mmol, 1.25 equiv) and 3,4-dichloroiodobenzene (46, 0.770 g, 2.82 mmol) were dissolved in triethylamine (10.0 mL) and acetonitrile (10 mL). Palladium acetate (6 mg, 0.028 mmol, 0.01 equiv) was added. The yellow solution was heated at 100 °C in a thick-walled glass container for 20 h. The reaction was cooled to room temperature, upon which precipitation occurred to give a brown solid. The solid was filtered and washed with water. The solid was then dissolved in hot DMF to give an orange liquid which was filtered through Celite to remove Pd(0). The resulting hot DMF filtrate was diluted with an equal volume of water, upon which precipitation oc-curred. The yellow solid was filtered off and dried in vacuo (6.9 g, 85%) 1H NMR (400 MHz, DMSO-d6) δ 7.92–7.84 (mult, 4H), 7.73 (d, J = 1.9 Hz, 1H), 7.56 (d, J = 8.3 Hz, 1H), 7.44 (dd, J1 = 1.9 Hz, J1 = 8.3 Hz, 1H), 6.55 (d, J = 15.9 Hz, 1H), 6.50–6.43 (mult, 1H), 4.35 (d, J = 4.1 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.6, 137.1, 134.4, 131.7, 131.4, 130.6, 129.7, 128.7, 128.5, 128.2, 126.9, 126.3. HRMS calcd for C17H11Cl2NO2, 331.0167; found, 332.01368 [M+H]+.
6.2.30. (E)-3-(3,4-Dichlorophenyl)prop-2-en-1-amine (48)
Compound 47 (0.660 g, 1.99 mmol) was suspended in ethanol (7.5 mL) and hydrazine hydrate (0.1 mL, 2.0 mmol, 1.1 equiv) was added. The solution was refluxed (heating dissolved to give an orange solution) overnight to give a yellow precipitate. The suspension was cooled to room temperature, and the precipitate was filtered off and washed with ethanol. The yellow solid was then suspended in water, and 1.0 N NaOH was added until the solid mostly dissolved. DCM was added and the layers were separated. The organics were washed with water, dried over MgSO4, and concentrated in vacuo to give a yellow residue (0.221 g, 55%). 1HNMR (400 MHz, CDCl3) δ 7.42 (d, J = 2.2 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.17 (dd, J1 = 8.5 Hz, J2 = 2.1 Hz, 1H), 6.41 (d, J = 15.9 Hz, 1H), 6.31 (mult, J1 = 16.0 Hz, J2 = 5.1 Hz, 1H), 3.47 (d, J = 5.4 Hz, 2H), 1.23 (br s, 2H). 13C NMR (150 MHz, CDCl3) δ 137.6, 133.7, 130.6, 128.2, 127.2, 125.6, 44.3. m/z (APCI) calcd for C9H9Cl2N, 202.0; found, 202.1[ M]+.
General method for preparation of amides 49–60, exemplified by 49.
6.2.31. N-(2-(3,4-Dichlorophenylamino)ethyl)-3-(4-(methylsulfonamido)phenyl)propanamide (49)
Carboxylic acid 30 (0.700 g, 2.88 mmol) was dissolved in DMF (30.0 mL) and cooled to 0°C. To this solution, DMAP (0.352 g, 2.28 mmol, 1.1 equiv), and EDCI (0.552 g, 2.88 mmol, 1.0 equiv) were added to give a clear suspension. After stirring for 45 min, amine 33 (0.590 g, 2.88 mmol, 1.0 equiv) in THF (5.0 mL) was added dropwise. The mixture was warmed to room temperature and stirred for 12 h. The mixture was concentrated in vacuo, and the resulting residue was partitioned between 1.0 N HCl, and EtOAc. After extracting with EtOAc (2×), the combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude material was purified by taking the residue up in DCM and stirring. Immediately a white powder precipitated, which was collected by filtration (0.920 g, 74%). 1H NMR (400 MHz, CD3OD) δ 7.14–7.10 (mult, 5H), 6.72 (d, J = 2.9 Hz, 1H), 6.51 (dd, J1 = 8.9 Hz, J2 = 2.6 Hz, 1H), 3.27 (t, 2H), 3.10 (t, J = 6.4 Hz, 2H), 2.88 (s, 3H), 2.87 (t, J = 6.5 Hz, 2H), 2.46, (t, J = 6.4 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 175.8, 150.0, 138.7, 138.3, 131.7, 130.5, 122.3, 114.4, 113.6, 44.0, 39.8, 39.2, 39.0, 32.3. HRMS calcd for C18H21Cl2N3O3S, 429.0681; found, 431.08143 [M+H]+. Anal. (C18H21Cl2N3O3S) C, H, N.
6.2.32. N-(3,4-Dichlorocinnamyl)-3-(4-(methylsulfonamido)-phenyl)propanamide (50)
Compound 50 was obtained using carboxylic acid 30 and amine 48. The crude material was purified using silica gel chromatography (100% EtOAc) to give yellow crystals (1.05 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 8.11 (br t, J = 5.7 Hz, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.57 (d, J = 8.6 Hz, 2H), 7.38 (dd, J1 = 8.6 Hz, J2 = 1.9 Hz, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.11 (d, J = 8.6 Hz, 2H), 6.37–6.34 (mult, 2H), 3.83 (t, J = 5.1 Hz, 2H), 2.80 (t, J = 7.6 Hz, 2H), 2.92 (s, 3H), 2.41 (t, J = 8.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.2, 137.5, 137.0, 136.2, 131.4, 130.7, 130.1, 129.4, 129.1, 127.9, 127.3, 126.2, 120.2, 39.0, 36.9, 30.4. HRMS calcd for C19H20Cl2N2O3S, 426.0672; found, 427.06418 [M+H]+. Anal. (C19H20Cl2N2O3S) C, H, N.
6.2.33. N-(4-(3-(4-(3,4-Dichlorophenyl)piperazin-1-yl)-3-oxopropyl)phenyl)methanesulfonamide (51)
Compound 51 was obtained using carboxylic acid 30 and 1-(3,4-dichlorophenyl)piperazine. The crude material was purified using silica gel chromatography (100% EtOAc) to give an off-white foam (0.320 g, 28%). 1H NMR (600 MHz, CDCl3) δ 7.37 (s, 1H), 7.34 (d, J = 8.6 Hz, 1H), 7.15 (s, 4H), 6.88 (d, J = 2.4 Hz, 1H), 6.67 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 3.71 (t, J = 4.8 Hz, 2H), 3.51 (t, J = 4.8 Hz, 2H), 3.06 (t, J = 5.2 Hz, 2H), 3.01 (t, J = 5.2 Hz, 2H), 2.92 (t, J = 7.6 Hz, 2H), 2.91 (s, 3H), 2.62 (t, J = 7.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 170.9, 150.4, 138.5, 135.4, 133.1, 130.8, 129.9, 123.2, 121.6, 118.0, 116.0, 49.1, 49.0, 45.3, 41.5, 39.3, 34.9, 30.9. HRMS calcd for C20H23Cl2N3O3S, 455.0837; found, 456.09066 [M+H]+. HPLC (a) 99.5% (c) 99.6%.
6.2.34. N-(2-(3,4-Dichlorophenylamino)ethyl)-2-(4-(meth-ylsulfonamido)phenoxy)acetamide (52)
Compound 52 was obtained using carboxylic acid 29 and amine 33. The crude material was purified using silica gel chromatography (100% EtOAc) to give a white powder (0.970 g, 55%). 1H NMR (400 MHz, acetone-d6) δ 7.28 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 8.9 Hz, 2H), 6.79 (d, J = 2.9 Hz, 1H), 6.61 (dd, J1 = 8.8 Hz, J2 = 2.9 Hz, 1H), 4.47 (s, 2H), 3.78 (s, 1H), 3.49 (quint, J = 6.3 Hz, 2H), 3.28 (quint, J = 6.3 Hz, 2H), 2.89 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ 169.3, 156.3, 149.7, 132.9, 131.5, 129.0, 124.4, 118.5, 116.4, 113.9, 113.5, 68.4, 43.8, 38.9, 38.7. HRMS calcd for C17H19Cl2N3O4S, 431.0473; found: 432.05383 [M+H]+. Anal. (C17H19Cl2N3O4S) C, H, N.
6.2.35. N-(3,4-Dichlorocinnamyl)-2-(4-(methylsulfonamido)-phenoxy)acetamide (53)
Compound 53 was obtained using carboxylic acid 29 and amine 48. The crude material was purified using silica gel chromatogra-phy (2 EtOAc/1 hexanes) to give a white solid (0.247, 71%). 1H NMR (300 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.40 (br t, J = 5.7 Hz, 1H), 7.68 (d, J = 1.9 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.39 (dd, J1 = 8.2 Hz, J2 = 1.9 Hz, 1H), 7.17 (d, J = 9.0 Hz, 2H), 8.96 (d, J = 9.0 Hz, 2H), 6.41–6.39 (mult, 2H), 4.52 (s, 2H), 3.94–3.92 (mult, 2H), 2.89 (s, 3H). 13C NMR (75 MHz, DMSO-d6) d 167.6, 154.9, 137.5, 131.5, 131.4, 130.4, 129.7, 129.5, 127.9, 127.4, 126.2, 123.0, 115.4, 67.2, 39.0. HRMS calcd for C18H18Cl2N2O4S, 428.0364; found, 427.0296 [M−H]+. Anal. (C18H18Cl2N2O4S) C,H,N.
6.2.36. N-(3-(3,4-Dichlorophenyl)propyl)-2-(4-(methylsulfonamido)phenoxy)acetamide (54)
Compound 54 was obtained using carboxylic acid 29 and amine 43. The crude material was purified using silica gel chromatogra-phy (3 EtOAc/1 hexanes) to give a yellow oil (0.380 g, 43%). 1H NMR (400 MHz, CDCl3) δ 7.37 (br s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.26–7.23 (mult, 3H), 7.00 (dd, J1 = 8.1 Hz, J2 = 2.2 Hz, 1H), 6.89 (d, J = 8.8 Hz, 2H), 6.67 (br t, J = 5.71 Hz, 1H), 4.46 (s, 2H), 3.38 (q, J = 6.7 Hz, 2H), 2.95 (s, 3H), 2.60 (t, J = 7.3 Hz, 2H), 1.87 (quint, J = 7.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 168.4, 155.3, 141.6, 132.4, 131.2, 130.5, 130.4, 130.1, 128.0, 124.3, 115.8, 67.7, 39.1, 38.7, 34.4, 30.9. HRMS calcd for C18H20Cl2N2O4S, 430.0521; found, 431.05898 [M+H]+. HPLC (a) 99.7% (b) 98.6%
6.2.37. N-(2-(3,4-Dichlorophenoxy)ethyl)-2-(4-(methylsulfonamido)phenoxy)acetamide (55)
Compound 55 was obtained using carboxylic acid 29 and amine 37.The crude material was purified using silica gel chromatography (2 EtOAc/1 hexanes) to give a white foam (0.366 g, 41%). 1HNMR (300 MHz, CDCl3) δ 7.32 (s, 1H), 7.23 (d, J = 9.0 Hz, 2H), 7.05 (br t, J = 5.7 Hz, 1H), 6.96 (d, J = 2.8 Hz, 1H), 6.89 (d, J = 9.0 Hz, 2H), 6.73 (dd, J1 = 9.0 Hz, J2 = 2.8 Hz, 1H), 4.50 (s, 2H), 4.04 (t, J = 5.2 Hz, 2H), 3.76 (q, J = 5.5 Hz, 2H), 2.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 168.6, 157.5, 155.2, 133.0, 131.1, 130.9, 124.6, 124.2, 116.6, 115.8, 114.5, 67.7, 67.2, 39.1, 38.6. HRMS calcd for C H + 17 18Cl2N2O5S, 432.0313; found, 433.03824 [M+H]. Anal. (C18H18Cl2N2O4S) C, H, N.
6.2.38. N-(2-(3,4-Dichlorophenylthio)ethyl)-2-(4-(methylsulfonamido)phenoxy)acetamide (56)
Compound 56 was obtained using carboxylic acid 29 and amine 40. The crude material was purified using silica gel chromatography (3 EtOAc/1 hexanes) to give a white foam (0.389 g, 69%). 1H NMR (600 MHz, CDCl3) δ 7.44 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H), 7.29 (br s, 1H), 7.24 (d, J = 8.6 Hz, 2H), 7.19 (dd, J1 = 8.3 Hz, J2 = 2.4 Hz, 1H), 6.98 (br t, J = 5.7 Hz, 1H), 6.88 (d, J = 9.0 Hz), 4.45 (s, 2H), 3.58 (quart, J = 6.2 Hz, 2H), 3.10 (t, J = 6.7 Hz, 2H), 2.96 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 168.6, 155.3, 135.6, 133.2, 131.2, 131.0, 130.9, 130.8, 128.8, 124.4, 115.9, 67.7, 39.2, 39.1, 38.5, 33.3. HRMS calcd for C17H18Cl2N2O4S2, 448.0085; found, 449.01536 [M+H]+. Anal. (C17H18Cl2N2O4S2) C, H, N.
6.2.39. N-(4-(2-(4-(3,4-Dichlorophenyl)piperazin-1-yl)-2-oxoethoxy)phenyl)methanesulfonamide (57)
Compound 57 was obtained using carboxylic acid 29 and 1-(3,4-dichlorophenyl)piperazine. Upon addition of 1.0 N HCl a white powder precipitated out which was obtained by filtration and washing with hexanes (0.775 g, 85%). 1H NMR (600 MHz, DMSO-d6) δ 9.39 (s, 1H), 7.42 (d, J = 9.1 Hz, 1H), 7.17 (d, J = 2.3 Hz, 1H), 7.13 (d, J = 9.1 Hz, 2H), 6.96 (dd, J1 = 9.1 Hz, J2 = 2.3 Hz, 1H), 6.92 (d, J = 9.1 Hz, 2H), 4.84 (s, 2H), 3.27–3.18 (mult, 4H), 2.88 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.9, 155.3, 150.4, 131.5, 131.2, 130.5, 123.2, 120.0, 116.6, 115.6, 115.3, 59.8, 47.7, 47.4, 43.7, 40.7, 38.7. HRMS calcd for C19H21Cl2N3O4S, 457.0630; found, 458.07034 [M+H]+. Anal. (C19H21Cl2N3O4S −0.8H2O) C, H, N.
6.2.40. N-(3,4-Dichlorophenethyl)-4-(4-(methylsulfonamido)-phenyl)butanamide (58)
Compound 58 was obtained using carboxylic acid 31 and 3,4-dichlorophenylethylamine. The crude material was purified using silica gel chromatography (50% EtOAc/50% hexanes to 100% EtOAc gradient) to give a white foam which solidified in vacuo (0.571 g, 68%). 1H NMR (600 MHz, CDCl3) δ 7.57 (s, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.23 (d, J = 1.9 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.1 Hz, 2H), 6.99 (dd, J1 = 8.1 Hz, J2 = 1.9 Hz, 1H), 5.82 (br t, J = 6.2 Hz, 1H), 3.44 (quart, J = 6.2 Hz, 2H), 2.92 (s, 3H), 2.73 (t, J = 7.1 Hz, 2H), 2.52 (t, J = 7.1 Hz, 2H), 2.11 (t, J = 7.1 Hz, 2H), 1.84 (quint, J = 8.1 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 173.3, 139.4, 138.9, 135.2, 132.5, 130.9, 130.7, 130.6, 129.7, 128.5, 121.6, 40.4, 39.3, 35.9, 35.0, 34.6, 27.3. HRMS calcd for C19H22Cl2N2O3S, 428.0728; found, 429.08027 [M+H]+. Anal. (C19H22Cl2N2O3S) C, H, N.
6.2.41. N-(3-(3,4-Dichlorophenylamino)propyl)-2-(4-(methyl-sulfonamido)phenoxy)acetamide (59)
Compound 59 was obtained using carboxylic acid 29 and amine 34. The crude material was purified using silica gel chromatogra-phy (2 EtOAc/1 hexanes to 100% EtOAc gradient) to give a white foam. Trituration with MeOH give a white solid obtained by filtra-tion (0.177 g, 64%). 1H NMR (600 MHz, CD3OD) δ 7.22 (d, J = 9.1 Hz, 2H), 7.14 (d, J = 9.1 Hz, 1H), 6.97 (d, J = 9.1 Hz, 2H), 6.68 (d, J = 2.9 Hz, 1H), 6.48 (dd, J1 = 9.1 Hz, J2 = 2.9 Hz, 1H), 4.50 (s, 2H), 3.38 (mult, 2H), 3.05 (t, J = 6.7 Hz, 2H), 2.87 (s, 3H), 1.81 (quint, J = 6.7 Hz, 2H). 13C NMR (150 MHz, CD3OD) δ 171.3, 156.9, 150.3, 133.6, 133.3C, 125.0, 119.4, 116.8, 114.5, 113.7, 68.6, 42.0, 38.9, 38.0, 29.8. HRMS calcd for C18H21Cl2N3O4S, 445.0630; found, 446.07031 [M+H]+. Anal. (C18H21Cl2N3O4S) C, H, N.
6.2.42. N-(3-(3,4-Dichlorophenylamino)propyl)-3-(4-(methylsulfonamido)phenyl)propanamide (60)
Compound 60 was obtained using carboxylic acid 31 and amine 34. The crude material was purified using silica gel chromatography (2 EtOAc/1 hexanes to 100% EtOAc gradient) to give a white foam which solidified in vacuo (0.182 g, 40%). 1H NMR (300 MHz, CD3OD) δ 7.99 (br s, 1H), 7.21–7.14 (mult, 5H), 6.68 (d, J = 2.3 Hz, 1H), 6.48 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 3.23–3.19 (mult, 2H), 2.95 (t, J = 6.7 Hz, 2H), 2.89 (s, 3H), 2.47 (t, J = 7.6 Hz, 2H), 1.68 (quint, J = 6.7 Hz, 2H). 13C NMR (150 MHz, CD3OD) δ 174.0, 148.9, 137.5, 136.4, 132.3, 130.4, 129.2, 120.9, 118.0, 113.1, 112.3, 40.5, 37.8, 37.6, 36.7, 31.0, 28.5. HRMS calcd for C19H23Cl2N3O3S, 443.0837; found, 444.09059 [M+H]+. Anal. (C19H23Cl2N3O3S) C, H, N.
6.2.43. tert-Butyl N-2-(4-nitrophenoxy)ethylcarbamate (62)
4-Nitrophenol (61, 1.9 g, 14 mmol, 1.1 equiv), tert-butyl N-2-hydroxyethylcarbamate (2.0 mL, 13.0 mmol, 1.0 equiv) and triphenylphosphine (4.47 g, 18.0 mmol, 1.4 equiv) were dissolved in 60 mL of dry THF. To this mixture, DIAD (3.6 mL, 18.0 mmol, 1.4 equiv) was added dropwise at 0 °C. The reaction was allowed to slowly warm to room temperature and stirred for 16 h. The resulting mixture was concentrated in vacuo and the remaining yellow-orange residue was dissolved in EtOAc, treated with 1.0 N NaOH and washed with water and brine. The aqueous layer was extracted once more with EtOAc. The organic layers were combined, dried over MgSO4 and volatiles removed in vacuo. The remaining yellow residue was purified using silica gel chromatography in 2:1 EtOAc/hexanes to yield 62 (3.0 g, 82%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 9.3 Hz, 2H), 6.96 (d, J = 9.3 Hz, 2H), 4.95 (br s, 1H), 4.11 (t, J = 5.4 Hz, 2H), 3.56 (t, J = 5.4 Hz, 2H), 1.45 (s, 9H).
6.2.44. tert-Butyl N-2-(4-aminophenoxy)ethylcarbamate (63)
Nitro compound, 62, (3.0 g, 11.0 mmol, 1.0 equiv) was dissolved in 20 mL of MeOH. 5% palladium on carbon catalyst (0.6 g, 10 wt %) was added to the reaction mixture. The suspension was hydrogenolyzed using a balloon for 16 h. The reaction mixture was filtered over a pad of Celite and washed with DCM, EtOAc, and MeOH. The volatiles were removed in vacuo to yield 63 (2.7 g, 100%) as a brown-red oil. 1H NMR (300 MHz, CD3OD) δ 6.88 (mult, 4H), 3.90 (t, J = 5.7 Hz, 2H), 3.36 (t, J = 5.7 Hz, 2H), 1.41 (s, 9H).
6.2.45. tert-Butyl N-2-(4-(methylsulfonamido)phenoxy)ethylcarbamate (64)
Aniline 63, (2.5 g, 10 mmol, 1.0 equiv) was suspended in DCM (30 mL). Diisopropylethylamine (2 mL, 12 mmol, 1.2 equiv) was added to the reaction mixture and stirred for 15 min. The reaction mixture was cooled to 0 °C and methanesulfonyl chloride (0.9 mL, 11 mmol, 1.1 equiv) was added dropwise to the solution. The reaction was warmed to room temperature and stirred for 10 h. The reaction was quenched with water and extracted with DCM (3×). The organic layers were combined and washed with brine and water, dried over MgSO4, and concentrated in vacuo. The remaining brown residue was purified using silica gel chromatography (1:1 EtOAc/hexanes) to yield an off-white solid (0.5 g, 15%). 1H NMR (300 MHz, CD3OD) δ 7.17 (d, J = 6.9 Hz, 2H), 6.89 (d, J = 6.9 Hz, 2H), 3.96 (t, J = 5.4 Hz, 2H), 3.39 (t, J = 3.5 Hz, 2H), 2.85 (s, 3H), 1.42 (s, 9H). 13C NMR (75 MHz, CD3OD) δ 123.96, 115.07, 67.00, 41.73, 39.77, 37.44, 27.54. HRMS calcd for C14H22N2O5S, 330.1249; found, 331.13245 [M+H]+.
6.2.46. N-(4-(2-Aminoethoxy)phenyl)methanesulfonamide hydrochloride (65)
Sulfonamide 64 (0.5 g, 2 mmol, 1.0 equiv), was dissolved in DCM (10 mL). Trifluoroacetic acid (5 mL) was added to the solution and the reaction mixture was stirred for 3 h at room temperature. The reaction was quenched with water and extracted with DCM. The organics were washed with brine and then dried over MgSO4. Volatiles were removed in vacuo to yield 0.17 g of a brown oil. The free base was converted to the HCl salt by bubbling HCl (g) through a solution of the amine dissolved in EtOAc. The resulting precipitate was filtered to yield a purple solid (0.2 g, 49%). 1H NMR (300 MHz, CD3OD) δ 7.13 (d, J = 9 Hz, 2H), 6.91 (d, J = 9 Hz, 2H), 4.12 (t, J = 4.8 Hz, 2H), 3.28 (t, J = 3.6 Hz, 2H).
6.2.47. 2-Chloro-N-(3,4-dichlorophenyl)acetamide (66)
3,4-Dichloroaniline (32, 2.0 g, 12 mmol, 1.0 equiv) and triethylamine (6.8 mL, 49 mmol, 4 equiv) were suspended in DCM (50 mL). The mixture was cooled to 0 °C and chloroacetyl chloride (0.98 mL, 12 mmol, 1 equiv) was added dropwise. Immediate evolution of gas occured upon addition of the acid chloride as well as a notable color change from red-orange to dark brown. The reaction was warmed to room temperature and stirred for 2 h. The reaction mixture was washed with 1 M HCl. The aqueous layer was reextracted with DCM. The organics were combined and dried over MgSO4. The organics were removed and the solid was concentrated in vacuo to give an off-white solid (2.9 g, 99%). 1H NMR (300 MHz, CDCl3) δ 8.28 (br s, 1H), 7.81 (mult, 1H), 7.41 (mult, 2H), 4.21 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 130.8, 121.9, 119.4, 43.0.
6.2.48. N-(3,4-Dichlorophenyl)-2-(2-(4-(methylsulfonamido)-phenoxy)ethylamino) acetamide (67)
Compound 65 (0.26 g, 1 mmol, 1.9 equiv) was suspended in THF (20.0 mL). Triethylamine (0.2 mL, 1 mmol, 2 equiv) was added to the mixture and 66 (0.15 g, 0.6 mmol, 1 equiv) was added after 10 min. The reaction mixture was refluxed and stirred for 16 h. The reaction mixture was basified with NaHCO3 and extracted with DCM (2×). The organic layers were washed with water and brine, and then dried over MgSO4, and concentrated in vacuo. The resulting free base was converted to the HCl salt by bubbling HCl (g) through a solution of the substrate in EtOAc. The volatiles were removed in vacuo to yield an orange solid (0.2 g, 76%). 1H NMR (300 MHz, CD3OD) δ 7.85 (d, J = 2.4 Hz, 1H), 7.43 (mult, 1H), 7.38 (mult, 1H), 7.11 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 4.40 (mult, 2H), 3.93 (s, 2H), 3.20 (t, J = 3.6 Hz, 2H) 2.87 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 165.8, 155.9, 137.5, 132.1, 131,8, 130.5, 127.2, 123.8, 123.3, 121.3, 115.2, 67.4, 54.7, 39.1, 37.6. HRMS calcd for C17H19Cl2N3O4S, 431.0473; found, 432.05423 [M+H]+. Anal. (C17H19Cl2N3O4S) C, H, N.
General method for preparation of amines 68 and 69, exemplified by 68.
6.2.49. N-(4-(3-(2-(3,4-Dichlorophenylamino)ethylamino)-propyl)phenyl)methanesulfonamide HCl (68)
Amide 49 (0.500 g, 1.2 mmol) was dissolved in THF (30.0 mL). After cooling to 0 °C, a solution of lithium aluminum hydride (2.0 M solution in THF, 2.3 mL, 4.6 mmol, 4.0 equiv) was added dropwise. After stirring for 10 min at 0 °C, the ice bath was removed and the reaction mixed was warmed to room temperature and stirred for 12 h. The mixture was diluted with DCM and water to give an emulsion. Rochelle’s salt (satd) was added and the mixture stirred for 20 min before filtering over a pad of Celite. The resulting liquid was separated, and the organics were washed with brine, dried over MgSO4, and concentrated in vacuo to give a white foam. The free base was converted to the HCl salt by bubbling HCl (g) through a solution of substrate dissolved in ethanol. The white powder precipitated out and was collected by filtration (0.358 g, 74%). 1H NMR (300 MHz, CDCl3) δ 7.20–7.11 (mult, 5H), 6.69 (d, J = 2.8 Hz, 1H), 6.46 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 4.36 (br s, 1H), 3.16 (mult, 2H), 3.00 (s, 3H), 2.88 (t, J = 6.2 Hz, 2H), 2.66 (t, J = 7.1 Hz, 4H), 1.86–1.79 (mult, 3H). 13C NMR (75 MHz, CDCl3) δ 148.1, 139.4, 134.8, 132.8, 130.7, 129.7, 121.6, 119.7, 113.9, 112.9, 49.0, 48.2, 43.2, 39.3, 32.9, 31.5. HRMS calcd for C18H22Cl2N3O2S, 429.0681; found, 431.08143 [M+H]+. Anal. (C18H23Cl2N3O2S·0.45 HCl) C, H, N.
6.2.50. N-(4-(2-(2-(3,4-Dichlorophenylamino)ethylamino)-ethoxy)phenyl)methanesulfonamide HCl (69)
Compound 69 was obtained from amide 52 (0.486 g, 50%). 1H NMR (300 MHz, CDCl3) δ 7.16 (dd, J1 = 8.9 Hz, J2 = 2.4 Hz, 3H), 6.81 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 2.8 Hz, 1H), 6.46 (dd, J1 = 8.6 Hz, J2 = 2.6 Hz, 1H), 4.06 (t, J = 5.0 Hz, 2H), 3.23 (t, J = 5.2 Hz, 2H), 3.05 (t, J = 5.0 Hz, 2H), 2.98 (t, J = 5.2 Hz, 2H), 2.93 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 157.1, 147.9, 132.9, 130.7, 129.7, 124.8, 119.8, 115.5, 113.9, 112.9, 67.2, 48.3, 48.0, 43.1, 39.1. HRMS calcd for C17H21Cl2N3O3S, 417.0681; found, 418.07543 [M+H]+. Anal. (C17H21Cl2N3O3S·HCl) C, H, N.
General method for preparation of imidazolinones 70 and 71, exemplified by 70.
6.2.51. N-(4-(3-(3-(3,4-Dichlorophenyl)-2-oxoimidazolidin-1-yl)propyl)phenyl)methanesulfonamide (70)
Amine 68 (0.113 g, 0.27 mmol) was dissolved in THF (10.0 mL). To this solution 1,1-carbonyldimidazole (0.048 g, 0.30 mmol, 1.1 equiv) was added, and the mixture was stirred at room temperature for 12 h. The solution was concentrated in vacuo and the residue was taken up in ethyl acetate, washed with brine, dried over Na2SO4, and concentrated in vacuo to give a colorless oil. The crude material was purified using silica gel chromatography (100% EtOAc) to give a white foam (0.070 g, 58%). 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H), 7.16 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.6 Hz, 2H), 7.02 (s, 1H), 6.64 (d, J = 2.9 Hz, 1H), 6.41 (dd, J1 = 8.5 Hz, J2 = 2.9 Hz, 1H), 3.64 (t, J = 6.0 Hz, 2H), 3.40–3.36 (mult, 4H), 2.97 (s, 3H), 2.57 (t, J = 7.3 Hz, 2H), 1.26 (t, J = 7.3 Hz, 2H). 13C (75 MHz, CDCl3) δ 152.6, 147.1, 137.3, 136.8, 135.6, 130.9, 129.5, 121.6, 118.0, 113.6, 112.5. HRMS calcd for C19H21Cl2N3O3S, 441.0681; found, 442.07527 [M+H]+. HPLC (a) 99.1% (b) 99.6%.
6.2.52. N-(4-(2-(3-(3,4-Dichlorophenyl)-2-oxoimidazolidin-1-yl)ethoxy)phenyl)methanesulfonamide (71)
Compound 71 was obtained using amine 69 (white foam, 0.100 g, 56%). 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 2.5 Hz, 1H), 7.30 (dd, J1 = 8.9 Hz, J2 = 2.5 Hz, 1H), 7.25 (d, J = 8.9 Hz, 1H), 7.12 (d, J = 8.9 Hz, 2H), 6.77 (d, J = 8.9 Hz, 2H), 4.05–4.02 (mult, 2H), 3.70–3.59 (mult, 6H), 2.84 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 157.4, 156.8, 140.0, 132.6, 130.4, 130.0, 125.5, 124.6, 118.9, 116.6, 115.4, 67.4, 38.9. HRMS calcd for C18H19Cl2N3O4S, 444.0473; found, 444.05451 [M]+. HPLC (a) 98.3% (b) 97.6%.
Acknowledgements
The authors are grateful to Drs. S. Nakanishi, P. Seeburg, and S. F. Heinemann for cDNAs encoding the glutamate receptor subunits. We thank Kimberly Vellano for excellent technical assistance. This work was supported in part by the NINDS (NS036654, ST; NS036604, RD).
Footnotes
Several of the authors (C.A.M., Y.A.T., L.J.W., S.J.M., R.D., S.F.T., D.C.L.) are inventors of Emory University owned patent-pending technology associated with these compounds, or have an equity position in companies actively seeking to license these compounds (D.C.L., S.F.T., R.D.).
References and notes
- 1.Dingledine R, Borges K, Bowie D, Traynelis SF. Pharmacol. Rev. 1999;51:7. [PubMed] [Google Scholar]
- 2.Mayer ML, Armstrong N. Annu. Rev. Physiol. 2004;66:161. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- 3.Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Crit. Rev. Neurobiol. 2004;16:187. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 4.Wang CX, Shuaib A. Curr. Drug Targets CNS Neurol. Disord. 2005;4:143. doi: 10.2174/1568007053544183. [DOI] [PubMed] [Google Scholar]
- 5.Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. Ann. Neurol. 1988;24:543. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- 6.Dirnagl U, Iadecola C, Moskowitz MA. Trends Neurosci. 1999;22:391. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 7.Whetsell WO., Jr. J. Neuropathol. Exp. Neurol. 1996;55:1. doi: 10.1097/00005072-199601000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Lipton SA. Curr. Opin. Neurol. Neurosurg. 1993;6:588. [PubMed] [Google Scholar]
- 9.Henneberry RC. Neurobiol. Aging. 1989;10:611. doi: 10.1016/0197-4580(89)90149-8. [DOI] [PubMed] [Google Scholar]
- 10.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Nat. Neurosci. 2005;8:1051. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 11.Beal MF. Ann. Neurol. 1992;31:119. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 12.Olney JW, Wozniak DF, Farber NB. Arch. Neurol. 1997;54:1234. doi: 10.1001/archneur.1997.00550220042012. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Fan M, Icton CD, Chen N, Leavitt BR, Hayden MR, Murphy TH, Raymond LA. Neurobiol. Aging. 2003;24:1113. doi: 10.1016/j.neurobiolaging.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, Bird ED. J. Neurol. Sci. 1992;108:80. doi: 10.1016/0022-510x(92)90191-m. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez A. M. Estrada, Mejia-Toiber J, Massieu L. Arch. Med. Res. 2008;39:265. doi: 10.1016/j.arcmed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Fan MM, Raymond LA. Prog. Neurobiol. 2007;81:272. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Loftis JM, Janowsky A. Pharmacol. Ther. 2003;97:55. doi: 10.1016/s0163-7258(02)00302-9. [DOI] [PubMed] [Google Scholar]
- 18.Dunah AW, Wang Y, Yasuda RP, Kameyama K, Huganir RL, Wolfe BB, Standaert DG. Mol. Pharmacol. 2000;57:342. [PubMed] [Google Scholar]
- 19.Hallett PJ, Standaert DG. Pharmacol. Ther. 2004;102:155. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. J. Clin. Psychopharmacol. 2008;28:631. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- 21.Dickenson AH, Chapman V, Green GM. Gen. Pharmacol. 1997;28:633. doi: 10.1016/s0306-3623(96)00359-x. [DOI] [PubMed] [Google Scholar]
- 22.Dickenson AH, Sullivan AF. Neuropharmacology. 1987;26:1235. doi: 10.1016/0028-3908(87)90275-9. [DOI] [PubMed] [Google Scholar]
- 23.Sandkuhler J, Liu X. Eur. J. Neurosci. 1998;10:2476. doi: 10.1046/j.1460-9568.1998.00278.x. [DOI] [PubMed] [Google Scholar]
- 24.Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Neuropharmacology. 1999;38:611. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 25.Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7104. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodward RM, Huettner JE, Guastella J, Keana JF, Weber E. Mol. Pharmacol. 1995;47:568. [PubMed] [Google Scholar]
- 27.Jarvis MF, Murphy DE, Williams M, Gerhardt SC, Boast CA. Synapse. 1988;2:577. doi: 10.1002/syn.890020603. [DOI] [PubMed] [Google Scholar]
- 28.Lehmann J, Hutchison AJ, McPherson SE, Mondadori C, Schmutz M, Sinton CM, Tsai C, Murphy DE, Steel DJ, Williams M, et al. J. Pharmacol. Exp. Ther. 1988;246:65. [PubMed] [Google Scholar]
- 29.Perin-Dureau F, Rachline J, Neyton J, Paoletti P. J. Neurosci. 2002;22:5955. doi: 10.1523/JNEUROSCI.22-14-05955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallagher MJ, Huang H, Pritchett DB, Lynch DR. J. Biol. Chem. 1996;271:9603. doi: 10.1074/jbc.271.16.9603. [DOI] [PubMed] [Google Scholar]
- 31.McCauley JA, Theberge CR, Romano JJ, Billings SB, Anderson KD, Claremon DA, Freidinger RM, Bednar RA, Mosser SD, Gaul SL, Connolly TM, Condra CL, Xia M, Cunningham ME, Bednar B, Stump GL, Lynch JJ, Macaulay A, Wafford KA, Koblan KS, Liverton NJ. J. Med. Chem. 2004;47:2089. doi: 10.1021/jm030483s. [DOI] [PubMed] [Google Scholar]
- 32.Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, Decosta DL, Ducat MF, Dumont ML, Fox CB, Mena EE, Menniti FS, Nielsen J, Pagnozzi MJ, Richter KEG, Ronau RT, Shalaby IA, Stemple JZ, White WF. J. Med. Chem. 1995;38:3138. doi: 10.1021/jm00016a017. [DOI] [PubMed] [Google Scholar]
- 33.Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. J. Pharmacol. Exp. Ther. 1997;283:1285. [PubMed] [Google Scholar]
- 34.Barta-Szalai G, Borza I, Bozo E, Kiss C, Agai B, Proszenyak A, Keseru GM, Gere A, Kolok S, Galgoczy K, Horvath C, Farkas S, Domany G. Bioorg. Med. Chem. Lett. 2004;14:3953. doi: 10.1016/j.bmcl.2004.05.053. [DOI] [PubMed] [Google Scholar]
- 35.Borza I, Kolok S, Gere A, Agai-Csongor E, Agai B, Tarkanyi G, Horvath C, Barta-Szalai G, Bozo E, Kiss C, Bielik A, Nagy J, Farkas S, Domany G. Bioorg. Med. Chem. Lett. 2003;13:3859. doi: 10.1016/s0960-894x(03)00708-x. [DOI] [PubMed] [Google Scholar]
- 36.Curtis NR, Diggle HJ, Kulagowski JJ, London C, Grimwood S, Hutson PH, Murray F, Richards P, Macaulay A, Wafford KA. Bioorg. Med. Chem. Lett. 2003;13:693. doi: 10.1016/s0960-894x(02)01060-0. [DOI] [PubMed] [Google Scholar]
- 37.Gladstone DJ, Black SE, Hakim AM. Stroke. 2002;33:2123. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- 38.Lipton SA. J. Alzheimers Dis. 2004;6:S61. doi: 10.3233/jad-2004-6s610. [DOI] [PubMed] [Google Scholar]
- 39.Robinson DM, Keating GM. Drugs. 2006;66:1515. doi: 10.2165/00003495-200666110-00015. [DOI] [PubMed] [Google Scholar]
- 40.Gogas KR. Curr. Opin. Pharmacol. 2006;6:68. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Chazot PL. Curr. Med. Chem. 2004;11:389. doi: 10.2174/0929867043456061. [DOI] [PubMed] [Google Scholar]
- 42.Waxman EA, Lynch DR. Neuroscientist. 2005;11:37. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- 43.Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M. Nat. Neurosci. 2001;4:164. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- 44.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Neuron. 1994;12:529. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 45.Standaert DG, Testa CM, Young AB, Penney JB., Jr. J. Comp. Neurol. 1994;343:1. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- 46.Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. J. Comp. Neurol. 1994;347:150. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 47.Chenard BL, Menniti FS. Curr. Pharm. Des. 1999;5:381. [PubMed] [Google Scholar]
- 48.Nikam SS, Meltzer LT. Curr. Pharm. Des. 2002;8:845. doi: 10.2174/1381612024607072. [DOI] [PubMed] [Google Scholar]
- 49.Borza I, Domany G. Curr. Top. Med. Chem. 2006;6:687. doi: 10.2174/156802606776894456. [DOI] [PubMed] [Google Scholar]
- 50.Tahirovic YA, Geballe M, Gruszecka-Kowalik E, Myers SJ, Lyuboslavsky P, Le P, French A, Irier H, Choi WB, Easterling K, Yuan H, Wilson LJ, Kotloski R, McNamara JO, Dingledine R, Liotta DC, Traynelis SF, Snyder JP. J. Med. Chem. 2008;51:5506. doi: 10.1021/jm8002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finlayson K, Witchel HJ, McCulloch J, Sharkey J. Eur. J. Pharmacol. 2004;500:129. doi: 10.1016/j.ejphar.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 52.Jamieson C, Moir EM, Rankovic Z, Wishart G. J. Med. Chem. 2006;49:5029. doi: 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- 53.Prata JV, Clemente DS, Prabhakar S, Lobo AM, Murato I, Branco PS. J. Chem. Soc., Perkin Trans. 1. 2002:513. [Google Scholar]
- 54.Perillo I, Caterina MC, López J, Salerno A. Synthesis. 2004:851. [Google Scholar]
- 55.Miller JF, Brieger M, Furfine ES, Hazen RJ, Kaldor I, Reynolds D, Sherrill RG, Spaltenstein A. Bioorg. Med. Chem. Lett. 2005;15:3496. doi: 10.1016/j.bmcl.2005.05.129. [DOI] [PubMed] [Google Scholar]
- 56.Poindexter GS, Owens DA, Dolan PL, Woo E. J. Org. Chem. 1992;57:6257. [Google Scholar]
- 57.Kao LC, Stakem FG, Patel BA, Heck RF. J. Org. Chem. 1982;47:1267. [Google Scholar]
- 58.Gordon MS. J. Am. Chem. Soc. 1969;91:3122. [Google Scholar]
- 59.Williams K. Mol. Pharmacol. 1993;44:851. [PubMed] [Google Scholar]
- 60.Ilyin VI, Whittemore ER, Guastella J, Weber E, Woodward RM. Mol. Pharmacol. 1996;50:1541. [PubMed] [Google Scholar]
- 61.Kew JN, Trube G, Kemp JA. J. Physiol. 1996;497:761. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kew JN, Trube G, Kemp JA. Br. J. Pharmacol. 1998;123:463. doi: 10.1038/sj.bjp.0701634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallagher MJ, Huang H, Lynch DR. J. Neurochem. 1998;70:2120. doi: 10.1046/j.1471-4159.1998.70052120.x. [DOI] [PubMed] [Google Scholar]
- 64.Wong E, Ng FM, Yu CY, Lim P, Lim LH, Traynelis SF, Low CM. Protein Sci. 2005;14:2275. doi: 10.1110/ps.051509905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marinelli L, Cosconati S, Steinbrecher T, Limongelli V, Bertamino A, Novellino E, Case DA. ChemMedChem. 2007;2:1498. doi: 10.1002/cmdc.200700091. [DOI] [PubMed] [Google Scholar]
- 66.Mony L, Krzaczkowski L, Leonetti M, Le Goff A, Alarcon K, Neyton J, Bertrand HO, Acher F, Paoletti P. Mol. Pharmacol. 2009;75:60. doi: 10.1124/mol.108.050971. [DOI] [PubMed] [Google Scholar]
- 67.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. J. Neurosci. 1998;18:6163. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]