

Abstract
Lecithin:cholesterol acyltransferase (LCAT) is an enzyme that first hydrolyzes the sn-2 position of phospholipids, preferentially a diacylphosphocholine, and then transfers the fatty acid to cholesterol to yield a cholesteryl ester. HDL ApoA-I is the principal catalytic activator for LCAT. Activity of LCAT on nascent or lipid-poor HDL particles composed of phospholipid, cholesterol and ApoA-I allows the maturation of HDL particles into lipid-rich spherical particles that contain a core of cholesteryl ester surrounded by phospholipid and ApoA-I on the surface. This article reviews the recent progress in elucidating structural aspects of the interaction between LCAT and ApoA-I. In the last decade, there has been considerable progress in understanding the structure of ApoA-I and the central helices 5, 6, and 7 that are known to activate LCAT. However, much less information has been forthcoming describing the 3D structure and conformation of LCAT required to catalyze two separate reactions within a single monomeric peptide.
Keywords: ApoA-I, cholesterol, cholesteryl ester, HDL, lecithin:cholesterol acyltransferase, phospholipid, protein structure, reverse cholesterol transport
Lecithin:cholesterol acyltransferase & the formation of plasma cholesteryl esters
It has been almost 8 years since reviews covering the activation of lecithin:cholesterol acyltransferase (LCAT), EC 2.3.1.43, also called phosphatidylcholine-sterol acyltransferase, by HDL ApoA-I were published [1,2]. While reviews covering HDL ApoA-I structure continue to be published yearly [3–7], studies covering LCAT activation via HDL ApoA-I helices 5, 6 and 7 have only recently gained the attention of a number of investigators. Therefore, our emphasis will be to review the recent advances in both LCAT structure and its interaction with the central helices of ApoA-I.
Lecithin:cholesterol acyltransferase synthesizes HDL cholesteryl esters by transferring a phospholipid-derived fatty acid to cholesterol when bound to ApoA-I [8]. Unique among enzymes, LCAT is responsible for catalyzing two distinct steps in the formation of plasma-derived cholesteryl esters. First, LCAT hydrolyzes an sn-2 fatty acid from phosphatidylcholine to form an acyl enzyme intermediate, while in the second step the fatty acid is transferred to cholesterol to form a cholesteryl ester. Functionally, LCAT is the major source of plasma-derived cholesteryl esters and is responsible for transforming nascent or lipid-poor HDL into spherical HDL containing a core of cholesteryl ester with an exterior composed of ApoA-I and phospholipid. The second step of the reaction has been shown to be reversible and may play a role in regulating cholesterol ester production [9], while sphingomyleins are known to inhibit the overall process [10–12]. ApoA-I, the principal protein component of HDL, is a potent activator of LCAT, and if not present little to no spherical HDL is formed [1,13]. Therefore, LCAT and ApoA-I appear to be essential for maintaining whole-body cholesterol homeostasis and essential components of reverse cholesterol transport, a process that is critical for removing cholesterol from the body [14].
Structure of LCAT
The LCAT enzyme is a 416 amino acid protein with a molecular mass of approximately 47 kDa. LCAT undergoes post-translational glycosylation adding four N-linked and two O-linked complex glycans [15,16], giving a final molecular weight of approximately 58 kDa [17]. The specific role of the glycan chains has not been identified, but they are known to be important for LCAT activity [18]. Several mutagenesis studies have either reported inhibition, no effect or enhancement of LCAT reactivity, compared with wild-type LCAT, after replacing Asn residues within LCAT with amino acids that are not recognized for glycosylation [19–22]. Mutants without Asn residues at position 84 or 384 were unstable and lost catalytic activity [17], suggesting that glycosylation was essential for conformational stability of LCAT.
Assuming that LCAT belongs to the α/β hydrolase fold family, Peelman et al. proposed a structure using threading methods that aligned the sequence of LCAT with solved 3D structures [23]. The final structure, based on fitting half of the LCAT sequence, suggested that it has seven β-strands connected together with loops and four α-helices. Among other defined structural elements in LCAT are two sets of disulfide bridges between Cys50–Cys74 and Cys313–Cys356. The Cys50–Cys74 disulfide was reported to be essential for the binding of LCAT to lipoprotein surfaces [24] and appears to be located in the putative ‘lid’ region that may cover the active site [25]. Peelman reported that deletion of amino acids at positions 56 to 68 gave a mutant LCAT that was not active with any substrate [25].
The first five N-terminal amino acids are highly conserved and Vickaryous et al. demonstrated that deletion mutations of these hydrophobic amino acids had a dramatic effect on LCAT activity [26]. Their scheme involved preparing mutant LCAT molecules lacking the first amino acid, then the first two amino acids, and so on, until they had deleted the first five amino acids. Deletion of the first amino acid through the first five N-terminal amino acids caused an almost complete loss of ApoA-I cofactor-dependent LCAT-catalyzed phospholipase activity and cholesterol esterification, or α-LCAT activity. More extensive N-terminal deletion caused an almost complete loss of phospholipase activity. Mutants lacking the first N-terminal amino acid could still esterify LDL-C (β-LCAT activity), but all other deletions resulted in a significant loss in all cholesterol esterification α- and β-LCAT activity. N-terminal deletions of the first and the first and second N-terminal amino acids did not affect binding of LCAT to recombinant HDL (rHDL) [26], similar to reports for other LCAT mutants [27]. The loss of esterification activity suggests that these deletions cause conformational change(s) that reduce or eliminate the access of the phospholipid substrate to the catalytic site [26], but not the affinity of LCAT for rHDL. Peelman et al. demonstrated that the N-terminal conserved basic residues, Arg80, Arg147 and Asp145, if altered, would reduce LCAT activity and suggested that the mutations prevented intra-molecular salt-bridge formation, which is essential for LCAT activity [28]. Another substitution in this region, the mutant Thr123 to Ile was also reported to be inactive with HDL [29,30]. Murray et al. found that amino acids at positions 121 to 136 of LCAT were highly conserved among six different species and that LCAT activity is sensitive to mutations in this region [31]. They explored this observation by measuring antibody binding to this region when LCAT was bound to a hydrophobic surface or to plasma HDL coated onto microtiter plates. They found that the amino acid region at position 121 to 136 is unavailable for binding a polyclonal antibody specific for this region [31] when LCAT is bound to lipid. Based on predictions that the amino acid region at position 154–171 of LCAT could form an amphiphthic α-helix, Peelman et al. demonstrated that the synthetic peptide had a strong affinity for phospholipids [32]. Increasing the hydrophobicity increased lipid affinity, while increasing the hydrophilicity decreased lipid affinity. These results support their hypothesis that the 154–171 region participates in the interaction of LCAT with lipid surfaces.
Mechanism of LCAT catalysis
Investigations of LCAT’s active site have revealed only one nucleophilic amino acid, Ser181 [33,34], involved in catalysis, through the systematic mutation of LCAT serines. Further analysis has suggested that the catalytic site is composed of a catalytic triad [35,36], containing Asp345, His377 and Ser181, respectively [37]. However, the some-what poor homology with other serine lipases and esterases has hindered more detailed predictions of LCAT’s structure. Nevertheless, assuming that LCAT did belong to the α/β hydrolase fold family, Peelman et al. proposed a structure using threading methods that aligned the sequence of LCAT with solved 3D structures [23]. These studies also identified Phe103 and Leu182 as putative oxyanion hole residues [23]. Subsequent studies identified Trp61, believed to be part of the putative lid region, as essential for catalytic activity [25]. Using the proposed model for LCAT, Vanloo et al. showed that three natural mutations, Asn131 to Asp, Thr123 to Ile, and Asn391 to Ser, are located within 12 Å of one another. Natural and engineered mutations at Thr123 substantially reduced LCAT activity with rHDL. Other mutations caused a moderate reduction in LCAT activities as an acyltransferase and a phospholipase or PLA2 [37]. Replacement of either Tyr120 or Phe382 with Ala resulted in a substantial reduction of rHDL-dependent acyltransferase activity. The model depicted by Peelman suggests that these two residues at positions 120 and 382 may lie close to one another [37].
LCAT is required for HDL maturation
ApoA-I, a 28 kDa protein, is synthesized by the liver and small intestine and plays a key role in the formation, metabolism and catabolism of HDL [38]. The plasma concentration of HDL is highly correlated with protection against the development of coronary artery disease, even in patients with very low LDL-C levels [39]. Lipidation of ApoA-I by the ATP-binding cassette transporter A1 (ABCA1) is essential for the formation of plasma HDL particles [40–42]. Lipid-free or lipid-poor ApoA-I acquire small amounts of phospholipid and cholesterol from membrane-bound ABCA1, generating several classes of nascent HDL particles [43] carrying one or more molecules of ApoA-I [44–46].
Maturation of HDL requires a second lipidation step, the synthesis of HDL cholesteryl esters catalyzed by LCAT [47], which provides a hydrophobic core by transforming lipidated, ABCA1-derived HDLs into spherical lipid-rich HDLs. In humans, if either ABCA1 or LCAT are inactive, the plasma concentrations of HDL ApoA-I are low because lipid-poor HDL is rapidly removed from the circulation [13,48]. These two ApoA-I lipidation steps are crucial to the formation of mature plasma HDL and whole-body cholesterol homeostasis [49–51]. Based on molecular models, an illustration showing the activation of LCAT [52] is shown in Figure 1, both before its docking (Figure 1A), and after docking (Figure 1B), onto a 96 Å diameter rHDL particle [53]. LCAT is shown in yellow. The rHDL is composed of 150 molecules of palmitoyloleoylphosphatidylcholine (POPC) and two molecules of ApoA-I. The conformation of LCAT depicted in Figure 1 was derived from a model containing amino acids at position 9 through position 420 of the mature protein [52]. Figure 1B illustrates that after LCAT docks or binds to ApoA-I helix 6 (purple), the LCAT domain corresponding to amino acids at positions 121–136 (blue) are shielded and are no longer accessible, as suggested by Murray et al., owing to their interaction with the surface of the rHDL [31]. LCAT belongs to the α/β hydrolase fold family and there is a ‘gate or lid’ that may be involved in controlling substrate specificity. For neutral lipases, Dolinsky et al. suggest that in an aqueous environment the lid covers the active site [54]. When the enzyme contacts a hydrophobic surface, such as a lipid, the lid opens making the catalytic site accessible to the lipid substrate. X-ray crystal analysis of substrates bound to bacterial lipases, such as Pseudomonas aeruginosa lipase that is in the same α/β hydrolase fold family, show that this ‘gate’ must move aside for the enzyme to bind and then react with lipid [55]. Jonas proposed a simple hypothetical model for the actions of LCAT on rHDL in which LCAT first interacts with the lipid bilayer and subsequently interacts with ApoA-I to activate its catalytic function [56]. Consistent with Jonas’s hypothetical model, Figure 1 shows the lipid-binding region, indicated by the arrow, interacting with phospholipid while part of LCAT interacts with ApoA-I helix 6.
Figure 1. Lecithin:cholesterol acyltransferase (yellow) and recombinant HDL [53] composed of 150 molecules of palmitoyloleoylphosphatidyl choline and two molecules of ApoA-I (green, orange and purple).
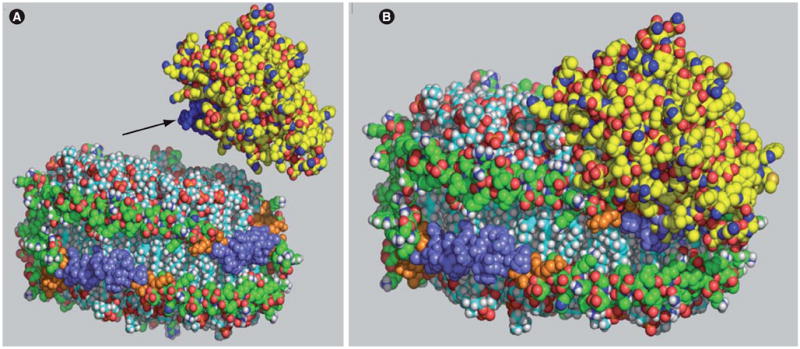
(A) Before docking and (B) after docking with ApoA-I helix 6 (purple). For clarity, those amino acids of ApoA-I that were reported to participate in lecithin:cholesterol acyltransferase (LCAT) activation, positions 140–178, are shown in orange. Helix 6 residues in the center of this region, amino acids at position 143–164, are shown in purple, while all other ApoA-I amino acids are shown in green. Murray et al. suggested that the LCAT region corresponding to amino acids at position 121 to 136, highlighted in blue (A), was shielded when LCAT binds lipid [31]. An arrow points to this region. The conformation of LCAT shown contains amino acids at position 9 through 420 of the proform of LCAT [52] and is available at ModBase [201] with the primary database link P04180. The structure was rendered with MacPyMOL [116,202].
From the conformation of ApoA-I on discs and the estimated size of LCAT it would appear that as many as two LCAT molecules could bind at one time to rHDL, however, there is presently no experimental evidence supporting the simultaneous binding of two LCAT molecules. Because most kinetic studies are conducted under conditions where the LCAT concentration is much lower, approximately 105-fold lower, than the rHDL concentration, statistical considerations suggest that there is only a small probability that two molecules of LCAT to simultaneously reside on a single disc.
Structural domains of ApoA-I
The primary structure of ApoA-I consists of 243 amino acids with secondary structural motifs that are similar to other apolipoproteins. Lipid-free ApoA-I has been shown to have at least two regions or domains, N-terminal (position 1–187) and C-terminal (position 188–243), and each is reported to have unique interactions with lipids and proteins. The solution structure of lipid-free ApoA-I has been described as ‘molten globular’ [57], a state where the α-helical regions usually remain intact but β-sheets and connecting regions are more disordered. In molten globular proteins the hydrophobic regions are believed to face inward, with the hydrophilic faces interacting with the aqueous environment. Recent studies have shown that the N-terminal domain appears to be more compact than originally thought [58]. These findings, along with the well known propensity of lipid-free ApoA-I to oligomerize [59], has led to the suggestion that ApoA-I may possess a more well-ordered structure [58] than has been inferred from its description as a ‘molten globular’ structure.
The first 43 residues of the N-terminal region are believed to be relatively disordered, but residues at positions 8–33 have been identified as a G* amphipathic α-helical region [60]. Amino acids at positions 44–243, comprising approximately 80% of the protein, are composed of a series of ten tandem amphipathic α-helical repeating units with considerable amino acid conservation. Eight repeats contain 22 amino acids, while repeats 3 and 9 are composed of 11 amino acids. Seven of the ten repeats are separated by the helix-breaking amino acid proline [61]. These ‘breaks’ have been suggested to impart flexibility to the protein allowing it to ‘bend’ around the periphery of HDL particles [62,63]. Two crystal forms of ApoA-I have been studied by x-ray crystallography. The first report was for an ApoA-I mutant that lacked the first 43 amino acids [62] and crystallized in a saddle conformation. This report stimulated additional studies into the structure of ApoA-I carried by rHDL. A later report by Ajees et al. on native ApoA-I showed that full-length, lipid-free ApoA-I crystallized as a four-helix bundle, similar to what has been reported for ApoE [64–66].
The type A amphipathic α-helix [67], first described by Segrest in 1974 [68], is a unique feature of lipoproteins. Each helical repeat of ApoA-I has one face enriched in hydrophobic amino acids while the opposing face is enriched with negatively charged amino acids. To enhance lipid solubility, the hydrophobic face of each amphipathic helix interacts with the phospholipid acyl chains in HDL, while the negatively charged hydrophilic face of each helix supports solvation by the aqueous phase. The function of the highly conserved positively charged amino acids that lie between the ‘faces’ of the helices remains speculative. It has been suggested that these positively charged residues interact with the negatively charged oxygen of the phospholipid head group [68,69]; this is termed the snorkel hypothesis. Although NMR evidence from a model amphipathic peptide associated with dodecylphosphatidylcholine (DDPC) micelles does not support the hypothesis [70], an NMR study of ApoC-II associated with DDPC micelles was consistent with the snorkel hypothesis [71].
Functional properties of ApoA-I helices 5, 6 & 7
Studies employing a variety of techniques have suggested that the central region of ApoA-I is essential for binding and activation of LCAT [13,72–74]. Analysis of the amino acid conservation among species within ApoA-I helices 5, 6 and 7 (or residues at positions 121–142, 143–164 and 165–186, respectively) has identified a large number of highly conserved, charged amino acids in this region. It has been hypothesized that these residues function by either forming electrostatic interactions directly with LCAT or forming intrahelical salt bridges within the ApoA-I central domain that interact with LCAT [75,76]. In Figure 1A, a 96 Å diameter rHDL composed of 150 molecules of POPC and two molecules of ApoA-I (green, orange and purple) is shown [53]. The ApoA-I region corresponding to position 140–170 is colored in orange, with helix 6 amino acids at positions 143–164 colored in purple, and all other ApoA-I amino acids are shown in green. Based on structural models of LCAT, amino acids at positions 9–420 of the pro-form of LCAT [52] are shown.
In support of these models, Maiorano et al. suggested that ApoA-I helices 5, 6 and 7 (amino acids at positions 130–174) were important for the activation of LCAT since they were more solvent exposed when compared with other helices within lipid-bound ApoA-I [77]. Sorci-Thomas et al. demonstrated that removing either helix 6 or 7 (amino acids at position 143–164) [13,72,73], or by replacing helix 6 with helix 10 (amino acids at position 220–241) [78], or by inverting helix 6’s hydrophobic face [78], caused a reduction in LCAT binding to rHDL, as well as reducing the apparent Vmax of the reaction. Sviridov et al. also explored the role of ApoA-I helix 6, specifically amino acids position 140–150, on rHDL’s ability to activate LCAT [79]. Their results suggested that amino acids at position 140–150 were essential for LCAT activation [79].
All of these models depend on determining the 3D conformation of lipid-bound ApoA-I. Several advances have occurred over the past 10 years as experimental methods have been refined and used to investigate the conformation of ApoA-I on lipidated particles, including: mass spectrometry [6,53,80–83], electron paramagnetic resonance [84], fluorescence resonance energy transfer (FRET) [58,84–86], and NMR [87]. New insights into how the conformation of ApoA-I adapts to increases in lipid content have come from molecular dynamics simulations [88–91]. There is considerable agreement among the various reports. For example, in all of the models the central region of lipid-bound ApoA-I assumes an antiparallel ‘double belt’ with a helix 5 to 5 registry, that is, the central amphipathic helices 5 on each ApoA-I strand are situated side-by-side on the edge of the particle. Both Wu et al. [83] and Martin et al. [84] suggest that the two ApoA-I monomers do not wrap completely around the edge of the lipid particle as a smooth belt and present evidence for additional protein structures that appear to be important for LCAT activation. The ‘solar flares’ model proposed by Wu et al. was derived from the protruding loops consisting of residues at position 159–178 (parts of helices 6 and 7) [83]. This region displayed significantly reduced exchange in the presence of LCAT [83], suggesting that the presence of the enzyme binding to this site blocked exchange. Although the concept for these loops within helices 5, 6 and 7 (amino acids at position 130–174) was first suggested by Maiorano et al. [77], Martin et al. [84] refined the analysis using a series of ApoA-I variants with cysteines engineered into the loop region. They found a single loop region protruding from the rest of the rHDL, located between amino acids at position 133–146 (portions of helices 5 and 6) through the use of nitroxide spin probes and FRET analysis. The effect of changes in registry on LCAT activation have not been systematically studied, but cysteine substitution mutants of ApoA-I have the ability to ‘lock’ the relationship of the two strands in other than a 5 to 5 registry. Studies using rHDL made from natural mutations, such as ApoA-IMilano [92] and ApoA-IParis [93], and engineered mutants [94] have suggested that these mutant ApoA-Is are poorer substrates for LCAT than wild-type ApoA-I.
The principal differences among the proposed 3D models of lipid-bound ApoA-I are related to the conformation of the C- and N-terminal ends [3]. Bhat et al. proposed a lipid-bound conformation for ApoA-I on 96 Å diameter POPC rHDL (75 POPC/ApoA-I) using chemical crosslinking and mass spectrometry [82]. In this study they discovered the generation of two crosslinked ApoA-I dimers with unique electrophoretic mobility. These two dimers corresponded to distinct intermolecular crosslinks that involve either terminus or the central region of the protein and this information helps identify the regions where crosslinking has occurred [82]. Their studies suggested that the N- and C-terminal ends were folded back and interact with each other forming a ‘buckle’ [53,82]. Bhat et al. later demonstrated that 80 Å diameter POPC rHDL assumed a conformation similar to that proposed for 96 Å diameter rHDL, except that the N- and C-terminal ends appeared to be further folded onto the ‘belt’ [53]. Later analyses have indicated that the ends on 96Å rHDL show some flexibility and that the N-terminal end can also fold back over the C-terminal end. Wu et al. employed hydrogen-deuterium exchange (H/DX) with mass spectrometry to study ApoA-I structure on approximately 104 Å POPC rHDL (100 POPC/ApoA-I) [83]. Solvent accessibility of different regions was assessed across the entire sequence of ApoA-I. The H/DX data were combined with the ‘double belt’ model [95] as a starting point for computational modeling of ApoA-I structure. The ‘solar-flares’ model predicts that the N-termini of two ApoA-I molecules form a symmetric, globular domain with amino acids at position 14–18 of both ApoA-I’s interacting. This model contrasts with the ‘belt buckle’ model where the C- and N-terminal ends fold back onto the edge of the disc. Recently, molecular dynamics simulations were used to rigorously test the ‘solar flares’ model and found that the model was not stable with these simulations [91].
Other recent studies have shown that the diameter of rHDL particles plays a highly significant role in LCAT activation. Cavigiolio et al. demonstrated that the catalytic efficiency, Vmax/Km(Apparent), for LCAT increases in proportion to the rHDL particle diameter [96]. The increase in catalytic efficiency was primarily due to decreases in the binding of LCAT to the rHDL or Km. However, the lowest apparent Km was associated with rHDL particles having the largest diameter, which contain three to four ApoA-I monomers per particle [96]. Since composition, lipid packing and the number of ApoA-I molecules per particle vary as the rHDL size changes, the interplay of each of these variables on LCAT activation will likely be the subject of future studies.
The size of rHDL particles is an important determinant of LCAT activation and ApoA-I helices 7 and 8 have been shown to act as determinants of HDL subclass distribution [97]. Human ApoA-I binds almost equally well to both HDL2, an approximately 102 Å diameter particle, and HDL3, an approximately 84 Å diameter particle. However, murine ApoA-I (unlike the human ApoA-I sequence that has proline punctuation between helical regions 7 and 8) shows greater affinity for HDL3 compared with HDL2 [97]. Systematically replacing amino acids at positions 184–190 with a seven amino acid transition sequence from each of the seven proline-punctuated interhelical regions from human ApoA-I to give mutant ApoA-Is, provided evidence for fourfold difference in affinities for HDL2 compared with HDL3 that depended on which proline-punctuated sequence was used [98]. These results suggest that the effect-specific ‘helix-breaking’ regions are important for determining the size of the particle, and thus its ability to activate LCAT.
Oxidation of HDL ApoA-I alters its ability to activate LCAT
Consistent with the LCAT-activation properties of amino acids at positions 140 to 150 within helix 6, Shao et al. reported that the oxidation of one of the three methionine residues in ApoA-I – Met148 – to a sulfoxide completely obliterates LCAT activity on both rHDL and HDL3 [99]. These conclusions are supported by other reports showing that mild oxidation of ApoA-I severely retards both HDL remodeling [100], as well as its 3D structure [101]. In addition, earlier work on the effects of methionine oxidation also suggests that this process produces serious structural changes in ApoA-I conformation, as determined by altered chromatographic mobility [102–104] or denaturation with guanidine hydrochloride [105]. In 2005, Panzenbock et al. published a review of methionine sulfoxide formation in oxidized HDL and concluded that owing to the large structural and functional consequences associated with this modification, it could be used as a surrogate marker for atherosclerosis in humans [106]. Since that time, other investigators have shown that oxidation of ApoA-I’s methionines appear to be more frequent in the plasma from diabetic patients [107] and from the plasma of mice with hepatocellular carcinoma [108].
In addition to the formation of methionine sulfoxide, the modification of other ApoA-I residues has been systematically studied to determine their effects on ApoA-I function [109]. ApoA-I lysine, methionine, tryptophan and tyrosine residues were all studied to determine their susceptibility to myeloperoxidase modification. From these studies it was determined that loss of ApoA-I function was mediated primarily through the alteration of tryptophan residues [109], while the modification of methionine residues Met86 and Met112 may actually play a protective role, as previously suggested by Garner et al. [106,110]. Recent studies with myoglobin have suggested that although both methionine and tryptophan are oxidized by hypochlorite/myeloperoxidase, methionine appears to be somewhat more susceptible to oxidation [111].
Conformation of ApoA-I on spherical HDL
Structural studies of lipidated ApoA-I have emphasized the use of rHDL that can be prepared with homogenous size and composition. Borhani et al. suggested that ApoA-I floats on the lipid surface of spherical HDL [62] with the hydrophobic faces of each helix interacting with the fatty acid acyl chains [69,112]. The α-helix content, tryptophan fluorescence and antibody binding of ApoA-I on 93 Å diameter POPC spheres were reported to be similar to that of 96 Å POPC rHDL, suggesting that the secondary structure of ApoA-I was similar on both [113]. Although spherical HDL particles made in vitro carried three to four molecules of ApoA-I, FRET analysis of cholesteryl ester-containing HDL spheres prepared from dye-labeled ApoA-I POPC rHDL particles showed that the labeled helical repeats were significantly farther apart than those on original rHDL [85]. Similar findings have also been reported from studies employing chemical crosslinking [114]. However, the assignment of specific distance relationships between the helices was not possible [85]. Recently, HDL with a cholesteryl ester core has been the subject of theoretical computations using a model particle composed of two ApoA-Is, 56 POPC and 16 molecules of cholesteryl oleate. These analyses suggested a shape that resembles a prolate ellipsoid [88], not unlike that reported for smaller HDL particles [89]; moreover, they suggested that the cholesterol component of the cholesteryl oleate core was in contact with ApoA-I and that this contact may contribute to the stabilization of the particle.
Future perspective
Future studies on the physiologic roles of LCAT should cover several different topics. Our first suggestion is a more intense analysis of the interaction between LCAT and ApoA-I. There are three facets to these studies, the first of which is to refine the structure of ApoA-I by defining the conformation its N- and C-terminal ends and obtaining a detailed conformation of the central looped region. The second issue is to more specifically define where LCAT interacts with, and is activated by, ApoA-I. The last issue in this set, and likely the most difficult, is to determine the 3D structure of LCAT so that the overall relationship between LCAT, ApoA-I and the lipid surface can be defined. As these issues are resolved using well-defined rHDL prepared by cholate dialysis, they should be followed up by the analysis of these interactions using small HDL generated by ABCA1.
A final understanding of the 3D relationship between LCAT, ApoA-I and phospholipid will come when we know how ApoA-I interacts with phospholipid in rHDL and small HDL particles from ABCA1. As alluded to in the text there is good theoretical justification of a phospholipid–ApoA-I interaction, which is described by the snorkel hypothesis. The snorkel hypothesis elegantly explains how the protein and phospholipid could form more thermodynamically stable particles by the combination of hydrophobic binding between lipid acyl chains and hydrophobic faces of the amphipathic helices and ionic interactions between the charged phospholipid head group and charged amino acids. The hypothesis has been studied using NMR, but at this time there have been mixed results. Resolution might be achieved by obtaining an x-ray structure of phospholipid–ApoA-I particles or by further analysis using more sophisticated NMR techniques.
HDL has been shown to carry oxidized lipids and reverse cholesterol transport may be a principal pathway for removing oxidized lipids from circulation when LCAT remodels ABCA1-derived particles. Previous studies have demonstrated that the lipids of HDL and LDL are equally susceptible to in vitro oxidation [115]. It will be important to determine how the transport of oxidized lipids does not promote the oxidation of HDL phospholipids.
Executive summary
Cause
Lipid bound ApoA-I activates the plasma enzyme lecithin:cholesterol acyltransferase (LCAT), which catalyzes the conversion of cholesterol to cholesteryl ester. Without this conversion, mature spherical HDL particles are not formed and excess peripheral tissue cholesterol is less efficiently transported to the liver for excretion from the body.
Consequence
Maturation of plasma HDL requires a second lipidation step catalyzed by LCAT allowing the synthesis of HDL cholesteryl esters that provide a hydrophobic core, and transforms small, lipid-poor particles to spherical lipid-rich HDLs. If either ABCA1 or LCAT are inactive, the plasma concentrations of HDL ApoA-I are low because lipid-poor HDL is rapidly removed from the circulation. These two ApoA-I lipidation steps are crucial to the formation of mature plasma HDL and whole-body cholesterol homeostasis.
Conclusion
While catalytic components of LCAT have been established, the structure of LCAT and the mechanism describing its activation have not been experimentally determined. The precise structure of lipid-bound ApoA-I is also presently under active investigation. To date, the majority of studies suggest that LCAT amino acids at position 121–136 may interact with lipid-bound ApoA-I within helix 6 amino acids at position 143–164.
Footnotes
Financial & competing interests disclosure
These studies were supported by grants from the National Heart, Lung and Blood Institute, NIH (HL-49373 and HL-64163 to Mary Sorci-Thomas) and an International HDL Pfizer Award to Michael J Thomas.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Mary G Sorci-Thomas, Department of Pathology, Wake Forest University School of Medicine, Medical Center Blvd, Winston-Salem, NC 27157-1016, USA, Tel.: +1 336 716 2147, Fax: +1 336 716 6279, msthomas@wfubmc.edu.
Shaila Bhat, Department of Pathology, Lipid Sciences Research Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA, Tel.: +1 336 716 6062, Fax: +1 336 716 6279, bhat@wfubmc.edu.
Michael J Thomas, Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA, Tel.: +1 336 716 2313, Fax: +1 336 716 6279, mthomas@wfubmc.edu.
Bibliography
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta. 2000;1529:245–256. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- 2.Lima VLM, Coelho LCBB, Kennedy JF, Owen JS, Dolphin PJ. Lecithin-cholesterol acyltransferase (LCAT) as a plasma glycoprotein: an overview. Carbohydrate Polymers. 2004;55:179–191. [Google Scholar]
- 3.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL ApoA-I: implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson WS, Thompson TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:9143–9149. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- 5.Thomas MJ, Bhat S, Sorci-Thomas MG. The use of chemical cross-linking and mass spectrometry to elucidate the tertiary conformation of lipid-bound apolipoprotein A-I. Curr Opin Lipidol. 2006;17:214–220. doi: 10.1097/01.mol.0000226111.05060.f4. [DOI] [PubMed] [Google Scholar]
- 6.Davidson WS, Silva RA. Apolipoprotein structural organization in high density lipoproteins: belts, bundles, hinges and hairpins. Curr Opin Lipidol. 2005;16:295–300. doi: 10.1097/01.mol.0000169349.38321.ad. [DOI] [PubMed] [Google Scholar]
- 7.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 9.Sorci-Thomas M, Babiak J, Rudel LL. Lecithin-cholesterol acyltransferase (LCAT) catalyzes transacylation of intact cholesteryl esters. Evidence for the partial reversal of the forward LCAT reaction. J Biol Chem. 1990;265:2665–2670. [PubMed] [Google Scholar]
- 10.Subbaiah PV, Horvath P, Achar SB. Regulation of the activity and fatty acid specificity of lecithin-cholesterol acyltransferase by sphingomyelin and its metabolites, ceramide and ceramide phosphate. Biochemistry. 2006;45:5029–5038. doi: 10.1021/bi0600704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolin DJ, Jonas A. Sphingomyelin inhibits the lecithin-cholesterol acyltransferase reaction with reconstituted high density lipoproteins by decreasing enzyme binding. J Biol Chem. 1996;271:19152–19158. doi: 10.1074/jbc.271.32.19152. [DOI] [PubMed] [Google Scholar]
- 12.Rye KA, Hime NJ, Barter PJ. The influence of sphingomyelin on the structure and function of reconstituted high density lipoproteins. J Biol Chem. 1996;271:4243–4250. doi: 10.1074/jbc.271.8.4243. [DOI] [PubMed] [Google Scholar]
- 13.Sorci-Thomas MG, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 14.Cucuianu M, Coca M, Hancu N. Reverse cholesterol transport and atherosclerosis. A mini review. Rom J Intern Med. 2007;45:17–27. [PubMed] [Google Scholar]
- 15•.Lacko AG, Reason AJ, Nuckolls C, et al. Characterization of recombinant human plasma lecithin:cholesterol acyltransferase (LCAT): N-linked carbohydrate structures and catalytic properties. J Lipid Res. 1998;39:807–820. FAB mass spectrometry and linkage analysis showed that the N-linked glycans present on recombinant lecithin: cholesterol acyltransferase (LCAT) were primarily triantennary and tetraantennary structures. LCAT activities of plasma LCAT were determined using lipoprotein substrates. [PubMed] [Google Scholar]
- 16.Schindler PA, Settineri CA, Collet X, Fielding CJ, Burlingame AL. Site-specific detection and structural characterization of the glycosylation of human plasma proteins lecithin:cholesterol acyltransferase and apolipoprotein D using HPLC/electrospray mass spectrometry and sequential glycosidase digestion. Protein Sci. 1995;4:791–803. doi: 10.1002/pro.5560040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosman J, Jonas A. Deletion of specific glycan chains affects differentially the stability, local structures, and activity of lecithin-cholesterol acyltransferase. J Biol Chem. 2001;276:37230–37236. doi: 10.1074/jbc.M104326200. [DOI] [PubMed] [Google Scholar]
- 18.Miller KR, Wang J, Sorci-Thomas M, Anderson RA, Parks JS. Glycosylation structure and enzyme activity of lecithin:cholesterol acyltransferase from human plasma, HepG2 cells, and baculoviral and Chinese hamster ovary cell expression systems. J Lipid Res. 1996;37:551–561. [PubMed] [Google Scholar]
- 19.Qu SJ, Fan HZ, Blanco-Vaca F, Pownall HJ. Effects of site-directed mutagenesis on the N-glycosylation sites of human lecithin:cholesterol acyltransferase. Biochemistry. 1993;32:8732–8736. doi: 10.1021/bi00085a002. [DOI] [PubMed] [Google Scholar]
- 20.Hill JS, Wang X, McLeod R, Pritchard PH. Lecithin:cholesterol acyltransferase: role of N-linked glycosylation in enzyme function. Biochem J. 1993;294 (Pt 3):879–884. doi: 10.1042/bj2940879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill JS, Pritchard PH. Role of N-linked glycosylation of lecithin:cholesterol acyltransferase in lipoprotein substrate specificity. Biochim Biophys Acta. 1995;1254:193–197. doi: 10.1016/0005-2760(94)00183-y. [DOI] [PubMed] [Google Scholar]
- 22.Francone OL, Evangelista L, Fielding CJ. Lecithin-cholesterol acyltransferase: effects of mutagenesis at N-linked oligosaccharide attachment sites on acyl acceptor specificity. Biochim Biophys Acta. 1993;1166:301–304. doi: 10.1016/0005-2760(93)90110-u. [DOI] [PubMed] [Google Scholar]
- 23••.Peelman F, Vinaimont N, Verhee A, et al. A proposed architecture for lecithin cholesterol acyl transferase (LCAT): identification of the catalytic triad and molecular modeling. Protein Sci. 1998;7:587–599. doi: 10.1002/pro.5560070307. The most complete analysis of LCAT structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adimoolam S, Jonas A. Identification of a domain of lecithin-cholesterol acyltransferase that is involved in interfacial recognition. Biochem Biophys Res Commun. 1997;232:783–787. doi: 10.1006/bbrc.1997.6375. [DOI] [PubMed] [Google Scholar]
- 25.Peelman F, Vanloo B, Perez-Mendez O, et al. Characterization of functional residues in the interfacial recognition domain of lecithin cholesterol acyltransferase (LCAT) Protein Eng. 1999;12:71–78. doi: 10.1093/protein/12.1.71. [DOI] [PubMed] [Google Scholar]
- 26.Vickaryous NK, Teh EM, Stewart B, Dolphin PJ, Too CK, McLeod RS. Deletion of N-terminal amino acids from human lecithin:cholesterol acyltransferase differentially affects enzyme activity toward α- and β-substrate lipoproteins. Biochim Biophys Acta. 2003;1646:164–172. doi: 10.1016/s1570-9639(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 27.Jin L, Shieh JJ, Grabbe E, Adimoolam S, Durbin D, Jonas A. Surface plasmon resonance biosensor studies of human wild-type and mutant lecithin cholesterol acyltransferase interactions with lipoproteins. Biochemistry. 1999;38:15659–15665. doi: 10.1021/bi9916729. [DOI] [PubMed] [Google Scholar]
- 28.Peelman F, Vanloo B, Verschelde JL, et al. Effect of mutations of N- and C-terminal charged residues on the activity of LCAT. J Lipid Res. 2001;42:471–479. [PubMed] [Google Scholar]
- 29.Adimoolam S, Jin L, Grabbe E, Shieh JJ, Jonas A. Structural and functional properties of two mutants of lecithin-cholesterol acyltransferase (T123I and N228K) J Biol Chem. 1998;273:32561–32567. doi: 10.1074/jbc.273.49.32561. [DOI] [PubMed] [Google Scholar]
- 30•.Vanloo B, Peelman F, Deschuymere K, et al. Relationship between structure and biochemical phenotype of lecithin:cholesterol acyltransferase (LCAT) mutants causing fish-eye disease. J Lipid Res. 2000;41:752–761. Measured the esterase, phospholipase A2 and acyltransferase activity for natural and engineered mutants. The mutants seem to differentiate the different enzymic activities. [PubMed] [Google Scholar]
- 31.Murray KR, Nair MP, Ayyobi AF, Hill JS, Pritchard PH, Lacko AG. Probing the 121–136 domain of lecithin:cholesterol acyltransferase using antibodies. Arch Biochem Biophys. 2001;385:267–275. doi: 10.1006/abbi.2000.2154. [DOI] [PubMed] [Google Scholar]
- 32.Peelman F, Goethals M, Vanloo B, et al. Structural and functional properties of the 154–171 wild-type and variant peptides of human lecithin-cholesterol acyltransferase. Eur J Biochem. 1997;249:708–715. doi: 10.1111/j.1432-1033.1997.t01-2-00708.x. [DOI] [PubMed] [Google Scholar]
- 33.Francone OL, Fielding CJ. Structure–function relationships in human lecithin:cholesterol acyltransferase. Site-directed mutagenesis at serine residues 181 and 216. Biochemistry. 1991;30:10074–10077. doi: 10.1021/bi00106a002. [DOI] [PubMed] [Google Scholar]
- 34.Qu SJ, Fan HZ, Blanco-Vaca F, Pownall HJ. Effects of site-directed mutagenesis on the serine residues of human lecithin:cholesterol acyltransferase. Lipids. 1994;29:803–809. doi: 10.1007/BF02536246. [DOI] [PubMed] [Google Scholar]
- 35.Jauhiainen M, Dolphin PJ. Human plasma lecithin:cholesterol acyltransferase (LCAT). On the role of essential carboxyl groups in catalysis. Adv Exp Med Biol. 1991;285:71–75. doi: 10.1007/978-1-4684-5904-3_8. [DOI] [PubMed] [Google Scholar]
- 36.Jauhiainen M, Ridgway ND, Dolphin PJ. Aromatic boronic acids as probes of the catalytic site of human plasma lecithin-cholesterol acyltransferase. Biochim Biophys Acta. 1987;918:175–188. doi: 10.1016/0005-2760(87)90193-7. [DOI] [PubMed] [Google Scholar]
- 37.Peelman F, Verschelde JL, Vanloo B, et al. Effects of natural mutations in lecithin: cholesterol acyltransferase on the enzyme structure and activity. J Lipid Res. 1999;40:59–69. [PubMed] [Google Scholar]
- 38.Zannis VI, Chroni A, Krieger M. Role of ApoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 39.Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- 40.Timmins JM, Lee JY, Boudyguina E, et al. Targeted inactivation of hepatic ABCA1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of ApoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 42.Brunham LR, Singaraja RR, Hayden MR. Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu Rev Nutr. 2006;26:105–129. doi: 10.1146/annurev.nutr.26.061505.111214. [DOI] [PubMed] [Google Scholar]
- 43.Krimbou L, Hajj Hassan H, Blain S, et al. Biogenesis and speciation of nascent ApoA-I-containing particles in various cell lines. J Lipid Res. 2005;46:1668–1677. doi: 10.1194/jlr.M500038-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Doung PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to ApoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-β HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;8:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 46.Doung PT, Weibel GL, Lund-Katz S, Rothblat GH, Phillips MC. Characterization and properties of preβ-HDL particles formed by ABCA1-mediated cellular lipid efflux to ApoA-I. J Lipid Res. 2008;49:1006–1014. doi: 10.1194/jlr.M700506-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26:12–19. doi: 10.1161/01.ATV.0000194291.94269.5a. [DOI] [PubMed] [Google Scholar]
- 48.von Eckardstein A. Differential diagnosis of familial high density lipoprotein deficiency syndromes. Atherosclerosis. 2006;186:231–239. doi: 10.1016/j.atherosclerosis.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 49.Koukos G, Chroni A, Duka A, Kardassis D, Zannis VI. LCAT can rescue the abnormal phenotype produced by the natural ApoA-I mutations (Leu141Arg)Pisa. Biochemistry 46, and (Leu159Arg)FIN. 2007:10713–10721. doi: 10.1021/bi7003203. [DOI] [PubMed] [Google Scholar]
- 50.McPherson PA, Young IS, McEneny J. A dual role for lecithin:cholesterol acyltransferase (EC 2.3.1.43) in lipoprotein oxidation. Free Radic Biol Med. 2007;43:1484–1493. doi: 10.1016/j.freeradbiomed.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Asztalos BF, Schaefer EJ, Horvath KV, et al. Role of LCAT in HDL remodeling: investigation of LCAT deficiency states. J Lipid Res. 2007;48:592–599. doi: 10.1194/jlr.M600403-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Pieper U, Narayanan E, Sali A. ModBase Database P04180, Model ID ca6c601e79a82c444d6baa952581f64e. 2005. Model of lecithin-cholesterol acyltransferase. [Google Scholar]
- 53.Bhat S, Sorci-Thomas MG, Tuladhar R, Samuel MP, Thomas MJ. Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry. 2007;46:7811–7821. doi: 10.1021/bi700384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dolinsky VW, Gilham D, Alam M, Vance DE, Lehner R. Triacylglycerol hydrolase: role in intracellular lipid metabolism. Cell Mol Life Sci. 2004;61:1633–1651. doi: 10.1007/s00018-004-3426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nardini M, Lang DA, Liebeton K, Jaeger KE, Dijkstra BW. Crystal structure of Pseudomonas aeruginosa lipase in the open conformation. The prototype for family I.1 of bacterial lipases. J Biol Chem. 2000;275:31219–31225. doi: 10.1074/jbc.M003903200. [DOI] [PubMed] [Google Scholar]
- 56.Jonas A. Lecithin-cholesterol acyltransferase in the metabolism of high-density lipoproteins. Biochim Biophys Acta. 1991;1084:205–220. doi: 10.1016/0005-2760(91)90062-m. [DOI] [PubMed] [Google Scholar]
- 57.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brouillette CG, Dong WJ, Yang ZW, et al. Forster resonance energy transfer measurements are consistent with a helical bundle model for lipid-free apolipoprotein A-I. Biochemistry. 2005;44:16413–16425. doi: 10.1021/bi051018v. [DOI] [PubMed] [Google Scholar]
- 59.Yokoyama S, Tajima S, Yamamoto A. The process of dissolving apolipoprotein A-I in an aqueous buffer. J Biochem. 1982;91:1267–1272. doi: 10.1093/oxfordjournals.jbchem.a133811. [DOI] [PubMed] [Google Scholar]
- 60.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 61.Chan L. The apolipoprotein multigene family: structure, expression, evolution, and molecular genetics. Klin Wochenschr. 1989;67:225–237. doi: 10.1007/BF01717324. [DOI] [PubMed] [Google Scholar]
- 62.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci USA. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 64.Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HM. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci USA. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Vasudevan S, Sojitrawala R, et al. A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry. 2007;46:10722–10732. doi: 10.1021/bi700672v. [DOI] [PubMed] [Google Scholar]
- 67.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic α-helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 68.Segrest JP, Jackson RL, Morrisett JD, Gotto AM. A molecular theory of lipid–protein interactions in the plasma lipoproteins. FEBS Lett. 1974;38:247–258. doi: 10.1016/0014-5793(74)80064-5. [DOI] [PubMed] [Google Scholar]
- 69.Mishra VK, Palgunachari MN, Segrest JP, Anantharamaiah GM. Interactions of synthetic peptide analogs of the class A amphipathic helix with lipids. Evidence for the snorkel hypothesis. J Biol Chem. 1994;269:7185–7191. [PubMed] [Google Scholar]
- 70.Buchko GW, Treleaven WD, Dunne SJ, Tracey AS, Cushley RJ. Structural studies of a peptide activator of human lecithin-cholesterol acyltransferase. J Biol Chem. 1996;271:3039–3045. doi: 10.1074/jbc.271.6.3039. [DOI] [PubMed] [Google Scholar]
- 71.MacRaild CA, Howlett GJ, Gooley PR. The structure and interactions of human apolipoprotein C-II in dodecyl phosphocholine. Biochemistry. 2004;43:8084–8093. doi: 10.1021/bi049817l. [DOI] [PubMed] [Google Scholar]
- 72.Sorci-Thomas MG, Kearns MW, Lee JP. Apolipoprotein A-I domains involved in lecithin-cholesterol acyltransferase activation. Structure–function relationships. J Biol Chem. 1993;268:21403–21409. [PubMed] [Google Scholar]
- 73.Sorci-Thomas MG, Thomas MJ, Curtiss LK, Landrum M. Single repeat deletion in ApoA-I blocks cholesterol esterification and results in rapid catabolism of δ6 and wild-type ApoA-I in transgenic mice. J Biol Chem. 2000;275:12156–12163. doi: 10.1074/jbc.275.16.12156. [DOI] [PubMed] [Google Scholar]
- 74.McManus DC, Scott BR, Frank PG, Franklin V, Schultz JR, Marcel YL. Distinct central amphipathic α-helices in apolipoprotein A-I contribute to the in vivo maturation of high density lipoprotein by either activating lecithin-cholesterol acyltransferase or binding lipids. J Biol Chem. 2000;275:5043–5051. doi: 10.1074/jbc.275.7.5043. [DOI] [PubMed] [Google Scholar]
- 75.Roosbeek S, Vanloo B, Duverger N, et al. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin: cholesterol acyltransferase. J Lipid Res. 2001;42:31–40. [PubMed] [Google Scholar]
- 76.Alexander ET, Bhat S, Thomas MJ, et al. Apolipoprotein A-I helix 6 negatively charged residues attenuate lecithin: cholesterol acyltransferase (LCAT) reactivity. Biochemistry. 2005;44:5409–5419. doi: 10.1021/bi047412v. [DOI] [PubMed] [Google Scholar]
- 77.Maiorano JN, Jandacek RJ, Horace EM, Davidson WS. Identification and structural ramifications of a hinge domain in apolipoprotein A-I discoidal high-density lipoproteins of different size. Biochemistry. 2004;43:11717–11726. doi: 10.1021/bi0496642. [DOI] [PubMed] [Google Scholar]
- 78.Sorci-Thomas MG, Curtiss L, Parks JS, Thomas MJ, Kearns MW, Landrum M. The hydrophobic face orientation of apolipo-protein A-I amphipathic helix domain 143–164 regulates lecithin: cholesterol acyltransferase activation. J Biol Chem. 1998;273:11776–11782. doi: 10.1074/jbc.273.19.11776. [DOI] [PubMed] [Google Scholar]
- 79.Sviridov D, Hoang A, Sawyer WH, Fidge NH. Identification of a sequence of apolipoprotein A-I associated with the activation of lecithin:cholesterol acyltransferase. J Biol Chem. 2000;275:19707–19712. doi: 10.1074/jbc.M000962200. [DOI] [PubMed] [Google Scholar]
- 80.Davidson WS, Hilliard GM. The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles – a mass spectrometry study. J Biol Chem. 2003;278:27199–27207. doi: 10.1074/jbc.M302764200. [DOI] [PubMed] [Google Scholar]
- 81.Silva RA, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 82.Bhat S, Sorci-Thomas MG, Alexander ET, Samuel MP, Thomas MJ. Intermolecular contact between globular N-terminal fold and C-terminal domain of ApoA-I stabilizes its lipid-bound conformation: studies employing chemical cross-linking and mass spectrometry. J Biol Chem. 2005;280:33015–33025. doi: 10.1074/jbc.M505081200. [DOI] [PubMed] [Google Scholar]
- 83••.Wu Z, Wagner MA, Zheng L, et al. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. Innovative use of H/DX to derive structural data for ApoA-I bound to a lipid disc. LCAT binding site on ApoA-I identified. [DOI] [PubMed] [Google Scholar]
- 84••.Martin DD, Budamagunta MS, Ryan RO, Voss JC, Oda MN. Apolipoprotein A-I assumes a ‘looped belt’ conformation on reconstituted high density lipoprotein. J Biol Chem. 2006;281:20418–20426. doi: 10.1074/jbc.M602077200. EPR spectroscopy indicates extended helical secondary structure at amino acid 139 of ApoA-I bound to lipid discs. They propose a looped belt for amino acids 133–146. [DOI] [PubMed] [Google Scholar]
- 85.Li HH, Lyles DS, Pan W, Alexander E, Thomas MJ, Sorci-Thomas MG. Apo A-I structure on discs and spheres. Variable helix registry and conformational states. J Biol Chem. 2002;42:39093–39101. doi: 10.1074/jbc.M206770200. [DOI] [PubMed] [Google Scholar]
- 86.Li HH, Lyles DS, Thomas MJ, Pan W, Sorci-Thomas MG. Structural determination of lipid-bound ApoA-I using fluorescence resonance energy transfer. J Biol Chem. 2000;275:37048–37054. doi: 10.1074/jbc.M005336200. [DOI] [PubMed] [Google Scholar]
- 87•.Li Y, Kijac AZ, Sligar SG, Rienstra CM. Structural analysis of nanoscale self-assembled discoidal lipid bilayers by solid-state NMR spectroscopy. Biophys J. 2006;91:3819–3828. doi: 10.1529/biophysj.106.087072. The authors use solid-state NMR to study phospholipid nanodiscs using 200-amino acid lipid-binding domain of native, human ApoA-I. The results support a belt model for nanodisc structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88••.Catte A, Patterson JC, Bashtovyy D, et al. Structure of spheroidal HDL particles revealed by combined atomistic and coarse-grained simulations. Biophys J. 2008;94:2306–2319. doi: 10.1529/biophysj.107.115857. Investigated the conformation of ApoA-I in model spheroidal HDL particles using both atomistic and coarse-grained molecular dynamics simulations. Cholesteryl oleate was included in the modeling. The model suggests that the surface is predominantly ApoA-I and phospholipid while the cholesteryl ester may interact with ApoA-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Catte A, Patterson JC, Jones MK, et al. Novel changes in discoidal high density lipoprotein morphology: a molecular dynamics study. Biophys J. 2006;90:4345–4360. doi: 10.1529/biophysj.105.071456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shih AY, Denisov IG, Phillips JC, Sligar SG, Schulten K. Molecular dynamics simulations of discoidal bilayers assembled from truncated human lipoproteins. Biophys J. 2005;88:548–556. doi: 10.1529/biophysj.104.046896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shih AY, Sligar SG, Schulten K. Molecular models need to be tested: the case of a solar flares discoidal HDL model. Biophys J. 2008;94:L87–L89. doi: 10.1529/biophysj.108.131581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Calabresi L, Franceschini G, Burkybile A, Jonas A. Activation of lecithin cholesterol acyltransferase by a disulfide-linked apolipoprotein A-I dimer. Biochem Biophys Res Commun. 1997;232:345–349. doi: 10.1006/bbrc.1997.6286. [DOI] [PubMed] [Google Scholar]
- 93.Koukos G, Chroni A, Duka A, Kardassis D, Zannis VI. Naturally occurring and bioengineered ApoA-I mutations that inhibit the conversion of discoidal to spherical HDL: the abnormal HDL phenotypes can be corrected by treatment with LCAT. Biochem J. 2007;406:167–174. doi: 10.1042/BJ20070296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian S, Jonas A. Structural and functional properties of apolipoprotein A-I mutants containing disulfide-linked cysteines at positions 124 or 232. Biochim Biophys Acta. 2002;1599:56–64. doi: 10.1016/s1570-9639(02)00377-1. [DOI] [PubMed] [Google Scholar]
- 95.Segrest JP, Jones MK, Klon AE, et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 96•.Cavigiolio G, Shao B, Geier EG, Ren G, Heinecke JW, Oda MN. The interplay between size, morphology, stability, and functionality of high-density lipoprotein subclasses. Biochemistry. 2008;47:4770–4779. doi: 10.1021/bi7023354. Five subclasses of rHDL of different diameters were prepared with phospholipid, cholesterol and ApoA-I. The apparent-Km decreased with increasing particle diameter with the most dramatic change for the largest particles. Smaller diameter particles, 7.8 to 9.6 nm, carried only two molecules of ApoA-I while larger particles, 12.2 and 17.0 nm, carried from three to four molecules of ApoA-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reschly EJ, Sorci-Thomas MG, Davidson WS, Meredith SC, Reardon CA, Getz GS. Apolipoprotein A-I α-helices 7 and 8 modulate high density lipoprotein subclass distribution. J Biol Chem. 2002;277:9645–9654. doi: 10.1074/jbc.M107883200. [DOI] [PubMed] [Google Scholar]
- 98•.Carnemolla R, Ren X, Biswas TK, et al. The specific amino acid sequence between helices 7 and 8 influences the binding specificity of human apolipoprotein A-I for high density lipoprotein (HDL) subclasses: a potential for HDL preferential generation. J Biol Chem. 2008;283:15779–15788. doi: 10.1074/jbc.M710244200. The specific sequence between helices 7 and 8 influences the HDL subclass distribution suggesting that ApoA-I sequence may affect HDL size. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99••.Shao B, Cavigiolio G, Brot N, Oda MN, Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci USA. 2008;105:12224–12229. doi: 10.1073/pnas.0802025105. Oxidation of amino acids Met148 or Tryp72 resulted in an approximately 80% decrease in LCAT catalyzed esterification of cholesterol. They propose that oxidation of Met148 disrupts ApoA-I’s central loop, which is in the region of ApoA-I that activates LCAT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gao X, Jayaraman S, Gursky O. Mild oxidation promotes and advanced oxidation impairs remodeling of human high-density lipoprotein in vitro. J Mol Biol. 2008;376:997–1007. doi: 10.1016/j.jmb.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roberts LM, Ray MJ, Shih TW, Hayden E, Reader MM, Brouillette CG. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human ApoA-I. Biochemistry. 1997;36:7615–7624. doi: 10.1021/bi962952g. [DOI] [PubMed] [Google Scholar]
- 102.von Eckardstein A, Walter M, Holz H, Benninghoven A, Assmann G. Site-specific methionine sulfoxide formation is the structural basis of chromatographic heterogeneity of apolipoproteins A-I, C-II, and C-III. J Lipid Res. 1991;32:1465–1476. [PubMed] [Google Scholar]
- 103.Anantharamaiah GM, Hughes TA, Iqbal M, et al. Effect of oxidation on the properties of apolipoproteins A-I and A-II. J Lipid Res. 1988;29:309–318. [PubMed] [Google Scholar]
- 104.Nofer JR, von Eckardstein A, Assmann G. Mannitol prevents methionine sulphoxidation mediated electrophoretic heterogeneity of apolipoprotein A-I. Biomed Chromatogr. 1995;9:28–31. doi: 10.1002/bmc.1130090106. [DOI] [PubMed] [Google Scholar]
- 105.Sigalov AB, Stern LJ. Oxidation of methionine residues affects the structure and stability of apolipoprotein A-I in reconstituted high density lipoprotein particles. Chem Phys Lipids. 2001;113:133–146. doi: 10.1016/s0009-3084(01)00186-4. [DOI] [PubMed] [Google Scholar]
- 106.Panzenbock U, Stocker R. Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biochim Biophys Acta. 2005;1703:171–181. doi: 10.1016/j.bbapap.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 107.Brock JW, Jenkins AJ, Lyons TJ, et al. Increased methionine sulfoxide content of ApoA-I in Type 1 diabetes. J Lipid Res. 2008;49:847–855. doi: 10.1194/jlr.M800015-JLR200. [DOI] [PubMed] [Google Scholar]
- 108.Fernandez-Irigoyen J, Santamaria E, Sesma L, et al. Oxidation of specific methionine and tryptophan residues of apolipoprotein A-I in hepatocarcinogenesis. Proteomics. 2005;5:4964–4972. doi: 10.1002/pmic.200500070. [DOI] [PubMed] [Google Scholar]
- 109.Peng DQ, Brubaker G, Wu Z, et al. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol. 2008;28(11):2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII . J Biol Chem. 1998;273:6088–6095. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 111.Szuchman-Sapir AJ, Pattison DI, Ellis NA, Hawkins CL, Davies MJ, Witting PK. Hypochlorous acid oxidizes methionine and tryptophan residues in myoglobin. Free Radic Biol Med. 2008;45(6):789–798. doi: 10.1016/j.freeradbiomed.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 112.Hristova K, Wimley WC, Mishra VK, Anantharamiah GM, Segrest JP, White SH. An amphipathic α-helix at a membrane interface: a structural study using a novel X-ray diffraction method. J Mol Biol. 1999;290:99–117. doi: 10.1006/jmbi.1999.2840. [DOI] [PubMed] [Google Scholar]
- 113.Jonas A, Wald JH, Toohill KL, Krul ES, Kezdy KE. Apolipoprotein A-I structure and lipid properties in homogeneous, reconstituted spherical and discoidal high density lipoproteins. J Biol Chem. 1990;265:22123–22129. [PubMed] [Google Scholar]
- 114••.Silva RA, Huang R, Morris J, et al. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA. 2008;105(34):12176–12181. doi: 10.1073/pnas.0803626105. First report on the structure of ApoA-I bound to a 93Å diameter spherical HDL using chemical cross-linking and mass spectrometry. They report that the general structural organization was similar between discs and spheres. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thomas MJ, Chen QR, Zabalawi M, et al. Is the oxidation of high-density lipoprotein lipids different than the oxidation of low-density lipoprotein lipids? Biochemistry. 2001;40:1719–1724. doi: 10.1021/bi0022442. [DOI] [PubMed] [Google Scholar]
- 116.Delano ML. MacPyMOL: A PyMOL-based molecular graphis application for MacOS X. DeLano Scientific LLC; Palo Alto, CA, USA: 2007. [Google Scholar]
Website
- 101.ModBase. Database of comparative protein structure models. doi: 10.1093/bioinformatics/15.12.1060. http://modbase.compbio.ucsf.edu. [DOI] [PubMed]
- 102.MacPyMOL. Popular molecular visualization program. www.pymol.org.