

Abstract
Recently, we characterized the more severe nature of hearing loss in aged Type 2 diabetic human subjects. The current study prospectively assessed hearing abilities in middle age CBA/CaJ mice with Type 1 diabetes mellitus (T1DM) (STZ injection) or Type 2 diabetes mellitus (T2DM) (high fat diet), for a period of 6 months. Blood glucose, body weight and auditory tests (Auditory Brainstem Response-ABR, Distortion Product Otoacoustic Emissions-DPOAE) were evaluated at baseline and every 2 months. Tone and broadband noise-burst responses in the inferior colliculus were obtained at 6 months. Body weights of controls did not change over 6 months (~32g), but there was a significant (~5g) decline in the T1DM, while T2DM exhibited ~10g weight gain. Blood glucose levels significantly increased: 3 fold for T1DM, 1.3 fold for T2DM; with no significant changes in controls. ABR threshold elevations were found for both types of diabetes, but were most pronounced in the T2DM, starting as early as 2 months after induction of diabetes. A decline of mean DPOAE amplitudes was observed in both diabetic groups at high frequencies, and for the T2DM at low frequencies. In contrast to ABR thresholds, tone and noise thresholds in the inferior colliculus were lower for both diabetic groups. Induction of diabetes in middle-aged CBA/CaJ mice promotes amplification of age-related peripheral hearing loss which makes it a suitable model for studying the interaction of age-related hearing loss and diabetes. On the other hand, initial results of effects from very high blood glucose level (T1DM) on the auditory midbrain showed disruption of central inhibition, increased response synchrony or enhanced excitation in the inferior colliculus.
Keywords: CBA/CaJ mice, hearing loss, diabetes, auditory brainstem response, distortion product otoacoustic emissions, auditory midbrain, inferior colliculus
INTRODUCTION
Diabetes mellitus (DM) is a prevalent metabolic disease of middle age and older adults worldwide (about 7.0% and rising). There are two major types of diabetes: Type 1 diabetes mellitus (T1DM) and Type 2 diabetes mellitus (T2DM). It is estimated that 5-10% of Americans who are diagnosed with diabetes have T1DM. Most common is T2DM, affecting more than 20% of people over age 60 (American Diabetes Association 2006). Both types of DM can cause serious health complications that involve multiple organs and physiological systems. Many T2DM complications in older people are associated with natural aging, but they appear earlier in diabetic patients (Biessels et al. 2002).
Studies that have attempted to characterize hearing loss in diabetics show inconsistent results (for reviews see Tay et al. 1995; Fowler and Jones 1999; Maia and Campos 2005). Specifically, some studies find bilateral hearing loss at high frequencies for children with T1DM (Elamin et al. 2005), while others did not find hearing loss (Dalton et al. 1998; de Espana et al. Kudelska et al. 1995), or found hearing loss in middle age and older T2DM patients (Rózańska 2002; Diaz de Leon-Morales et al. 2005; Sakuta et al. 2007). However, Tay et al. (1995) showed hearing impairment at low and middle frequencies in diabetics. In contrast, Salvinelli et al. (2004) found no effect on hearing for either type of diabetes, and evoked otoacoustic emissions (OAEs) were not affected at most frequencies in T2DM subjects (de Leo et al., 1997; Erdem et al., 2003). However, Sasso et al. (1999) reported a significant drop in OAE amplitudes for diabetic subjects. To evaluate brainstem auditory function, auditory brainstem response (ABR) latencies were used (de Leo et al. 1997; Sasso et al. 1999), to show prolonged activation of central auditory pathways. Except for the ABR latency experiments, these human studies used simple, limited auditory measures that evaluated peripheral hearing.
Recently, we showed a significant effect on both peripheral and central hearing functions in older T2DM subjects by using a comprehensive test battery (Frisina et al. 2006). Peripheral hearing function was assayed by pure tones, otoacoustic emissions (transient and distortion product), and speech thresholds, revealing more significant losses in the T2DM group compared to age-matched controls. For the first time, effects of T2DM on hearing tests involving central auditory processing in aged human subjects were evaluated by measuring hearing in-noise (HINT) and suprathreshold gap detection thresholds. As for the inner ear findings, the T2DM group showed significantly worse performance on these tests compared to age matched non-diabetic controls, suggesting that the CNS is susceptible to the damaging effects of the diabetes conditions.
There are several animal models of diabetes: animals that develop diabetes by specific experimental procedures and animals that develop diabetes spontaneously, owing to a genetic predisposition (Mordes et al. 1981; Shafrir 1997.). Streptozotocin (STZ) induced diabetes is a T1DM model and is widely used for longer (6 months) duration studies of diabetes. For example, this T1DM model has been used to evaluate alteration of auditory function with ABRs. Threshold elevations and first-peak latency prolongation appeared in rodents (Biessels et al., 2001; Manschot et al. 2003; Biessels et al. 2005). Also, significant prolongation of auditory evoked potentials was reported for STZ-treated rats by 3 months of diabetes duration, suggesting changes in central auditory regions (Biessels et al. 1999). In genetically predisposed young adult rats (T2DM, 13 months of age) evaluation of auditory function showed elevation of ABR thresholds (Ishikawa et al. 1995) as compared to non-susceptible strains. A main limitation of these previous studies that attempted to identify the effect of diabetes on hearing function was that hearing ability of the rodent strains were not well characterized initially, making it difficult to identify effects of hearing impairment, diabetes, and chemical side effects (those induced by streptozotocin, alloxan).
The necessity of utilizing an appropriate mouse model for studying effects of diabetes (prevalent in older populations) on age related hearing loss is obvious from scientific and clinical perspectives. The most commonly used animal model of T1DM, STZ injection, may interfere with metabolism of inner ear hair cells as well as with auditory neurons in the brain. Another, important aspect of evaluating mouse models of diabetes is selection of an appropriate mouse strain to avoid genetic effects on presbycusis. For example, C57Bl/6 and DBA strains of mice have the ahl allele, inducing a rapid, high-frequency hearing loss (Willott and Carlson 1995; Willott et al. 2001), making them unsuitable for studies in middle age and old animals of the interaction of diabetes and presbycusis. Much evidence suggests that the CBA/CaJ serves as an excellent model for many cases of human presbycusis since it shows hearing loss that progresses on a time frame similar to human’s, when one corrects for the different absolute lifespans of mice and men. The age related hearing loss of the CBA/CaJ strain most likely corresponds to the sensory-neural type of human presbycusis (high-tone frequency loss with changes in central auditory regions) (e.g., Spongr et al. 1998; Frisina et al. 1998; Parham et al. 1999; Willott 2001; Jacobson et al. 2003).
Anatomical analysis of the cochlea has shown stria vascularis degeneration and outer hair cell (OHC) loss for both types of diabetes in humans (Fukushima et al. 2005; 2006). However, studies on diabetic animals reveal mixed results. For example, sometimes examination of diabetic animal models has found loss of OHCs (Nakae and Tachibana 1986; Rust et al. 1992; Triana et al. 1995; Raynor et al. 1995) and inner hair cells (IHC) (Nakae & Tachibana 1986), as well as changes in intermediate (Nakae and Tachibana 1986; Ishikawa et al. 1995) and marginal cells of the stria vascularis. Some authors reported degeneration of spiral ganglion cells (Raynor et al. 1995; Ishikawa et al. 1995) and thickening of the basement-membranes of capillaries (Smith et al. 1995). Others did not find any of these associations in diabetic animal models (Nageris et al. 1998).
The aim of the present investigation was to induce two types of diabetes in middle age CBA/CaJ mice and evaluate effects on age-related hearing loss using different hearing tests. The effect of a long duration (6 months) metabolic stress on the progression and time course of age-related hearing loss - presbycusis, was characterized by hearing assessments for both peripheral (cochlea) and central (inferior colliculus - IC) portions of the auditory pathway. Age-matched non-diabetic control mice were utilized for comparison with hyperglycemic T1DM mice (STZ induced) and for T2DM (high fat diet) to maximize effects for future studies of possible anatomical, genetic and neurochemical mechanisms of diabetes on hearing ability with age.
METHODS
Animals
Twelve month old CBA/CaJ males mice were used (N=36). The breeding pairs were purchased from Jackson Laboratories and subjects of the present study were bred and housed at the University of Rochester. Animals were kept under specific pathogen free (SPF) conditions in 12-hr light/dark cycle with chow and water ad libitum in a low noise environment. All procedures were approved by the University of Rochester Animal Care Committee.
Experimental design
Male mice were randomly divided into the 3 experimental groups: control, n=14, T1DM, n=11, and T2DM, n=11. Females were excluded from this study based on estrogen-related decreases in susceptibility to diet-induced obesity (Wade et al. 1985; Zhang et al. 2005). T1DM was induced by a single intraperitoneal injection (IP) of streptozotocin (STZ, at 200 mg/kg of body weight; Sigma Chemical, St. Louis, MO) dissolved in 0.1M citric buffer (pH=4.5) (Paik et al., 1980). T2DM was induced with a high fat, high simple carbohydrate, low fiber diet (Diet #5800-B, Test Diet, Richmond, IN) (Surwit et al., 1988). Controls were injected with citrate buffer and fed standard Purina rodent chow and water ad libitum. There was about a 20% mortality rate due to late middle age in the T2DM group, and about 50% in the control and T1DM groups. The rate of mortality went even higher during survival surgery prior to the IC recording due to age, obesity, hyperglycemia, and hyperlipidemia in T2DM (5 mice) and T1DM (2 mice). Whereas the control mice all survived the IC surgery. Data from the mice that died were not included in the longitudinal results or statistics, e.g., for body weights, glucose levels, ABRs, DPOAEs. For cross-sectional IC recordings, the total number of animals was fewer.
Blood glucose levels evaluation
Mice fasted for 4 hours before the blood glucose test. Blood samples were obtained by puncture of the tail vein, and blood glucose level was determined by using an automated One-Touch Ultra glucometer and glucose test strips (LifeScan, Milpitas, CA).
Hearing Tests
Distortion product otoacoustic emissions (DPOAEs) and ABR functional data were obtained for all three experimental groups at baseline, 2, 4 and 6 months, using procedures described in detail previously (Jacobson et al. 2003; Guimaraes et al. 2004; Tadros et al. 2007; Zhu et al. 2007). Briefly, mice were anesthetized with a mixture of ketamine/xylazine (Bedford Labs, Bedford, OH; Phoenix Scientific, St. Joseph, MO; 120 and 10 mg/kg body weight, respectively, IP injection). All recording sessions were completed in a soundproof acoustic chamber (IAC, Bronx, NY) lined with Sonex. Body temperature was maintained with a heating pad. ABRs were recorded with subcutaneous platinum needle electrodes placed at the vertex (noninverting input), right mastoid prominence (inverted input), and tail (indifferent site). Calibrated tone pips, 5 ms duration, 0.5 ms rise-fall time (phase alternating 90°) were utilized. Electroencephalographic (EEG) activity was differentially amplified (50 or 100 k; Grass [Quincy, MA] model P511 EEG amplifier), then input to an A/D converter (Tucker-Davis Technologies [TDT, Alachua, FL] AD1), and digitized at 50 kHz. Each averaged response was based on 300-500 stimulus repetitions recorded over 10-ms epochs. Contamination by muscle or cardiac activity was prevented by rejecting data epochs in which the single-trace EEG contained peak-to-peak amplitudes exceeding 50 μV. The threshold was defined as the first level that did not evoke a response to a measured frequency, i.e., no difference from the baseline. Peak-1 ABR amplitude, representing an auditory nerve response (Henry and Price 1994; Henry 2002), was also measured at 80 dB SPL for all animals.
Distortion-product otoacoustic emissions were accomplished with the TDT BioSig III system. Stimuli were digitally synthesized at 200 kHz using SigGen software applications. The DPOAE amplitudes (DP-grams) were obtained with frequency 1 intensity level (L1) = 65 dB and frequency 2 intensity level (L2) = 50 dB sound pressure level (SPL); and f1/f2= 1.25, using 8 points per octave covering a frequency range from 5.6 to 44.8 kHz (geometric mean [GM] frequency). Results for each animal were averaged for each test frequency, and then used to calculate group mean data.
Tone and broad band noise IC recordings
Surgery was performed under aseptic conditions as previously described in detail (Walton et al., 1998; Allen et al., 2003). Briefly, surgical attachment of a head post was performed under general anesthesia (Avertin, 200 μg/g, IP). A small tungsten wire was implanted through the skull near the head post to contact the surface of the brain, thus serving as the indifferent electrode. Dental cement and dental acrylic were used to stabilize the head post and indifferent electrode. Animals were allowed to completely recover from surgery overnight prior to the experiment. During experiments all mice were tranquilized (Chlorprothixene 2.5-6 g/kg), and a small craniotomy was made to expose the IC. A monopolar tungsten electrode (20-50 μm tip diameter) was lowered into the IC under stereotactic control. Neural activity was amplified (10,000x), band-pass filtered (10 Hz-3 kHz) using a TDT Bioamplifier (HS4/DB4), and then digitally sampled at 40 kHz using a TDT AD2. The recording duration for tones and noise was 15 ms and each near-field auditory evoked potential (NFAEP) response was averaged for 100 presentations and replicated once for each condition.
The near-field responses to tone bursts of 3, 6, 12, 16, 20, 24, 32, 36, and 48 kHz were measured using stimuli identical to those used for evoking the ABR. These stimuli were presented in 5 dB steps in order of decreasing level from 80 dB SPL and the thresholds were defined operationally as the first level that did not evoke a response, i.e., no difference from the baseline. Tone burst durations were set to 5 ms, with 1.0 ms cos2 rise-fall times. Analysis of the NFAEPs for tone bursts and noise were performed by using custom-designed software (National Instruments Austin, TX) to calculate the root mean square voltage (RMS) of response amplitude for each level (80 dB SPL to 0 dB SPL). Automated analyses were performed for 15 ms windows, capturing the entire response, starting at stimulus levels of 80 dB SPL.
Statistical analyses
Statistical analysis of longitudinal data across the time points and between groups was performed with 2-way analysis of variance (ANOVA) for repeated measures followed by Bonferroni post-hoc tests corrected for multiple comparisons, using Prism ® 4.0 (GraphPad Software, San Diego, CA). Differences were considered statistically significant for p<0.05. Comparisons among experimental groups for the end of the experiment were done by one-way ANOVA followed by Dunnett’s post-hoc tests whose power was corrected for multiple comparisons.
RESULTS
Induction of two types of diabetes in middle aged CBA/CaJ mice
There was no difference in baseline body weight (BW) among experimental groups (~32 g). The T1DM group demonstrated BW loss by the second month after the STZ injection. On the contrary, the average BW increased in T2DM after two months of being on the high fat diet. For both groups, the BW after 2 months remained about the same until animals were sacrificed. Changes in BW for both diabetic groups were significantly different from the control group and to their baselines (Fig. 1A). Main effects using two-way ANOVA demonstrated significant changes for subject groups (F=33.9; p<0.0001; df=2) and interaction of experimental groups over time (F=34.7; p<0.0001; df=6). The control group average BW was not affected during the study.
Figure 1.

Induction of different types of diabetes in middle age CBA/CaJ mice. A. Diabetes alters body weight: T1DM showed significant body weight loss, whereas T2DM showed significant body weight gain, with no changes in controls over the 6 month experimental timeframe. B. Diabetes elevates fasted glucose levels in middle aged CBA mice: T1DM showed significant elevation of fasted blood glucose level by 2 months, whereas T2DM showed significant elevation of fasted blood glucose by 4 months. The control group showed no changes. Differences were compared to baseline. Data presented as mean ± SEM. Asterisks indicate significant differences: * p < 0.05, ** p< 0.01, *** p<0.001. n, number of mice in each subject group.
Both diabetic groups exhibited elevation of fasting blood glucose by 6 months. However, the T1DM group showed dramatic (~ 250%) elevation by 2 months, that remained the same thereafter until the end of the experiment (Fig. 1B). Induction of T2DM revealed elevation of fasting glucose levels (~30%) by 4 months compared to controls. Statistical analyses by two-way ANOVA showed significant main effects for subject groups (F=67.53; p<0.0001; df=2), time (F=40.17; p<0.0001; df=3) and interaction of experimental groups over time (F=22.84; p<0.0001; df=6). Bonferroni post hoc tests confirmed that the T1DM group had significantly higher levels of glucose by 2 months, while the T2DM by 6 months (Fig. 1B) compared to baseline.
Auditory brainstem response
Figure 2, A -F represents ABR thresholds for all experimental groups. Two-way ANOVA revealed that the T2DM group had highly elevated ABR thresholds (i.e. 10-25 dB) at all ABR frequencies (3, 6, 12, 24, 32, and 48 kHz). On the other hand, controls and T1DM showed trends towards higher ABR threshold elevations, typical for the late middle age progression of presbycusis. In addition, the T2DM group displayed a statistically significant peak-1 amplitude drop (Fig. 3) compared to baseline (t=3.10, p<0.01, df=3), that was more prominent than in the other two subject groups. Thus, T2DM promoted elevation of ABR thresholds, and declines in P1 amplitudes, that were significantly greater than in age matched controls. The T1DM group had slightly higher peak 1 amplitudes as compared to both T2DM and controls, but this difference did not reach statistical significance.
Figure 2.

Diabetes elevates ABR thresholds in aging CBA/CaJ mice. (A-F) ABR thresholds at 3, 6, 12, 24, 32, and 48 kHz, respectively. Longitudinal ABR threshold elevations were compared by two-way, repeated measures ANOVA relative to the baseline starting thresholds. A significant main effect of subject groups was found, and significant changes were found at all frequencies for post-hoc pairwise comparisons with the baseline thresholds for the T2DM subject group. Data presented as mean ± SEM. * p < 0.05, ** p< 0.01, *** p< 0.001. n, number of mice in each group.
Figure 3.
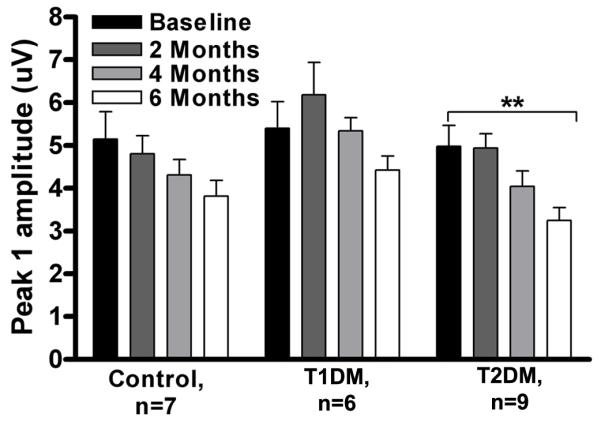
Influence of two types of diabetes on declining ABR peak-1 amplitudes with 12 kHz stimuli at 80 dB SPL. The peak-1 amplitude declined with age in all three subject groups; however, it only dropped significantly in T2DM by 6 months. Longitudinal data presented as mean ± SEM. ** p< 0.01 compared to baseline. n, number of mice in each group.
Distortion product otoacoustic emissions
To compare the three subject groups, the DPOAE levels were plotted, along with average noise floors, as a function of f2 frequency (Fig. 4A). Overall, there was a main effect of experimental groups (F= 20.21, p<0.0001, df= 2) and of DPOAE frequency (F=73.55, p<0.0001, df=24). A general decrease in DPOAE amplitude was observed mostly at high frequencies for both DM types. To compare subject groups and times, the DPOAE frequencies were grouped into three frequencies bands: low (5.6-10 kHz), middle (11-22 kHz) and high (23-45 kHz), with the response amplitudes averaged across mice in each group for each frequency, then the responses were averaged across frequencies within the band (Figs. 4B-D). For low frequencies only the T2DM group showed a significant drop in DP-amplitude at 6 months compared to baseline (Fig. 4B). The largest change in amplitude drop between baseline vs. 6 months occurred in the high frequency band (Fig. 4D). The control group had no statistically significant changes in DPOAE amplitudes within the 6 month experiment.
Figure 4.

Diabetes induces drops in DPOAE amplitude levels in middle age CBA/CaJ mice. A) Provides cross sectional data of DPOAE amplitude level as a function of the f2 frequency (kHz) at 6 months. B) Longitudinal data for low frequencies (5.6-10 kHz); C) Middle frequencies (11-22 kHz); and D) High frequencies (23-45 kHz). The two-way repeated-measures ANOVA showed significantly smaller DPOAE amplitudes by 6 months for T2DM at low frequencies, and for both diabetic groups at high frequencies, compared to the starting longitudinal baseline. Data presented as mean ±SEM. * p < 0.05, ** p< 0.01, *** p< 0.001. n, number of mice in each group.
Near-field Auditory Evoked Potential (NFAEP) to Tones and Noise in the Inferior Colliculus
Tone and broadband noise evoked neurophysiological responses were measured directly from dorsal and ventral IC locations for all three subject groups at the end of the 6 month experimental period. Figure 5A-B shows the growth in the NFAEP as a function of sound intensity for the controls in dorsal and ventral IC. T1DM showed higher IC response amplitudes in both locations in the IC (Figs. 5C-D), while T2DM exhibited similar response magnitude (Figs. 5E-F) as controls. The IC amplitude responses were plotted as a function of frequency for all experimental groups in dorsal and ventral locations in the IC (Figs. 6A-B). Overall there was a statistically significant effect of experimental groups on mean IC response magnitudes in both locations as determined by two-way ANOVA (Table 1). T1DM had considerably larger magnitude of the response at both locations in the IC. Bonferroni post hoc tests revealed that T1DM had significantly larger responses at all but one of the measured frequencies for ventral IC, while only for 36 kHz for dorsal IC, relative to controls (Table 1). The responses to the noise were also higher for the T1DM for both IC locations (Fig. 6).
Figure 5.

Tone and broadband noise-burst burst intensity function differences in the IC at 6 months. A) Dorsal, and B) Ventral IC NFAEP response amplitude RMS values for the control group. C) and D) IC response amplitudes for T1DM. E) and F) response amplitudes values for T2DM. Tone-burst frequencies from 3 to 48 kHz and noise burst responses are indicated by key symbols. Error bars are not shown due to a large variability in amplitude of responses, which resulted in overlapping standard errors. The insert in figure B shows representative ventral IC response to noise burst stimuli at 80 dB SPL. PP - positive peak, PN - negative peak. Scale bar = 50μV.
Figure 6.

Diabetes alters intensity coding by elevating responses to broad band noise and tone bursts in the IC at 80 dB SPL. A) Dorsal and B) Ventral IC NFAEP response amplitudes (RMS). T1DM showed higher IC response amplitudes at both locations in the IC, and this difference was statistically significant for experimental groups as determined by a two-way ANOVA (dorsal IC: F= 33.44, p<0.0001, df =2; ventral IC: F=38.47, p<0.0001, df=2). Post hoc Bonferroni tests showed that T1DM was significantly different vs. the control group at 3, 20, 24, 32, 36 and 48 kHz in the ventral IC, and at 36 kHz for dorsal IC. Data presented as mean ±SEM. n, number of mice in each group.
Table 1. Statistical comparisons of main effects of IC neurophysiological responses (thresholds and amplitudes) of the three subject groups.
| Dorsal IC | Ventral IC | |||
|---|---|---|---|---|
| Amplitude | Threshold | Amplitude | Threshold | |
| Main effect - Subject group | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 |
| F = 33.44 | F =24.50 | F = 38.47 | F = 36.04 | |
|
| ||||
| Bonferroni post hoc tests | T1DM vs Control | T1DM vs Control | T1DM vs Control | T1DM vs Control |
| 36 kHz, p<0.05 | 32 kHz, p<0.05 | 3 kHz, p<0.01 | 6 kHz, p<0.05 | |
| T2DM vs Control | 20 kHz, p<0.01 | 20 kHz, p<0.05 | ||
| 48 kHz, p<0.05 | 24 kHz, p<0.01 | 24 kHz, p<0.05 | ||
| 32 kHz, p<0.05 | 36 kHz, p<0.05 | |||
| 36 kHz, p<0.01 | T2DM vs Control | |||
| 48 kHz, p<0.05 | 20kHz, p<0.05 | |||
| 48 kHz, p<0.001 | ||||
Two Way ANOVA main effects: degrees of freedom, df=2.
Figure 7 summarizes the mean tone and noise thresholds for both IC locations (Figs. 7A-B) and ABR thresholds to tones and noise at 6 months (measured about 5 days before the IC recordings, Fig. 7C). T1DM and T2DM groups exhibited about 15-25 dB better thresholds in both IC locations compared to controls (Figs. 7A-B). There was a statistically significant effect of experimental groups on mean thresholds in both locations by two-way ANOVA (Table 1). Post hoc Bonferroni tests in dorsal IC showed that controls had significantly higher thresholds vs. T1DM for 32 kHz, and vs. T2DM for 48 kHz; while in ventral IC, controls had significant differences for 6, 20, 24, and 36 kHz vs. T1DM; and 20 and 48 kHz vs. T2DM (Table 1). Interestingly, in contrast to the IC thresholds, ABR thresholds were elevated for both types of diabetes as compare to controls (F= 8.66, p<0.001, df=2). Post hoc Bonferroni tests showed that ABR thresholds in controls were significantly lower at 48 kHz (p<0.05) compared to T2DM. In sum, auditory midbrain (IC) responses had significantly higher magnitudes for T1DM (predominantly in ventral IC). In addition, even though ABR thresholds were elevated in the diabetics, IC thresholds were lower for both type of diabetes, compared to their age-matched control mice.
Figure 7.

Diabetes increases thresholds assayed by ABRs, but decreases broad-band noise and tone burst thresholds in the IC. A) Dorsal and B) Ventral IC NFAEP thresholds; and C) ABR thresholds, recorded at 6 months following the start of the experiment (2-5 days prior to the IC recordings). Both diabetic subject groups showed significantly lower thresholds in both IC locations, while ABR thresholds were elevated compared to the control group, particularly at high frequencies. Statistically significant effects for subject groups was determined by two-way ANOVA (dorsal IC: F= 24.50, p<0.0001, df =2; ventral IC: F=36.04, p<0.0001, df=2). Post hoc Bonferroni tests in dorsal IC showed that controls had significantly higher thresholds vs. T1DM for 32 kHz, and vs. T2DM for 48 kHz. In ventral IC, controls were significantly different for 6, 20, 24, 32 and 36 kHz vs. T1DM, and 20 and 48 kHz vs. T2DM. Significant elevation of ABR thresholds for both types of diabetes was verified by two-way ANOVA (F= 8.66, p<0.001, df=2). Post hoc Bonferroni tests showed that ABR thresholds in the T2DM group were significantly higher at 48 kHz compared to controls. Data presented as mean ±SEM. n, number of mice in each group. For the ABR data, number of mice in each group was the same as in Fig. 2.
DISCUSSION
The results indicate that both types of diabetes mellitus cause exacerbation of hearing loss in aging CBA/CaJ mice. However, it is important to note that progression of hearing loss in the two types of diabetes has different characteristics. In particular, the ABR thresholds and the ABR peak-1 amplitude (Henry and Price 1994; Henry 2002) was changed significantly only in T2DM. In contrast to this peripheral hearing loss, the IC showed increases in response magnitudes in T1DM (Fig. 6) and better (lower) IC thresholds for both diabetic groups compared to the age-matched controls (Fig. 7). The effects of Type 2 diabetes on the peripheral auditory system of aging CBA/CaJ mice resemble effects recently reported in older Type 2 diabetic human subjects (Frisina et al. 2006).
Induction of T1DM and T2DM and Hearing Loss
Effects of duration of diabetes on auditory function and cochlear anatomical alterations were studied in rats where diabetes was induced by STZ injection (Smith et al. 1995; Raynor et al. 1995), in genetically predisposed strains (Mordes and Rossini 1981; Ishikawa et al. 1995; McQueen et al. 1999), or using a high fat diet (Rust et al. 1992). However, these studies did not include the relationships of age, diabetes, and hearing loss. Ishikawa et al. (1995) studied the effect of T2DM on the WBN/Kob strain of rats that are susceptible to diabetes at about 12 months (early middle age). The authors used a different strain of Wistar rats as age-matched controls to compare anatomical alterations in the cochlea. None of the studies compared effect of different types of diabetes on auditory function. There are several transgenic mouse models that display both diabetes/obesity and sensorineural hearing loss phenotypes. In particular, homozygotes for the tubby mutation develop obesity, retinal degradation and progressive hearing loss, via mutations in the gene coding for the high-affinity thiamine transporter Slc19a2, which underlies the clinical syndrome known as thiamine-responsive megaloblastic anemia, which is characterized by anemia and diabetes (Johnson et al. 2006; Liberman et al. 2006). Mutations in WFS1 that cause autosomal recessive Wolfram syndrome also cause diabetes and sensorineural hearing impairment at low frequencies (Cryns et al. 2003). Mutation of ALMS1 the gene that causes Alstrom syndrome in humans, leads in mice to neurosensory deficits after 8 months of age and metabolic defects including obesity, hyperinsulinemia and T2DM (Collin et al. 2005).
Physiological Hearing Changes: Humans and Previous Animal Studies
In some studies the evaluation of hearing impairment and diabetes in human subjects showed a gradually progressive, bilateral, high frequency hearing loss by using pure tone audiometry (Taylor and Irwin 1978; Kurien et al. 1989; Parving et al. 1990; Ferrer et al. 1991; Elamin et al. 2005; Sakuta et al. 2007). However others did not find any association of hearing loss with diabetes (Dalton et al. 1998; Gratton and Vazquez 2003; Salvinelli et al. 2004; Gates and Mills 2005). Yet, in some cases authors reported not only high frequency but also low frequency hearing loss in diabetic patients (Taylor and Irwin 1978; Tay et al. 1995; Kurien et al. 1989; Frisina et al. 2006). These human data are in agreement with our ABR threshold results where the T2DM group exhibited significant threshold elevation at all frequencies, whereas controls revealed only a lesser high frequency hearing loss with age (Fig. 2). The findings in non-diabetic (control) CBA/CaJ mice are similar to sensory-neuronal type human presbycusis when high frequency hearing loss is predominant (Kim et al. 2002; Jacobson et al. 2003). Longitudinal studies on diabetes susceptible WBN/Kob rats showed mean ABR threshold elevation compared to age-matched Wistar rats (Ishikavwa et al. 1995) but no detailed analysis of frequency effects was presented.
Effects of diabetes on otoacoustic emissions showed lower amplitudes in diabetic human subjects relative to control groups in some studies (Sasso et al. 1999; Frisina et al. 2006), while others showed no effect (de Leo et al. 1997) or mixed results (Erdem et al. 2003). The present study found that DPOAE amplitudes were significantly lower at higher frequencies for both types of diabetes, but only T2DM had reduced DPOAE responses at low frequencies (Figs. 4B-D). In the present investigation, the T2DM model was created through induced obesity. Thus, the contributions of hyperglycemia, insulin resistance, and hyperlipidemia could influence outer hair cell damage or other cochlear pathologies such as stria vascularis or fibrocyte damage, as a result of metabolic alterations (Hsueh et al. 2007).
For central auditory function, STZ-induced diabetes in rats (less then 5 months) showed no effect on ABR interpeak latencies, but increased peripheral (I and II) and central (IV) peak latencies (Notvest and Inserra 1987; Rubini et al. 1992). For longer duration of STZ induced diabetes (over 6 months), prolongation of central transmission times (increased late interpeak latencies) were observed in rats (Biessels et al. 1999, 2001; Manschot et al. 2003). Similarly, in diabetic human subjects ABR latencies were longer compared to their controls (Tay et al. 1995; de Leo et al. 1997, Diaz de Leon Morales et al. 2005). Donald et al. (1981) reported reduction of wave-1 ABR amplitude in diabetic humans, prolongation of wave 5 latency, and interwave 1-5 and 1-3 latency prolongations. In the present report, we did not find any effect of either type of diabetes on peak-1 latencies (data not shown). However, the amplitude of peak 1 was significantly reduced in T2DM by 6 months. T1DM showed increased magnitude of peak 1 by the second month that was followed by a decline towards the end of the experiment (Fig. 3). Interestingly, in an STZ-induced diabetic rat model, peak-1 latencies decreased significantly by the second month after induction of pathology, and reached the level of age matched non-diabetic controls by 6 months (Biessels et al. 1999). Frisina et al. (2006) used a comprehensive battery to evaluate the effects of T2DM on auditory processing in humans (HINT, supra-threshold gap detection), and showed effects of diabetes on both peripheral and central auditory function in aged subjects. The present investigation used NFAEP responses directly from the midbrain (IC) in aged diabetic CBA/CaJ mice, and the results indicated significantly lower thresholds in IC for both types of diabetes (Fig. 7). In addition, T1DM had higher response magnitudes in both locations of the IC (Fig. 6).
Thus, our data indicate declines in peripheral auditory processing, but T1DM increases neuronal activity in the central auditory system at the level of the auditory midbrain. This seemingly paradoxical finding might be attributable to a diabetes-induced acceleration of age-related hearing loss, involving a selective loss of inhibition at the level of the brainstem, including the IC. The latter has been reported for age-related hearing loss in rats and mice, particularly for the most prominent brainstem inhibitory neurotransmitter, GABA (Caspary et al. 1990, 1995, 1999; Willott and Carlson 1995; Willott et al. 1994, 2001, Frisina and Walton, 2006). An alternative explanation could be an enhancement of excitatory synchrony or excitatory drive resulting from brainstem circuitry alterations. Future studies are required to more fully understand mechanisms of auditory brainstem function in aged diabetic subjects.
Summary and Conclusions
Induction of diabetes significantly accelerates hearing impairment in middle aged CBA/CaJ male mice assayed by ABR thresholds and DPOAE levels over a period of 6 months. Interestingly, auditory midbrain (IC) responses had a higher magnitude for T1DM, suggesting an increased age-linked selective loss of inhibition or increase in excitation at that level of the auditory brainstem. In line with this, both types of diabetes exhibited lower IC thresholds, compared to their age-matched controls, despite having higher ABR thresholds, which reflect auditory periphery sound sensitivity. Larger IC response magnitudes in the T1DM suggests differential, complex mechanisms for the acceleration of age-related hearing loss in the two types of diabetes investigated in the present report. The results of the present investigation also show that both types of diabetes accelerate different aspects of age related hearing changes in middle age CBA/CaJ mice, which may serve as valuable models to probe underling metabolic alterations and discover the anatomical and neurochemical pathologies of diabetes that negatively impact auditory and sensory processing.
Acknowledgments
We thank John Housel for collecting the ABR data, and Enza Daugherty for project assistance, and three anonymous reviewers for very helpful critiques. This research was funded by NIH Grant P01 AG09524 from the National Institute on Aging, and P30 DC05409 from the National Institute on Deafness & Communication Disorders.
Abbreviations
- ANOVA
Analysis of variance
- ABR
Auditory brainstem responses
- BW
Body weight
- DPOAEs
Distortion product otoacoustic emissions
- DM
Diabetes mellitus
- EEG
Electro-encephalography
- GM
Geometric mean
- HINT
Hearing-in-noise test
- IC
Inferior colliculus
- IHC
Inner hair cells
- IP
Intraperitoneal injection
- NFAEP
Near field Auditory Evoked Potential
- OAEs
Otoacoustic emissions
- OHC
Outer hair cells
- RM
Repeated measures
- RMS
Root mean square voltage
- IAC
Soundproof acoustic chamber
- SPL
Sound pressure level
- SPF
Specific pathogen free
- STZ
Streptozotocin
- T1DM
Type 1 diabetes mellitus
- T2DM
Type 2 diabetes mellitus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen PD, Burkard RF, Ison JR, Walton JP. Impaired gap encoding in aged mouse inferior colliculus at moderate but not high stimulus levels. Hear. Res. 2003;186:17–29. doi: 10.1016/s0378-5955(03)00300-9. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association Standards of Medical Care in Diabetes. Diabetes care. 2006;29:4–42. doi: 10.2337/dc06-0805. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Cristino NA, Rutten GJ, Hamers FP, Erkelens DW, Gispen WH. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats. Course of development and effects of insulin treatment. Brain. 1999;122:757–68. doi: 10.1093/brain/122.4.757. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Smale S, Duis SE, Kamal A, Gispen WH. The effect of gamma-linoleic acid-alpha-lipoic acid on functional deficits in the peripheral and central nervous system of streptozotocin-diabetic rats. J. Neurol. Sci. 2001;182:99–106. doi: 10.1016/s0022-510x(00)00456-1. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, ter Laak MP, Kamal A, Gispen WH. Effects of the Ca2+ antagonist nimodipine on functional deficits in the peripheral and central nervous system of streptozotocin-diabetic rats. Brain Res. 2005;21:86–93. doi: 10.1016/j.brainres.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH. Ageing and diabetes: implications for brain function. Eur. J. Pharmacol. 2002;44:1–14. doi: 10.1016/s0014-2999(02)01486-3. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Holder TM, Hughes LF, Milbrandt JC, McKernan RM, Naritoku DK. Age-related changes in GABAa receptor subunit composition and function in rat auditory system. Neuroscience. 1999;93:307–312. doi: 10.1016/s0306-4522(99)00121-9. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Milbrandt JC, Helfert RH. Central auditory aging: GABA changes in the inferior colliculus. Exp. Gerontol. 1995;30:349–360. doi: 10.1016/0531-5565(94)00052-5. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Raza A, Lawhorn Armour BA, Pippin J, Arneric SP. Immunocytochemical and neurochemical evidence for age-related loss of GABA in the inferior colliculus: implications for neural presbycusis. J. Neurosci. 1990;10:2363–2372. doi: 10.1523/JNEUROSCI.10-07-02363.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, Naggert JK. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum. Mol. Genet. 2005;14:2323–33. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns K, Thys S, Van Laer L, Oka Y, Pfister M, Van Nassauw L, Smith RJ, Timmermans JP, Van Camp G. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells. Histochem. Cell Biol. 2003;119:247–56. doi: 10.1007/s00418-003-0495-6. [DOI] [PubMed] [Google Scholar]
- Dalton DS, Cruickshanks KJ, Klein R, Klein BE, Wiley TL. Association of NIDDM and hearing loss. Diabetes Care. 1998;21:1540–4. doi: 10.2337/diacare.21.9.1540. [DOI] [PubMed] [Google Scholar]
- de Espana R, Biurrun O, Lorente J, Traserra J. Hearing and diabetes. ORL. J. Otorhinolaryngol Relat Spec. 1995;57:325–7. doi: 10.1159/000276774. [DOI] [PubMed] [Google Scholar]
- Di Leo MA, Di Nardo W, Cercone S, Ciervo A, Lo Monaco M, Greco AV, Paludetti G, Ghirlanda G. Cochlear dysfunction in IDDM patients with subclinical peripheral neuropathy. Diabetes Care. 1997;20:824–8. doi: 10.2337/diacare.20.5.824. [DOI] [PubMed] [Google Scholar]
- Diaz de Leon-Morales LV, Jauregui-Renaud K, Garay-Sevilla ME, Hernandez-Prado J, Malacara-Hernandez JM. Auditory impairment in patients with type 2 diabetes mellitus. Arch .Med. Res. 2005;36:507–10. doi: 10.1016/j.arcmed.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Donald MW, Bird CE, Lawson JS, Letemendia FJ, Monga TN, Surridge DH, Varette-Cerre P, Williams DL, Williams DM, Wilson DL. Delayed auditory brainstem responses in diabetes mellitus. J. Neurol. Neurosurg. Psychiatry. 1981;44:641–4. doi: 10.1136/jnnp.44.7.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin A, Fadlallah M, Tuevmo T. Hearing loss in children with type 1 diabetes. Indian Pediatr. 2005;42:15–21. [PubMed] [Google Scholar]
- Erdem T, Ozturan O, Miman MC, Ozturk C, Karatas E. Exploration of the early auditory effects of hyperlipoproteinemia and diabetes mellitus using otoacoustic emissions. Eur. Arch. Otorhinolaryngol. 2003;260:62–6. doi: 10.1007/s00405-002-0519-1. [DOI] [PubMed] [Google Scholar]
- Ferrer JP, Biurrun O, Lorente J, Conget JI, de Espana R, Esmatjes E, Gomis R. Auditory function in young patients with type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 1991;11:17–22. doi: 10.1016/0168-8227(91)90136-2. [DOI] [PubMed] [Google Scholar]
- Fowler PD, Jones NS. Diabetes and hearing loss. Clin. Otolaryngol. 1999;24:3–8. doi: 10.1046/j.1365-2273.1999.00212.x. [DOI] [PubMed] [Google Scholar]
- Frisina ST, Mapes F, Kim S, Frisina DR, Frisina RD. Characterization of hearing loss in aged type II diabetics. Hear. Res. 2006;211:103–13. doi: 10.1016/j.heares.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisina RD, Walton JP. Age-related structural and functional changes in the cochlear nucleus. Hear. Res. 2006;217:216–33. doi: 10.1016/j.heares.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Frisina RD, Walton JP, Lynch-Armour MA, Byrd JD. Inputs to a physiologically characterized region of the inferior colliculus of the young adult CBA mouse. Hear Res. 1998;115:61–81. doi: 10.1016/s0378-5955(97)00176-7. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Cureoglu S, Schachern PA, Kusunoki T, Oktay MF, Fukushima N, Paparella MM, Harada T. Cochlear changes in patients with type 1 diabetes mellitus. Otolaryngol Head Neck Surg. 2005;133:100–6. doi: 10.1016/j.otohns.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Cureoglu S, Schachern PA, Paparella MM, Harada T, Oktay MF. Effects of type 2 diabetes mellitus on cochlear structure in humans. Arch. Otolaryngol. Head. Neck. Surg. 2006;132:934–8. doi: 10.1001/archotol.132.9.934. [DOI] [PubMed] [Google Scholar]
- Gates GA, Mills JH. Presbycusis. Lancet. 2005;366:1111–20. doi: 10.1016/S0140-6736(05)67423-5. [DOI] [PubMed] [Google Scholar]
- Gratton MA, Vazquez AE. Age-related hearing loss: current research. Curr. Opin. Otolaryngol. Head. Neck Surg. 2003;11:367–71. doi: 10.1097/00020840-200310000-00010. [DOI] [PubMed] [Google Scholar]
- Guimaraes P, Zhu X, Cannon T, Kim S, Frisina RD. Sex differences in distortion product otoacoustic emissions as a function of age in CBA mice. Hear. Res. 2004;192:83–89. doi: 10.1016/j.heares.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Henry KR. Sex- and age-related elevation of cochlear nerve envelope response (CNER) and auditory brainstem response (ABR) thresholds in C57BL/6 mice. Hear. Res. 2002;170:107–115. doi: 10.1016/s0378-5955(02)00391-x. [DOI] [PubMed] [Google Scholar]
- Henry KR, Price JM. Amplitude enhancement is seen in the cochlear nerve but not at, or before, the afferent synapse. Hear. Res. 1994;79:190–196. doi: 10.1016/0378-5955(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Hsueh W, Abel ED, Breslow JL, Maeda N, Davis RC, Fisher EA, Dansky H, McClain DA, McIndoe R, Wassef MK, Rabadán-Diehl C, Goldberg IJ. Recipes for creating animal models of diabetic cardiovascular disease. Circ. Res. 2007;100:1415–27. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Naito Y, Taniguchi K. Hearing impairment in WBN/Kob rats with spontaneous diabetes mellitus. Diabetologia. 1995;38:649–55. doi: 10.1007/BF00401834. [DOI] [PubMed] [Google Scholar]
- Jacobson M, Kim S, Romney J, Zhu X, Frisina RD. Contralateral suppression of distortion-product otoacoustic emissions declines with age: a comparison of findings in CBA mice with human listeners. Laryngoscope. 2003;113:1707–13. doi: 10.1097/00005537-200310000-00009. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091:79–88. doi: 10.1016/j.brainres.2006.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-H, Frisina DR, Frisina RD. Effects of age on contralateral suppression of distortion product otoacoustic emissions in human listeners with normal hearing. Audiol. Neurootol. 2002;7:348–357. doi: 10.1159/000066159. [DOI] [PubMed] [Google Scholar]
- Kurien M, Thomas K, Bhanu TS. Hearing threshold in patients with diabetes mellitus. J. Laryngol. Otol. 1989;103:164–168. doi: 10.1017/s0022215100108345. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Tartaglini E, Fleming JC, Neufeld EJ. Deletion of SLC19A2, the high affinity thiamine transporter, causes selective inner hair cell loss and an auditory neuropathy phenotype. J. Assoc. Res. Otolaryngol. 2006;7:211–7. doi: 10.1007/s10162-006-0035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia CA, Campos CA. Diabetes mellitus as etiological factor of hearing loss. Bras Otorhinolaryngol (Engl Ed) 2005;71:208–14. doi: 10.1016/S1808-8694(15)31312-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manschot SM, Gispen WH, Kappelle LJ, Biessels GJ. Nerve conduction velocity and evoked potential latencies in streptozotocin-diabetic rats: effects of treatment with an angiotensin converting enzyme inhibitor. Diabetes Metab. Res. Rev. 2003;19:469–77. doi: 10.1002/dmrr.401. [DOI] [PubMed] [Google Scholar]
- McQueen CT, Baxter A, Smith TL, Raynor E, Yoon SM, Prazma J, Pillsbury HC., 3rd Non-insulin-dependent diabetic microangiopathy in the inner ear. J. Laryngol. Otol. 1999;113:13–8. doi: 10.1017/s0022215100143051. [DOI] [PubMed] [Google Scholar]
- Mordes JP, Rossini AA. Animal models of diabetes. Am. J. Med. 1998;70:353–360. doi: 10.1016/0002-9343(81)90772-5. [DOI] [PubMed] [Google Scholar]
- Nageris B, Hadar T, Feinmesser M, Elidan J. Cochlear histopathologic analysis in diabetic rats. Am. J. Otol. 1998;19:63–5. [PubMed] [Google Scholar]
- Nakae S, Tachibana M. The cochlea of the spontaneously diabetic mouse. II. electron microscopic observations of non-obese diabetic mice. Arch. Otorhinolaryngol. 1986;243:313–316. doi: 10.1007/BF00460208. [DOI] [PubMed] [Google Scholar]
- Notvest RR, Inserra JJ. Tolrestat, an aldose reductase inhibitor, prevents nerve dysfunction in conscious diabetic rats. Diabetes. 1987;36:500–4. doi: 10.2337/diab.36.4.500. [DOI] [PubMed] [Google Scholar]
- Paik SG, Fleischer N, Shin SI. Insulin-dependent diabetes mellitus induced by subdiabetogenic doses of streptozotocin: obligatory role of cell-mediated autoimmune processes. Proc. Natl. Acad. Sci. USA. 1980;77:6129–33. doi: 10.1073/pnas.77.10.6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham K, Sun XM, Kim DO. Distortion product otoacoustic emissions in the CBA/J mouse model of presbycusis. Hear. Res. 1999;134:29–38. doi: 10.1016/s0378-5955(99)00059-3. [DOI] [PubMed] [Google Scholar]
- Parving A, Elberling C, Balle V, Parbo J, Dejgaard A, Parving HH. Hearing disorders in patients with insulin-dependent diabetes mellitus. Audiology. 1990;29:113–121. doi: 10.3109/00206099009072844. [DOI] [PubMed] [Google Scholar]
- Raynor EM, Carrasco VN, Prazma J, Pillsbury HC. An assessment of cochlear hair-cell loss in insulin-dependent diabetes mellitus diabetic and noise-exposed rats. Arch. Otolaryngol. Head. Neck. Surg. 1995;121:452–6. doi: 10.1001/archotol.1995.01890040074012. [DOI] [PubMed] [Google Scholar]
- Rózańska-Kudelska M, Chodynicki S, Kinalska I, Kowalska I. Hearing loss in patients with diabetes mellitus type II. Otolaryngol. Pol. 2002;56:607–10. [PubMed] [Google Scholar]
- Rubini R, Biasiolo F, Fogarolo F, Magnavita V, Martini A, Fiori MG. Brainstem auditory evoked potentials in rats with streptozotocin-induced diabetes. Diabetes Res. Clin. Pract. 1992;16:19–25. doi: 10.1016/0168-8227(92)90131-a. [DOI] [PubMed] [Google Scholar]
- Rust KR, Prazma J, Triana RJ, Michaelis OE, 4th, Pillsbury HC. Inner ear damage secondary to diabetes mellitus. II. Changes in aging SHR/N-cp rats. Arch. Otolaryngol. Head Neck Surg. 1992;118:397–400. doi: 10.1001/archotol.1992.01880040059010. [DOI] [PubMed] [Google Scholar]
- Sakuta H, Suzuki T, Yasuda H, Ito T. Type 2 diabetes and hearing loss in personnel of the Self-Defense Forces. Diabetes Res. Clin. Pract. 2007;75:229–34. doi: 10.1016/j.diabres.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Salvinelli F, Miele A, Casale M, Greco F, D’Ascanio L, Firrisi L, Trivelli M, Petitti T, Aloe L, Pozzilli P. Hearing Thresholds In Patients With Diabetes. The Internet Journal of Otorhinolaryngology. 2004;3(1) [Google Scholar]
- Sasso FC, Salvatore T, Tranchino G, Cozzolino D, Caruso AA, Persico M, Gentile S, Torella D, Torella R. Cochlear dysfunction in type 2 diabetes: a complication independent of neuropathy and acute hyperglycemia. Metabolism. 1999;48:346–50. doi: 10.1016/s0026-0495(99)90141-5. [DOI] [PubMed] [Google Scholar]
- Shafrir E. Diabetes in animals: contribution to the understanding of diabetes by study of its etiopathology in animal models. In: Porte D Jr, Sherwin RS, editors. Ellenberg and Rifkin’s diabetes mellitus; theory and practice. 5th ed. Appleton & Lange; Stamford: 1997. pp. 301–348. [Google Scholar]
- Smith TL, Raynor E, Prazma J, Buenting JE, Pillsbury HC. Insulin-dependent diabetic microangiopathy in the inner ear. Laryngoscope. 1995;105:236–40. doi: 10.1288/00005537-199503000-00002. [DOI] [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J. Acoust. Soc. Am. 1997;101:3546–53. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–7. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- Tadros SF, D’Souza M, Zettel ML, Zhu X, Lynch-Erhardt M, Frisina RD. Serotonin 2B receptor: upregulated with age and hearing loss in mouse auditory system. Neurobiol. Aging. 2007;28:1112–23. doi: 10.1016/j.neurobiolaging.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Tay HL, Ray N, Ohri R, Frootko NJ. Diabetes mellitus and hearing loss. Clin Otolaryngol. Allied, Sci. 1995;20:130–4. doi: 10.1111/j.1365-2273.1995.tb00029.x. [DOI] [PubMed] [Google Scholar]
- Taylor IG, Irwin J. Some audiological aspects of diabetes mellitus. J. Laryngol. Otol. 1978;92:99–113. doi: 10.1017/s0022215100085108. [DOI] [PubMed] [Google Scholar]
- Triana RJ, Suits GW, Garrison S, Prazma J, Brechtelsbauer PB, Michaelis OE, Pillsbury HC. Inner ear damage secondary to diabetes mellitus. I. Changes in adolescent SHR/N-cp rats. Arch. Otolaryngol. Head Neck Surg. 1991;117:635–40. doi: 10.1001/archotol.1991.01870180071014. [DOI] [PubMed] [Google Scholar]
- Wade GN, Gray JM, Bartness TJ. Gonadal influences on adiposity. Int. J. Obes. 1985;9(Suppl.1):83–92. [PubMed] [Google Scholar]
- Walton JP, Frisina RD, O’Neill WE. Age-related alteration in neural processing of silent gaps in the central nucleus of the inferior colliculus in the CBA mouse model of presbycusis. J. Neuroscience. 1998;18:2764–2776. doi: 10.1523/JNEUROSCI.18-07-02764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott JF, Carlson S. Modification of the acoustic startle response in hearing-impaired C57BL/6J mice: prepulse augmentation and prolongation of prepulse inhibition. Behav Neurosci. 1995;109:396–403. doi: 10.1037//0735-7044.109.3.396. [DOI] [PubMed] [Google Scholar]
- Willott JF, Carlson S, Chen H. Prepulse inhibition of the startle response in mice: relationship to hearing loss and auditory system plasticity. Behav. Neurosci. 1994;108:703–13. doi: 10.1037//0735-7044.108.4.703. [DOI] [PubMed] [Google Scholar]
- Willott JF, Chisolm TH, Lister JJ. Modulation of presbycusis: current status and future directions. Audiol. Neurootol. 2001;6:231–249. doi: 10.1159/000046129. [DOI] [PubMed] [Google Scholar]
- Zhang L, Reidy SP, Nicholson TE, Lee HJ, Majdalawieh A, Webber C, Stewart BR, Dolphin P, Ro HS. The role of AEBP1 in sex-specific diet-induced obesity. Mol. Med. 2005;11:39–47. doi: 10.2119/2005-00021.Ro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Vasilyeva ON, Kim S, Jacobson M, Romney J, Waterman MS, Tuttle D, Frisina RD. Auditory efferent feedback system deficits precede age-related hearing loss: Contralateral suppression of otoacoustic emissions in mice. J. Comp. Neurol. 2007;10:593–604. doi: 10.1002/cne.21402. [DOI] [PubMed] [Google Scholar]