

Abstract
Despite its widespread application in nanomedicine, poly(ethylene glycol) (PEG) is seldom used for covalent modification of ligands for G protein-coupled receptors (GPCRs) due to potential steric complications. In order to study the influence of PEG chains on the biological activity of GPCR ligands bound to a common macromolecular carrier, we prepared a series of G3 polyamidoamine (PAMAM) dendrimers derivatized with Alexa Fluor 488, varying numbers of PEG550/PEG750/PEG2000, and nucleoside moieties derived from the A2A adenosine receptor (AR) agonist CGS21680 (2-[4-(2-carboxylethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine). These dendrimer conjugates were purified by size exclusion chromatography and characterized by 1H NMR and MALDI MS. In radioligand binding assays, some PAMAM-PEG conjugates showed enhanced subtype-selectivity at the human A2A AR compared to monomeric ligands of comparable affinity. The functional potency was measured in the A2A AR-mediated activation of adenylate cyclase and inhibition of ADP-induced platelet aggregation. Interestingly, the dendrimer conjugate 10c bearing 11 PEG750 chains (out of theo. 32 amino end groups) and 14 nucleoside moieties was 5-fold more potent in A2A AR–mediated stimulation of cyclic AMP formation than 10d with four PEG2000 chains and 21 nucleosides, although the binding affinities of these two compounds were similar. Thus, a relatively small (≤10 nm) multivalent ligand 10c modified for water solubility maintained high potency and displayed increased A2A AR binding selectivity over the monomeric nucleosides. Longer PEG chains reduced affinity at the A2A AR. The current study demonstrates the feasiblity of using short PEG chains in the design of carriers that target ligand-receptor interactions.
Introduction
Nanocarriers such as polymeric/inorganic nanoparticles, liposomes, micelles, dendrimers, and nanotubes present a superior means to tackle various challenging issues in biomedical research, such as in vivo lability and insolubility of pharmaceutical substances (1). Despite the excellent functional improvements over small molecules as shown in numerous examples, the ultimate success of recruiting these unnatural nanostructures for therapy/diagnosis will depend on meeting the rigorous safety standards that apply to humans.
Dendritic scaffolds (2) offer a robust spatial control, particularly valuable when the imparted multivalency (3) can significantly enhance the desired effects. Previously, we employed polyamidoamine (PAMAM) dendrimers (4) to conjugate multiple copies of a nucleoside-based agonist CGS21680 (2-[4-(2-carboxylethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine, 1, Figure 1 (5)) of the A2A adenosine receptor (AR) (6), a member of the G protein-coupled receptor (GPCR) family, in an effort to enhance the therapeutic potential through a multivalent effect (3). Generally, small-molecule agonists of GPCRs bind primarily at their heptahelical transmembrane domains (7), to trigger various intracellular signaling pathways. Diseases related to the actions of extracellular adenosine include cancer, arthritis, thrombosis, ischemia, and sleep disorders. Here, we report a simple PEGylation (8) strategy to improve the bioavailability (9) of dendritic carriers for GPCR ligands, which did not impose significant sterics, and manifested potent biological activities in comparison to the monovalent species, using a third generation (G3) PAMAM dendrimer as an example.
Figure 1.
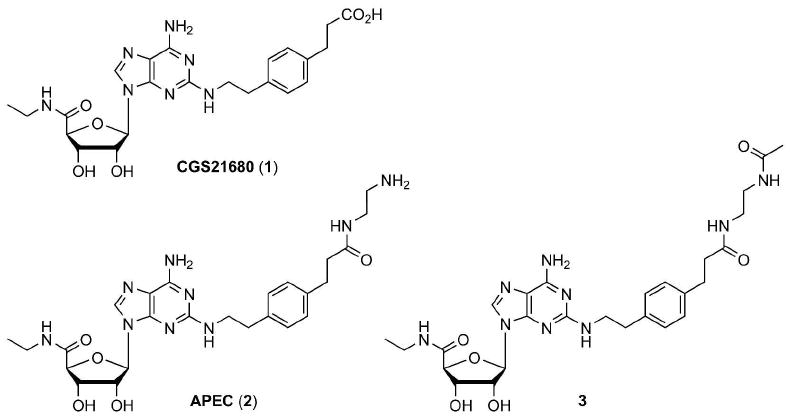
Structures of small molecule GPCR ligands (A2A AR agonists) 1-3 used as monomeric controls.
Experimental Procedures
Materials and Methods
Glassware was oven-dried and cooled in a desiccator before use. All reactions were carried out under a dry nitrogen atmosphere at room temperature. Solvents were purchased as anhydrous grade and used without further purification. Suppliers of the commercial compounds are listed as follows: amine-terminated G3 PAMAM dendrimer with an ethylenediamine core, poly(ethylene glycol) methyl ether (PEG, Mn = 550, 750, and 2000), acetic anhydride (Ac2O), and triethylamine were purchased from Aldrich; (benzotriazol-1-yl-oxy)tripyrrolidino-phosphonium hexafluorophosphate (PyBOP) was purchased from NovaBiochem; CGS21680 (1) hydrochloride hemihydrate was purchased from Tocris; Alexa Fluor 488 (AF488) 5-carboxylic acid 2,3,5,6-tetrafluorophenyl (TFP) ester (AF488-OTFP) was purchased from Invitrogen; dimethyl sulfoxide (DMSO)-d6 and D2O were purchased from Cambridge Isotope Laboratories. 2-[4-(2-Aminoethylaminocarbonylethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine (APEC, 2) bistrifluoroacetic acid was provided by NIMH Chemical Synthesis and Drug Supply Program. Compound 3 (11) and PEGylated PAMAM dendrimer conjugates (4a, 4b, 4c, and 4d (17)) were prepared as described before.
Preparative size exclusion chromatography (SEC) was performed on Bio-Beads® S-X1 beads (BIO-RAD, molecular weight (MW) operating range from 600-14000 Da), 200-400 mesh, with DMF (Aldrich 99.8%, anhydrous) as an eluent at ambient pressure.
NMR spectra were recorded on a Bruker DRX-600 spectrometer at 25.0 °C under an optimized parameter setting for each sample, unless otherwise mentioned. 1H NMR chemical shifts were measured relative to the residual solvent peak at 2.50 ppm in DMSO-d6 and at 4.80 ppm in D2O. Complete NMR peak assignments were made possible with 2D COSY and NOESY experiments. Each PAMAM dendrimer conjugate was not unimolecular, and thus, the characterization by NMR represented the average value from its polymeric distribution. The integration values were reported only for the peaks clearly resolved (i.e., with a relatively good baseline-separation) in the 1H NMR spectra and in two decimal places. Assignments of peaks for the PEGylated PAMAM dendrimer conjugates were based on the labeling method shown in Figure S2. A methylene peak of the G3 PAMAM dendrimer at 2.18 ppm in DMSO-d6 (“c”, see Figure S2, Supporting Information) was used as an internal standard (set at 120 H) for the integration, assuming 32 peripheral groups without any defects in the structure. Detailed methods used for the structural analysis of dendrimers by NMR in DMSO-d6 were described before (as Supporting Information (10, 17)). The stoichiometry and the average MWs of the PAMAM dendrimer conjugates determined as such are summarized in Table 1 and Figures S1 and S2 (Supporting Information).
Table 1.
Structural analysis of PAMAM dendrimer conjugates by 1H NMR and MALDI MS.
| cmpd | PEGa | NMRb | MALDI | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DHB matrix | THAP matrix | ||||||||||||||
| m | n | x | yc | z | MWd | Mne | Mwf | PDIg | zh | Mne | Mwf | PDIg | zh | ||
| 7a | 550 | 12.31 | 7.99 | 14.19 | 1 | 0 | 13021 | 10565 | 10974 | 1.04 | 0 | n.d.i | n.d.i | n.d.i | 0 |
| 7b | 550 | 12.64 | 14.34 | 7.45 | 1 | 0 | 16758 | 13472 | 13922 | 1.03 | 0 | 13520 | 13930 | 1.03 | 0 |
| 7d | 2000 | 43.89 | 4.61 | 19.32 | 1 | 0 | 17621 | 11844j | 12809j | 1.08j | 0 | 11875j | 12800j | 1.08j | 0 |
| 8c | 750 | 15.40 | 11.81 | 0 | 1 | 0 | 16325 | 11620 | 12033 | 1.04 | 0 | 11238 | 11637 | 1.04 | 0 |
| 8d | 2000 | 45.21 | 5.29 | 0 | 1 | 0 | 18470 | n.d.i | n.d.i | n.d.i | 0 | 9617j | 10444j | 1.09j | 0 |
| 9a | 550 | 12.34 | 8.17 | 14.28 | 1 | 1.20 | 13721 | 10800 | 11127 | 1.03 | 0.32 | 10911 | 11262 | 1.03 | N/A |
| 9b | 550 | 12.48 | 15.00 | 7.57 | 1 | 2.00 | 18026 | 14702 | 15457 | 1.05 | 3.19 | 15331 | 15831 | 1.03 | 3.95 |
| 9d | 2000 | 43.25 | 4.74 | 19.30 | 1 | 1.14 | 18294 | 12843j | 13329j | 1.04j | 1.08 | n.d.i | n.d.i | n.d.i | N/A |
| 10c | 750 | 15.75 | 11.32 | 0 | 1 | 14.05 | 22904 | 18575 | 19180 | 1.03 | 14.84 | 18848 | 19561 | 1.04 | 16.46 |
| 10d | 2000 | 41.16 | 5.60 | 0 | 1 | 20.51 | 27982 | 21607 | 22824 | 1.06 | N/A | 22493 | 23518 | 1.05 | 27.15 |
Mn of the commercial PEG monomethyl ether (Aldrich) originally used to prepare compound 4.
Based on 1H NMR integration determined in DMSO-d6. m = number of PEG repeat unit, n = number of PEG, x = number of acetamide, y = number of AF488, z = number of CGS21680 (see Scheme 1).
Based on the stoichiometry of addition.
Dendrimer conjugates were assumed to be made from the commercial PAMAM dendrimer with 32 peripheral amino groups and no structural defects. Counterions of sulfonate for AF488 were assumed to be triethylammonium groups.
Number-average molar mass.
Weight-average molar mass.
Polydispersity index.
Based on MALDI MS results. No matrix adduct was considered for the estimation. ΔMw between 9/10 and its precursor 7/8 was divided by the MW increase upon the covalent attachment of a CGS21680 ligand (481.5 Da) to the PAMAM amino group. Purification by SEC was assumed not to affect the MW distribution.
n.d. = not determined.
Mass range selected for the average MW calculations contained a part of the peak region corresponding to the half-size of each desired compound.
MALDI-TOF MS experiments were performed on an Applied Biosystems Voyager-DE STR spectrometer at the Mass Spectrometry Laboratory, University of Illinois. 2,5-Dihydroxybenzoic acid (DHB) or 2,4,6-trihydroxyacetophenone (THAP) was used as the matrix for the MALDI samples. Average MWs determined by MALDI are listed in Table 1 and the spectra are shown in Figures S7 and S8 (Supporting Information).
General Procedure for Acetylation of G3 PAMAM-PEG Dendrimer Conjugates
PEGylated G3 PAMAM dendrimer 4a, 4b, or 4d was dried in vacuo extensively, and then was dissolved in DMSO-d6. To this stirred dendrimer solution was added triethylamine, followed by a 0.317 M solution of Ac2O in DMSO-d6 (30.0 μL/1.00 mL, total volume). The final concentration of the dendrimer solution was ca. 0.5-1.1 mM. The reaction was stirred at room temperature for 21 h, then purified by SEC in DMF, and dried in vacuo to give dendrimer 5 as a colorless glassy solid.
Acetylated PAMAM-PEG Dendrimer 5a
PAMAM-PEG dendrimer 4a (17) (12.1 mg, 1.10 μmol) in DMSO-d6 (944 μL) was treated with triethylamine (8.10 μL, 58.1 μmol) and 0.317 M Ac2O solution in DMSO-d6 (48.5 μL, 15.4 μmol). The crude mixture was loaded on a SEC column (H 47 cm × O.D. 4.5 cm) to isolate the dendrimer conjugate 5a (9.28 mg). 1H NMR (600 MHz, DMSO-d6) δ 8.06–7.82 (m, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.23 (br s, 7.44H, NHPEG of major isomer), 6.81 (br s, 0.61H, NHPEG of minor isomer), 4.03 (t, 16.25H, J = 4.2 Hz, HhPEG), 3.62–3.38 (m, 389.27H, satellites J = 69.0 Hz, Hi, Hj, and Hk), 3.23 (s, 30.11H, Hl), 3.12–2.99 (m, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 118.64H, Hb and Hg), 2.42 (m, 60.10H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 41.73H, HhAc).
Acetylated PAMAM-PEG Dendrimer 5b
PAMAM-PEG dendrimer 4b (17) (11.3 mg, 0.746 μmol) in DMSO-d6 (1.28 mL) was treated with triethylamine (3.30 μL, 23.7 μmol) and 0.317 M Ac2O solution in DMSO-d6 (16.5 μL, 5.24 μmol). The crude mixture was loaded on a SEC column (H 40 cm × O.D. 3.0 cm) to isolate the dendrimer conjugate 5b (11.44 mg). 1H NMR (600 MHz, DMSO-d6) δ 8.04–7.81 (m, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.23 (br s, 12.70H, NHPEG of major isomer), 6.81 (br s, 1.14H, NHPEG of minor isomer), 4.03 (t, 27.47H, J = 4.1 Hz, HhPEG), 3.62–3.38 (m, 659.19H, satellites J = 70.7 Hz, Hi, Hj, and Hk), 3.23 (s, 47.25H, Hl), 3.12–2.99 (m, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 117.00H, Hb and Hg), 2.42 (m, 57.69H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 21.64H, HhAc).
Acetylated PAMAM-PEG Dendrimer 5d
PAMAM-PEG dendrimer 4d (17) (13.3 mg, 0.895 μmol) in DMSO-d6 (944 μL) was treated with triethylamine (7.70 μL, 55.2 μmol) and 0.317 M Ac2O solution in DMSO-d6 (48.0 μL, 15.2 μmol). The crude mixture was loaded on a SEC column (H 45 cm × O.D. 4.5 cm) to isolate the dendrimer conjugate 5d (13.28 mg). 1H NMR (600 MHz, DMSO-d6) δ 8.07–7.83 (m, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.24 (br s, 4.45H, NHPEG of major isomer), 6.82 (br s, 0.43H, NHPEG of minor isomer), 4.03 (t, 9.77H, J = 4.1 Hz, HhPEG), 3.62–3.38 (m, 795.81H, satellites J = 70.7 Hz, Hi, Hj, and Hk), 3.23 (s, 17.88H, Hl), 3.12–2.99 (m, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 114.51H, Hb and Hg), 2.42 (m, 58.04H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 57.24H, HhAc).
General Procedure for Conjugation of AF488
PEGylated G3 PAMAM dendrimer 5a, 5b, 5d, 4c, or 4d was dried in vacuo extensively, and then was dissolved in DMSO-d6. To this stirred dendrimer solution was added triethylamine, followed by a freshly prepared 11.3 mM solution of AF488-OTFP in DMSO-d6 (1.00 mg/100 μL). The final concentration of the dendrimer solution was ca. 0.4-1.0 mM. The reaction was stirred for 22 h protected from light, and then a portion of crude product 7/8 was transferred into another flask for the next step without any purification. The remaining reaction mixture was subjected to purification by SEC in DMF, and the column fractions corresponding to the first major fluorescent band were combined and dried in vacuo to give dendrimer 7/8 as a colored glassy solid.
PAMAM-PEG-AF488 Dendrimer 7a
PAMAM-PEG dendrimer 5a (9.77 mg, 0.804 μmol) in DMSO-d6 (1.53 mL) was treated with triethylamine (1.50 μL, 10.8 μmol) and 11.3 mM AF488-OTFP solution in DMSO-d6 (71.0 μL, 0.802 μmol). 1.10 mL of the crude reaction mixture was taken for the next step and the remainder was purified by SEC (H 23 cm × O.D. 2.5 cm) to isolate the dendrimer conjugate 7a (3.78 mg). The color of a 1.0 mM solution of 7a in water (HPLC grade, Aldrich) prepared for the biological assay was light yellow. 1H NMR (600 MHz, DMSO-d6) δ 8.04–7.81 (m, 76.71H, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.21 (br s, 8.38H, NHPEG of major isomer), 6.79 (br s, 0.85H, NHPEG of minor isomer), 4.03 (t, 15.98H, J = 4.2 Hz, HhPEG), 3.62–3.38 (m, 377.45H, satellites J = 70.0 Hz, Hi, Hj, and Hk), 3.23 (s, 30.54H, Hl), 3.13–2.99 (m, 182.37H, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 118.15H, Hb and Hg), 2.42 (m, 57.66H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 42.58H, HhAc).
PAMAM-PEG-AF488 Dendrimer 7b
PAMAM-PEG dendrimer 5b (11.2 mg, 0.702 μmol) in DMSO-d6 (1.44 mL) was treated with triethylamine (1.10 μL, 7.89 μmol) and 11.3 mM AF488-OTFP solution in DMSO-d6 (62.0 μL, 0.701 μmol). 1.00 mL of the crude reaction mixture was taken for the next step and the remainder was purified by SEC (H 38 cm × O.D. 3.0 cm) to isolate the dendrimer conjugate 7b (4.29 mg). The color of a 1.0 mM solution of 7b in water (HPLC grade, Aldrich) prepared for the biological assay was yellowish orange. 1H NMR (600 MHz, DMSO-d6) δ 8.42 (br s, 0.37H, HF), 8.22 (br s, 0.65H, HE), 8.04–7.79 (m, 69.52H, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.30 (br s, 0.68H, HD), 7.20 (br s, 14.83H, NHPEG of major isomer), 6.79 (br s, 1.32H, NHPEG of minor isomer), 6.45 (br s, 0.64H, HB/HC), 6.35 (br s, 0.55H, HB/HC), 4.03 (t, 28.68H, J = 4.2 Hz, HhPEG), 3.62–3.38 (m, 696.24H, satellites J = 70.2 Hz, Hi, Hj, and Hk), 3.23 (s, 48.74H, Hl), 3.11–2.99 (m, 188.79H, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 120.55H, Hb and Hg), 2.42 (m, 56.57H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 22.34H, HhAc).
PAMAM-PEG-AF488 Dendrimer 7d
PAMAM-PEG dendrimer 5d (12.9 mg, 0.788 μmol) in DMSO-d6 (1.63 mL) was treated with triethylamine (1.20 μL, 8.61 μmol) and 11.3 mM AF488-OTFP solution in DMSO-d6 (70.0 μL, 0.791 μmol). 1.16 mL of the crude reaction mixture was taken for the next step and the remainderwas purified by SEC (H 38 cm × O.D. 3.0 cm) to isolate the dendrimer conjugate 7d (4.52 mg). The color of a 1.0 mM solution of 7d in water (HPLC grade, Aldrich) prepared for the biological assay was light yellow. 1H NMR (600 MHz, DMSO-d6) δ 8.05–7.82 (m, 83.64H, NHG3, NHAc, NHG2, NHG1, and NHG0), 7.22 (br s, 5.69H, NHPEG of major isomer), 6.80 (br s, 0.68H, NHPEG of minor isomer), 4.03 (t, 9.22H, J = 4.2 Hz, HhPEG), 3.62–3.38 (m, 800.08H, satellites J = 70.9 Hz, Hi, Hj, and Hk), 3.24 (s, 21.04H, Hl), 3.12–2.99 (m, 198.41H, Hd, Hf, HfAc, HgAc, HfPEG, and HgPEG), 2.64 (m, 120.33H, Hb and Hg), 2.42 (m, 59.43H, He and Ha), 2.18 (m, 120.00H, Hc), 1.79 (s, 57.95H, HhAc).
PAMAM-PEG-AF488 Dendrimer 8c
PAMAM-PEG dendrimer 4c (17) (12.2 mg, 0.888 μmol) in DMSO-d6 (809 μL) was treated with triethylamine (2.85 μL, 20.5 μmol) and 11.3 mM AF488-OTFP solution in DMSO-d6 (78.7 μL, 0.889 μmol). 592 μL of the crude reaction mixture was taken for the next step and the remainder was purified by SEC (H 38 cm × O.D. 3.0 cm) to isolate the dendrimer conjugate 8c (3.37 mg). The color of a ca. 0.5 mM solution (not fully homogeneous) of 8c in water (HPLC grade, Aldrich) prepared for the biological assay was yellowish orange. 1H NMR (600 MHz, DMSO-d6) δ 8.17–7.80 (m, 71.97H, NHG3, NHG2, NHG1, and NHG0), 7.21 (br s, 11.31H, NHPEG of major isomer), 6.79 (br s, 1.68H, NHPEG of minor isomer), 4.03 (t, 23.61H, J = 4.1 Hz, HhPEG), 3.62–3.38 (m, 703.42H, satellites J = 70.4 Hz, Hi, Hj, and Hk), 3.23 (s, 44.08H, Hl), 3.11–2.99 (m, 182.07H, Hd, Hf, HfPEG, and HgPEG), 2.64 (m, 136.23H, Hb and Hg), 2.42 (m, 58.27H, He and Ha), 2.18 (m, 120.00H, Hc).
PAMAM-PEG-AF488 Dendrimer 8d
PAMAM-PEG dendrimer 4d (17) (7.63 mg, 0.512 μmol) in DMSO-d6 (1.15 mL) was treated with triethylamine (2.30 μL, 16.5 μmol) and 11.3 mM AF488-OTFP solution in DMSO-d6 (46.0 μL, 0.520 μmol). 850 μL of the crude reaction mixture was taken for the next step, and the remainder was purified by SEC (H 23 cm × O.D. 2.5 cm) to isolate the dendrimer conjugate 8d (3.44 mg). The color of a ca. 0.5 mM solution (not fully homogeneous) of 8d in water (HPLC grade, Aldrich) prepared for the biological assay was yellowish orange. 1H NMR (600 MHz, DMSO-d6) δ 8.13–7.80 (m, 69.31H, NHG3, NHG2, NHG1, and NHG0), 7.23 (br s, 7.21H, NHPEG of major isomer), 4.03 (t, 10.57H, J = 4.1 Hz, HhPEG), 3.62–3.38 (m, 945.12H, satellites J = 71.1 Hz, Hi, Hj, and Hk), 3.24 (s, Hl), 3.12–2.99 (m, 161.83H, Hd, Hf, HfPEG, and HgPEG), 2.64 (m, 132.91H, Hb and Hg), 2.42 (m, 56.76H, He and Ha), 2.19 (m, 120.00H, Hc).
PAMAM-PEG550-Ac-AF488-CGS21680 Dendrimer 9a
To a crude reaction mixture of fluorescent dendrimer 7a in DMSO-d6 (1.10 mL), was added triethylamine (3.50 μL, 25.1 μmol) and CGS21680 (1, as a hydrochloride hemihydrate form, 4.95 mg, 9.08 μmol). Subsequently, a freshly prepared 42.2 mM solution of PyBOP in DMSO-d6 (180 μL, 7.60 μmol) was added slowly to the stirred mixture of 7a. The reaction was stirred for 20 h, and the reaction mixture was loaded directly on a SEC column (H 43 cm × O.D. 4.5 cm) in DMF for purification, and the fractions containing the desired product were identified by 1H NMR. The first and last SEC fractions that were confirmed by NMR to contain minor amounts of the desired dendrimer were eliminated deliberately to reduce the polydispersity of the dendrimer conjugates. The SEC fractions selected as such were combined and dried in vacuo to give 5.55 mg of 9a′ (structure not shown). Unfortunately, the analysis of 9a′ by 1H NMR in DMSO-d6 indicated only ca. 0.38 CGS21680 groups were attached out of ca. nine available PAMAM amino groups. In an attempt to bring the CGS21680 attachment to completion, dendrimer 9a′, the isolated derivative of 7a, underwent another round of conjugation reaction, but this time under an exhaustive coupling condition. In a separate flask, CGS21680 (1, as a hydrochloride hemihydrate form, 23.3 mg, 42.8 μmol) in DMSO-d6 (978 μL) was treated with triethylamine (21.8 μL, 156 μmol) and PyBOP (20.3 mg, 39.1 μmol) [Solution A], and stirred for 15 min. Subsequently, dendrimer 9a′ (5.55 mg) in DMSO-d6 (190 μL) was treated with 220 μL (8.59 μmol) of Solution A, and the reaction mixture was stirred for 72 h protected from light. Another batch of pre-activated ester of CGS21680 (1, as a hydrochloride hemihydrate form, 19.6 mg, 36.0 μmol) was freshly prepared in DMSO-d6 (985 μL) by treating with triethylamine (14.8 μL, 106 μmol) and PyBOP (17.4 mg, 33.5 μmol) [Solution B], which was stirred for 12 min. The dendrimer mixture was treated with 120 μL (4.02 μmol) of Solution B and stirring continued for 39 h. The crude mixture was loaded on a SEC column (H 38 cm × O.D. 3.0 cm) for purification in DMF, and the fractions containing the desired compound were determined similarly by 1H NMR. The selected SEC fractions were combined and dried in vacuo to give dendrimer 9a (6.20 mg) as a glassy solid. The color of a 1.0 mM solution of 9a in water (HPLC grade, Aldrich) prepared for the biological assay was light yellow. 1H NMR (600 MHz, DMSO-d6) δ 8.04–7.80 (m, 80.15H, H8, H8Ad NHG3, NHAc, NHCGS, NHG2, NHG1, and NHG0), 7.22 (br s, 8.54H, NHPEG of major isomer), 7.15, 7.10 (d (each), 5.42H, H4 and H5), 6.79 (br s, 2.75H, H6Ad, NHPEG of minor isomer), 6.19 (s, 0.91H, H1), 5.84 (d, 0.86H, J = 6.5 Hz, H1′), 5.63, 5.56 (br s (each), 1.70H, H3′-OH and H2′-OH), 4.72 (m, 0.86H, H2′), 4.25 (s, H4′), 4.19 (s, 1.03H, H3′), 4.03 (t, 16.34H, J = 4.4 Hz, HhPEG), 3.62–3.38 (m, 388.25H, satellites J = 70.1 Hz, Hi, Hj, and Hk, one of H2), 3.23 (s, 30.93H, Hl), 3.13–2.99 (m, 190.69H, Hd, Hf, HfAc, HgAc, HfCGS, HgCGS, HfPEG, HgPEG, and H9), 2.76 (m, H3 and H6), 2.64 (m, 110.80H, Hb and Hg), 2.42 (m, 58.01H, He and Ha), 2.34 (t, H7), 2.18 (m, 120.00H, Hc), 1.79 (s, 42.84H, HhAc), 0.97 (t, 3.60H, J = 7.1 Hz, H10).
PAMAM-PEG550-Ac-AF488-CGS21680 Dendrimer 9b
The conjugation reaction was performed following a procedure similar to that described for 9a. To a crude reaction mixture of fluorescent dendrimer 7b in DMSO-d6 (1.00 mL), was added triethylamine (2.50 μL, 17.9 μmol) and CGS21680 (1, as a hydrochloride hemihydrate form, 3.50 mg, 6.42 μmol). Subsequently, a freshly prepared 42.2 mM solution of PyBOP in DMSO-d6 (129 μL, 5.45 μmol) was added slowly to the stirred mixture of 7b. The reaction was stirred for 20 h, then the reaction mixture was loaded directly on a SEC column (H 38 cm × O.D. 3.0 cm) in DMF and purified to give 8.82 mg of 9b′ (structure not shown). Unfortunately, the analysis of 9b′ by 1H NMR in DMSO-d6 indicated only ca. 1.24 CGS21680 groups were attached out of ca. 9 available PAMAM amino groups. Next, dendrimer 9b′ (8.82 mg) in DMSO-d6 (148 μL) was treated with 170 μL (6.64 μmol) of Solution A (vide supra, stirred for 12 min), and the reaction mixture was stirred for 72 h protected from light. Then, the dendrimer mixture was treated with 100 μL (3.35 μmol) of Solution B (vide supra, stirred for 11 min), and continued stirred for 39 h. The crude mixture was loaded on a SEC column (H 43 cm × O.D. 4.5 cm) for purification in DMF, and the fractions containing the desired compound were determined similarly by 1H NMR. The selected SEC fractions were combined and dried in vacuo to give dendrimer 9b (7.87 mg) as a glassy solid. The color of a 1.0 mM solution of 9b in water (HPLC grade, Aldrich) prepared for the biological assay was orange. 1H NMR (600 MHz, DMSO-d6) δ 8.86 (br s, 0.45H, NHAF), 8.43 (br s, 0.98H, HF), 8.21 (br s, HE), 8.04–7.80 (m, 74.60H, H8, H8Ad NHG3, NHAc, NHCGS, NHG2, NHG1, and NHG0), 7.30 (br s, 0.72H, HD), 7.21 (br s, 14.06H, NHPEG of major isomer), 7.14, 7.10 (d (each), 10.14H, H4 and H5), 6.79 (br s, 5.23H, H6Ad, NHPEG of minor isomer), 6.45 (br s, 0.69H, HB/HC), 6.35 (br s, 0.66H, HB/HC), 6.19 (s, 1.73H, H1), 5.84 (d, 1.72H, J = 5.9 Hz, H1′), 5.63, 5.56 (br s (each), 2.57H, H3′-OH and H2′-OH), 4.71 (m, 1.62H, H2′), 4.26 (s, 2.29H, H4′), 4.19 (s, 1.97H, H3′), 4.03 (br s, 29.99H, HhPEG), 3.62–3.38 (m, 720.52H, satellites J = 70.6 Hz, Hi, Hj, and Hk, one of H2), 3.23 (s, 51.65H, Hl), 3.19–2.99 (m, 189.36H, Hd, Hf, HfAc, HgAc, HfCGS, HgCGS, HfPEG, HgPEG, and H9), 2.76 (m, 18.44H, H3 and H6), 2.64 (m, 111.40H, Hb and Hg), 2.42 (m, 54.30H, He and Ha), 2.34 (t, 8.37H, H7), 2.18 (m, 120.00H, Hc), 1.79 (s, 22.70H, HhAc), 0.97 (t, 6.01H, J = 6.9 Hz, H10).
PAMAM-PEG2000-Ac-AF488-CGS21680 Dendrimer 9d
The conjugation reaction was performed following a procedure similar to that described for 9a. To a crude reaction mixture of fluorescent dendrimer 7d in DMSO-d6 (1.16 mL), was added triethylamine (2.70 μL, 19.4 μmol) and CGS21680 (1, as a hydrochloride hemihydrate form, 3.83 mg, 7.03 μmol). Subsequently, a freshly prepared 42.2 mM solution of PyBOP in DMSO-d6 (138 μL, 5.81 μmol) was added slowly to the stirred mixture of 7d. The reaction was stirred for 20 h, then the reaction mixture was loaded directly on a SEC column (H 41 cm × O.D. 4.5 cm) in DMF and purified to give 8.81 mg of 9d′ (structure not shown). Unfortunately, the analysis of 9d′ by 1H NMR in DMSO-d6 indicated only ca. 0.65 CGS21680 groups were attached out of ca. 7 available PAMAM amino groups. Next, dendrimer 9d′ (8.81 mg) in DMSO-d6 (300 μL) was treated with 180 μL (7.03 μmol) of Solution A (vide supra, stirred for 8 min), and the reaction mixture was stirred for 72 h protected from light. Then, the dendrimer mixture was treated with 100 μL (3.35 μmol) of Solution B (vide supra, stirred for 10 min) and stirring continued for 39 h. The crude mixture was loaded on a SEC column (H 38 cm × O.D. 4.5 cm) for purification in DMF, and the fractions containing the desired compound were determined similarly by 1H NMR. The selected SEC fractions were combined and dried in vacuo to give dendrimer 9d (8.82 mg) as a glassy solid. The color of a 1.0 mM solution of 9d in water (HPLC grade, Aldrich) prepared for the biological assay was light orange. 1H NMR (600 MHz, DMSO-d6) δ 8.87 (br s, NHAF), 8.43 (br s, HF), 8.05–7.81 (m, 83.00H, H8, H8Ad NHG3, NHAc, NHCGS, NHG2, NHG1, and NHG0), 7.22 (br s, 4.43H, NHPEG of major isomer), 7.14, 7.10 (d (each), 5.22H, H4 and H5), 6.80 (br s, 1.95H, H6Ad, NHPEG of minor isomer), 6.47 (br s, HB/HC), 6.36 (br s, HB/HC), 6.19 (s, 0.67H, H1), 5.84 (d, 0.68H, J = 6.7 Hz, H1′), 5.65, 5.58 (br s (each), 0.61H, H3′-OH and H2′-OH), 4.72 (m, 0.78H, H2′), 4.26 (s, 1.71H, H4′), 4.19 (s, 0.90H, H3′), 4.03 (t, 9.47H, J = 4.0 Hz, HhPEG), 3.62–3.39 (m, 810.85H, satellites J = 70.7 Hz, Hi, Hj, and Hk, one of H2), 3.24 (s, 20.59H, Hl), 3.14–2.99 (m, 190.63H, Hd, Hf, HfAc, HgAc, HfCGS, HgCGS, HfPEG, HgPEG, and H9), 2.76 (m, H3 and H6), 2.64 (m, 115.71H, Hb and Hg), 2.42 (m, 55.85H, He and Ha), 2.34 (t, H7), 2.18 (m, 120.00H, Hc), 1.79 (s, 57.89H, HhAc), 0.97 (t, 3.42H, J = 7.0 Hz, H10).
PAMAM-PEG750-AF488-CGS21680 Dendrimer 10c
CGS21680 (1, as a hydrochloride hemihydrate form, 14.7 mg, 27.0 μmol) in DMSO-d6 (242 μL) was treated with triethylamine (10.4 μL, 74.6 μmol) and PyBOP (12.9 mg, 24.9 μmol) [Solution C], and stirred for 8 min. Subsequently, in a separate flask, a crude reaction mixture of fluorescent dendrimer 8c in DMSO-d6 (592 μL) was treated with 150 μL (14.8 μmol) of Solution C, and the reaction mixture was stirred for 50 h protected from light. Then, the dendrimer mixture was treated with 220 μL (7.37 μmol) of Solution B (vide supra, stirred for 7 min) and stirring continued for 14 h. The crude mixture was loaded on a SEC column (H 38 cm × O.D. 4.5 cm) for purification in DMF, and the fractions containing the desired compound were determined similarly by 1H NMR. The selected SEC fractions were combined and dried in vacuo to give dendrimer 10c (9.98 mg) as a glassy solid. The color of a 1.0 mM solution of 10c in water (HPLC grade, Aldrich) prepared for the biological assay was intense orange. 1H NMR (600 MHz, DMSO-d6) δ 8.85 (br s, NHAF), 8.44 (br s, HF), 8.05, 8.02 (s (each), 36.49H, H8 and H8Ad), 7.93, 7.89 (br s, 47.73H, NHG3, NHCGS), 7.80 (br s, 25.82H, NHG2, NHG1, and NHG0), 7.20 (br s, 12.52H, NHPEG of major isomer), 7.13, 7.09 (d (each), 60.79H, H4 and H5), 6.80 (br s, 25.16H, H6Ad, NHPEG of minor isomer), 6.19 (s, 12.19H, H1), 5.85 (d, 13.94H, J = 6.1 Hz, H1′), 5.62, 5.56 (br s (each), 21.91H, H3′-OH and H2′-OH), 4.72 (m, 13.13H, H2′), 4.26 (s, 14.50H, H4′), 4.19 (s, 13.88H, H3′), 4.03 (br s, 22.64H, HhPEG), 3.61–3.38 (m, 704.59H, satellites J = 67.0 Hz, Hi, Hj, and Hk, one of H2), 3.23 (s, 63.37H, Hl), 3.17–2.99 (m, 209.06H, Hd, Hf, HfCGS, HgCGS, HfPEG, HgPEG, and H9), 2.76 (m, 76.79H, H3 and H6), 2.64 (m, 115.47H, Hb and Hg), 2.42 (m, 54.39H, He and Ha), 2.33 (t, 39.56H, J = 7.3 Hz, H7), 2.18 (m, 120.00H, Hc), 0.96 (br s, 42.16H, H10).
PAMAM-PEG2000-AF488-CGS21680 Dendrimer 10d
The conjugation reaction was performed following a procedure similar to that described for 9a. To a crude reaction mixture of fluorescent dendrimer 8d in DMSO-d6 (850 μL), was added triethylamine (5.10 μL, 36.6 μmol) and CGS21680 (1, as a hydrochloride hemihydrate form, 6.96 mg, 12.8 μmol). Subsequently, a freshly prepared 42.2 mM solution of PyBOP in DMSO-d6 (280 μL, 11.8 μmol) was added slowly to the stirred mixture of 7d. The reaction was stirred for 43 h, and the reaction mixture was loaded directly on a SEC column (H 43 cm × O.D. 4.5 cm) in DMF and purified to give 9.44 mg of 10d′ (structure not shown). Unfortunately, the analysis of 10d′ by 1H NMR in DMSO-d6 indicated only ca. 18.49 CGS21680 groups were attached out of ca. 26 available PAMAM amino groups. Next, dendrimer 10d′ (9.44 mg) in DMSO-d6 (300 μL) was treated with 170 μL (6.64 μmol) of Solution A (vide supra, stirred for 6 min), and the reaction mixture was stirred for 72 h protected from light. Then, the dendrimer mixture was treated with 100 μL (3.35 μmol) of Solution B (vide supra, stirred for 8 min) and stirring continued for 14 h. The crude mixture was loaded on a SEC column (H 38 cm × O.D. 3.0 cm) for purification in DMF, and the fractions containing the desired compound were determined similarly by 1H NMR. The selected SEC fractions were combined and dried in vacuo to give dendrimer 10d (8.39 mg) as a glassy solid. The color of a 1.0 mM solution of 10d in water (HPLC grade, Aldrich) prepared for the biological assay was intense red. 1H NMR (600 MHz, DMSO-d6) δ 8.05, 8.02 (s (each), 47.63H, H8 and H8Ad), 7.93, 7.89 (br s, 54.71H, NHG3, NHCGS), 7.80 (br s, 25.60H, NHG2, NHG1, and NHG0), 7.21 (br s, 7.03H, NHPEG of major isomer), 7.13, 7.09 (d (each), 85.97H, H4 and H5), 6.81 (br s, 36.71H, H6Ad, NHPEG of minor isomer), 6.19 (s, 19.05H, H1), 5.85 (d, 20.03H, J = 6.5 Hz, H1′), 5.62, 5.56 (br s (each), 35.34H, H3′-OH and H2′-OH), 4.72 (m, 18.36H, H2′), 4.26 (s, 20.93H, H4′), 4.19 (s, 18.93H, H3′), 4.02 (br s, 11.19H, HhPEG), 3.62–3.37 (m, 930.57H, satellites J = 73.3 Hz, Hi, Hj, and Hk, one of H2), 3.23 (s, 23.17H, Hl), 3.19–2.99 (m, 240.33H, Hd, Hf, HfCGS, HgCGS, HfPEG, HgPEG, and H9), 2.76 (m, 99.23H, H3 and H6), 2.64 (m, 110.31H, Hb and Hg), 2.41 (m, 55.64H, He and Ha), 2.33 (t, 54.85H, J = 7.7 Hz, H7), 2.18 (m, 120.00H, Hc), 0.96 (br s, 61.52H, H10).
Receptor binding and functional assays.1
Materials
[125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (I-AB-MECA; 2000 Ci/mmol) and other radioligands were purchased from Perkin-Elmer Life and Analytical Science (Boston, MA). [3H]2-chloro-N6-cyclopentyladenosine (CCPA) was a custom synthesis product (Perkin Elmer). Samples for binding and functional assays were prepared as follows: compounds 1, 2, and 3 as 5.0 mM stock solutions in DMSO (ACS spectroscopic grade, >99.9%, Aldrich); dendrimers 7a, 7b, 7d, 9a, 9b, 9d, 10c, and 10d as 1.0 mM stock solutions in water (HPLC grade, Aldrich). Achieving a complete dissolution of dendrimer 8c or 8d in water (1.0 mM) was difficult, and thus, experiments were not performed for these compounds. Stock solutions were stored frozen at -80 °C.
Cell culture and membrane preparation (18)
Chinese hamster ovary (CHO) cells stably expressing the recombinant human (h) A1, hA2B, or hA3 AR or human embryonic kidney (HEK)-293 cells stably expressing the recombinant hA2A AR were cultured in DMEM (Dulbecco's modified Eagle's medium) and F12 (1:1, v/v) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 μmol/mL glutamine, and 800 μg/mL geneticin. After harvest, homogenization, and suspension, cell membranes were centrifuged at 500×g for 10 min, and the pellet was resuspended in 50 mM Tris–HCl buffer (pH 7.4) containing 10 mM MgCl2 and 1 mM EDTA. The suspension was homogenized with an electric homogenizer for 10 s, and was then re-centrifuged at 20000×g for 20 min at 4 °C. The resultant pellets were resuspended in buffer in the presence of 3 Units/mL adenosine deaminase, and the suspension was stored at -80 °C until the binding experiments. The protein concentration was measured using the Bradford assay (19).
Binding assays at the hA1 and hA2A ARs
For binding to hA1 ARs (20, 21), membranes (40 μg/tube) from CHO cells stably expressing the receptor were incubated with [3H]N6-[(R)-phenylisopropyl]adenosine (R-PIA; 2 nM) or [3H]CCPA (0.5 nM) at 25 °C for 60 min in 50 mM Tris–HCl buffer (pH 7.4, 10 mM MgCl2) and increasing concentrations of the test ligands in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of N6-cyclopentyladenosine (CPA).
For hA2A AR binding (22), membranes (20 μg/tube) from HEK-293 cells stably expressing the receptor were incubated with [3H]CGS21680 (15 nM) at 25 °C for 60 min in 50 mM Tris–HCl buffer (pH 7.4, 10 mM MgCl2) and increasing concentrations of the test ligands in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of 5′-N-ethylcarboxamidoadenosine (NECA).
For hA3 AR binding (22), membranes (20 μg/tube) from CHO cells stably expressing the receptor were incubated with [125I]I-AB-MECA (0.2 nM) (23) at 37 °C for 60 min in 50 mM Tris–HCl buffer (pH 7.4, 10 mM MgCl2) in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of Cl-IB-MECA.
Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using an MT-24 cell harvester (Brandell, Gaithersburg, MD). Filters were washed three times with 9 mL ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter. IC50 values were converted to Ki values as described (24).
Cyclic AMP accumulation assay
CHO cells expressing the A2A or A2B AR were seeded in 24-well plates and incubated at 37 °C overnight. The following day the medium was removed and replaced with DMEM containing 50 mM HEPES, 10 μM rolipram, 3 U/mL adenosine deaminase, and increasing concentrations of agonists. The medium was removed, and the cells were lysed with 200 μL of 0.1 M HCl. 100 μL of the HCl solution was used in the Sigma Direct cAMP Enzyme Immunoassay following the instructions provided with the kit. The results were interpreted using a Bio-Tek ELx808 Ultra Microplate reader at 405 nm.
Statistical Analysis
Binding and functional parameters were calculated using Prism 4.0 software (GraphPAD, San Diego, CA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation (24). Data were expressed as the mean ± standard error. Statistical analysis was performed using Analysis of Variance (ANOVA) with post hoc test or Student's test where appropriate with p values less than 0.05 being considered significant.
Studies of human washed platelets
Preparation
Human washed platelets were prepared as previously described (25). Briefly, fresh blood obtained from healthy donors was centrifuged at 175×g for 15 min at 37 °C, platelet rich plasma was removed, and centrifuged at 1570×g for 15 min at 37 °C. The platelet pellet was washed twice in Tyrode's buffer (137 mM NaCl, 2 mM KCl, 12 mM NaHCO3, 0.3 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 5.5 mM glucose, 5 mM Hepes, pH 7.3) containing 0.35% human serum albumin, and finally resuspended at a density of 3×105 platelets/μL in the same buffer in the presence of 0.02 U/mL of the adenosine 5′-diphosphate (ADP) scavenger apyrase (adenosine 5′-triphosphate diphosphohydrolase, EC 3.6.1.5), a concentration sufficient to prevent desensitization of platelet ADP receptors during storage. Platelets were kept at 37 °C throughout all experiments.
Platelet aggregation studies.1
Aggregation was measured at 37 °C by a turbidimetric method in a dual-channel Payton aggregometer (Payton Associates, Scarborough, Ontario, Canada). The stock solutions for platelet aggregation and internalization studies were prepared as follows: compounds 1, 2, and 3 as 5.0 mM solutions in DMSO (ACS spectroscopic grade, >99.9%, Aldrich); dendrimers 7a, 7b, 7d, 9a, 9b, 9d, 10c, and 10d as 1.5 mM solutions in water (HPLC grade, Aldrich); dendrimers 8c and 8d as 750 μM solutions in water (HPLC grade, Aldrich). A small amount of DMSO in monomer was previously demonstrated not to affect the platelet aggregation (10). A 450 μL aliquot of platelet suspension was stirred at 1100 rpm and activated by addition of ADP (5 μM), in the presence or absence of various monomers/dendrimers and in the presence of human fibrinogen (0.8 mg/mL), in a final volume of 500 μL. The extent of aggregation was estimated quantitatively by measuring the maximum curve height above the baseline level.
Flow cytometry analysis.1
Human washed platelets (3×105 platelets/μL) were incubated with various dendrimers for 30 min at 37 °C. The platelet suspension was then centrifuged at 1900×g for 8 min at 37 °C and platelets were resuspended in Tyrode's buffer containing 0.35% human serum albumin and fixed with paraformaldehyde 2% for 15 min. A 5 μL aliquot of sample was then diluted with 500 μL PBS for flow cytometry analysis. Samples were analyzed using a Becton Dickinson FACScalibur flow cytometer and Cell Quest software (Becton Dickinson, San Jose, CA). All parameters were acquired on a logarithmic scale and 20,000 events were recorded in the platelet population.
Results and Discussion
Molecular Design
Our recent investigation showed that a relatively low degree of surface-modification (ca. ≤25%) by short polyethylene glycol monomethyl ether (PEG) chains (Mn = 550/750) sufficiently increased water-solubility and reduced the cytotoxicity of G3 PAMAM dendrimers (17). Here, our goal was to examine if this PEGylation principle could be effectively applied to our earlier dendrimer constructs bearing GPCR ligands that interact within the transmembrane domain of the receptor to induce biological effects. Accordingly, three G3 PAMAM dendrimers (theo. 32 end groups) containing relatively small weight portions of PEG that differ in number and size (7PEG550 (4a), 14PEG550 (4b), and 4PEG2000 (4d), Scheme 1) were initially selected for a comparative study. We planned partial substitution of the dendrimer with a fixed number of agonist ligands (CGS21680) and with all remaining free amino groups concealed by prior acetylation in order to precisely evaluate the influence of different PEG groups on receptor-mediated activity. A fluorophore was also attached to the dendrimers for detection in biological systems. In addition, preparation of an unacetylated fluorescent PAMAM-4PEG2000 derivative with an increased number of CGS21680 ligands was intended for determining if any multivalent effect is discernable with this PEGylated system. The stoichiometry in the substitution reactions of the amine-terminated G3 PAMAM dendrimers has been well controlled (10, 11, 17). To summarize, our original molecular design considered the following: 1) partial acetylation of three G3 PAMAM-PEG dendrimers to leave similar numbers of free amino groups; 2) attachment of 1 equiv of Alexa Fluor 488 (AF488) for fluorescent imaging; 3) conjugation of CGS21680 at all remaining amino groups.
Scheme 1.
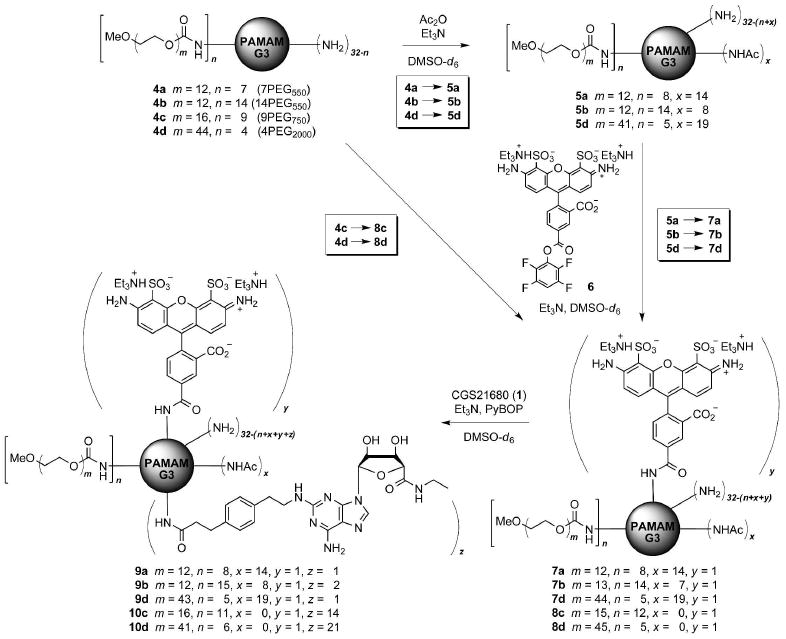
Synthetic route to PAMAM-PEG-CGS21680 dendrimer conjugates.
Synthesis
Acetylation of 4a, 4b, and 4d was achieved with a relatively efficient stoichiometric control to leave a similar fraction of free amino groups (31% for 5a; 35% for 5b; 25% for 5d) (Scheme 1). Purification by SEC in DMF after each step slightly shifted the composition (m and n) of each dendrimer mixture and its MW distribution as determined by 1H NMR integration (see Supporting Information) (17). Subsequently, each acetylated PAMAM-PEG dendrimer 5 was treated with an equimolar amount of AF488 as an activated ester (6). Some unreacted dye 6 was recovered during the SEC purification from all three reactions (5→7). In contrast, no free dye 6 was found when unacetylated dendrimer 4d was reacted similarly.
Next, the fluorescent dendrimers 7a, 7b, 7d, and 8d were treated with CGS21680 in slight excess for its conjugation to the PAMAM periphery via PyBOP-mediated coupling. Unfortunately, the conversion fraction in these reactions determined after SEC purification turned out to be low, being more prominent for the acetylated dendrimers 7 (0.38 CGS21680 attachments for 7a out of approximately nine remaining peripheral amino groups, 1.24 for 7b, 0.65 for 7d, and 18.5 for 8d by 1H NMR integration). The low coupling yield was in contrast to the full substitution of the G3 PAMAM amino ends with CGS21680 under the same reaction conditions (10). Obviously, the presence of an adjacent PEG chain, irrespective of its number and size, interfered sterically with the coupling reaction. Furthermore, we speculate that applying the partial acetylation at the PEGylated PAMAM surface may further phase-separate these unimolecular micelles intramolecularly (into microphase domains of hydrophobic core and hydrophilic PEG corona) to shrink the PAMAM surface and lower the reactivity of the remaining amino groups considerably. To overcome the low reactivity, the SEC-purified dendrimer mixture was further treated with two separate additions of the excess pre-activated ester of CGS21680 and was allowed to react for several additional days. Nevertheless, application of this exhaustive coupling condition did not substantially improve the total conversion for all cases. Among the three acetylated dendrimers, 7b with a lower proportion of acetamide groups showed a slightly better coupling efficiency. Since our goal was to systematically study the influence of adjacent PEG chains in eliciting biological activity through ligand-receptor interactions, attachment of CGS21680 earlier in the synthetic sequence was avoided in order to facilitate the stoichiometric control in PEG derivatization. Moreover, the activated form of PEG (as a p-nitrophenyl carbonate) potentially reacts with the hydroxy and the adenine amino groups of CGS21680. Another unacetylated fluorescent dendrimer analogue 10c with shorter PEG chains was prepared similarly from 4c (9PEG750), in order to compare the influence of PEG on the chemical reactivity and the biological effects between unacetylated dendrimer conjugates. Again, a complete conjugation of CGS21680 to 8c was not achieved.
Characterization by 1H NMR and MALDI-TOF MS
The final dendrimer conjugates 7–10 used for biological assays were characterized by analysis of 1H NMR integrals in DMSO-d6 (Table 1). NMR analysis of 9a, 9b, and 9d in D2O gave similar results; however, analysis of 10c and 10d in D2O was not feasible due to the significant peak broadening. This may have resulted from poor desolvation in D2O (26) of the relatively hydrophobic interior–CGS21680 moieties and the PAMAM domain. The average MW of each dendrimer was also determined by MALDI MS in two different matrices (DHB and THAP) to give somewhat underestimated values compared to those obtained by NMR (Table 1). Underestimation of MWs of similar PAMAM conjugates by MALDI was previously noted (10, 17). The number of CGS21680 groups (z) attached to each dendrimer calculated by MALDI was similar to (or slightly higher for 9b and 10d) NMR-estimated values.
Solution Conformation of Dendrimer Conjugates by NOESY
To predict the solution conformation of PAMAM-PEG-CGS21680 dendrimer conjugates (Figure 2) in the aqueous phase, 2D NOESY experiments were carried out for selected dendrimers 9b, 10c, and 10d in D2O (see Figures S4–S6, Supporting Information). Our previous investigation on various PAMAM-PEG conjugates (without ligands) suggested no backfolding of the PEG chains into the interior of the PAMAM domain (17). Intense NOE cross-peaks were found between the peaks from PAMAM and CGS21680 segments of dendrimers 10c and 10d with high-loading of CGS21680, but much weaker (or no) signals were detected from the corresponding cross-peaks (if any) of 9b. Related to this observation, it may be important to note the small difference in the chemical shift (Δδ ≈ 0.15 ppm) of the methyl group of the 5′-uronamide moiety of CGS21680 in 1D NMR in D2O, between the dendrimers with low-loading (0.97-0.98 ppm for 9a, 9b, and 9d) and high-loading (0.85 ppm for 10c; 0.82 ppm for 10d) of ligands. Hardly any NOE cross-peaks were observed between the peaks from CGS21680 and PEG regions in all three spectra. Detection of the NOE cross-peaks between those from PAMAM and PEG regions was difficult due to the severe overlapping of peaks. However, weak NOE cross-peaks between PAMAM and PEG peaks were indicated, particularly with dendrimer 10c, which were more vivid for the isolated PEG peaks (hPEG, i; see Figure S5, Supporting Information) close to the PAMAM region. Taken together, NOESY analysis suggests that CGS21680 ligands attached to the PEGylated PAMAM are more likely positioned in proximity to the PAMAM domain–by adhering to its surface or by partially backfolding into its interior void–rather than in the outer hydrophilic PEG layer in contact with the water molecules.
Figure 2.
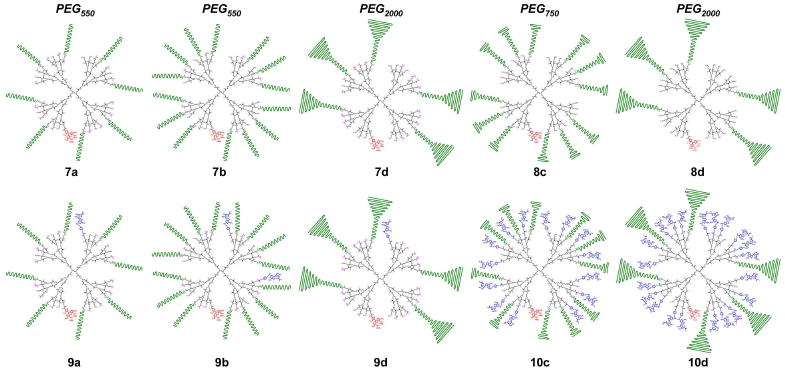
Structures of PEGylated PAMAM dendrimer conjugates 7–10 (black: PAMAM; green: PEG; pink: acetyl; red: AF488; blue: CGS21680).2
In Vitro Binding Assay at hARs
Next, the biological activities of these PEGylated PAMAM-CGS21680 conjugates 9 and 10 were examined by binding and functional assays in cell membranes (CHO or HEK-293 cells) expressing each hAR (Table 2) (11). Each of the four subtypes of ARs (A1, A2A, A2B, and A3) is associated with characteristic pathways of signaling, typically in a species-dependent manner (12). Recent evidence indicates the importance of secondary binding near the extracellular domain of a GPCR in enhancing selectivity and affinity of GPCR ligands (27). Here, we compared the binding and activity profiles of these five dendrimers (9 and 10, Figure 2) with dendritic controls (precursors without the ligand, 7, Figure 2) and three monomeric controls (1–3, Figure 1). APEC (2) is an analogue of CGS21680 (1) with an ethylenediamine extension at the distal unit (28) that exhibits a higher binding affinity at the hA2A AR compared to 1, but only three-fold selectivity in comparison to the hA3 AR. We also envisioned that 2, particularly with a free amino group, may serve as a rough topological mimic for some of the dendrimer-attached CGS21680 moieties which are adjacent to the unreacted PAMAM amino groups. Compound 3 (11) was intended as a monomeric mimic of the dendrimer-attached form of 1, because its terminal portion was identical to the linking segment in the dendrimer conjugates.
Table 2.
Binding and functional assay results of monomers and dendrimers at four subtypes of hARs.a
| cmpd | bindingb at ARs | activationc at ARs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| hA1 | hA2A | hA3 | hA2A | hA2B | |||||
| Ki [nM] | % inhibition | Ki [nM] | % inhibition | Ki [nM] | % inhibition | EC50 [nM] | % activation | % activation | |
| 1 | 5780 ± 590 | 24.9 ± 4.6 | 67 ± 19 | 84.7 ± 0.9 | 247 ± 79 | 59.8 ± 4.4 | 53.0 ± 16.0 | 100 ± 10 | 3.6 ± 6.3 |
| 2 | 168 ± 36 | 56.9 ± 9.1 | 12 ± 6 | 101 ± 1 | 38 ± 11 | 85.3 ± 3.2 | 8.2 ± 1.7 | 96.0 ± 6.0 | 5.5 ± 4.7 |
| 3 | 984 ± 223 | 36.9 ± 7.5 | 210 ± 19 | 82.1 ± 0.3 | 58 ± 24 | 85.2 ± 11.4 | 51.0 ± 8.0 | 103 ± 5 | -1.7 ± 5.0 |
| 7a | n.d.d | 5.5 ± 8.8 | n.d.d | 1.6 ± 7.4 | n.d.d | -13.4 ± 2.7 | n.d.d | 2.4 ± 1.7 | 5.3 ± 8.9 |
| 7b | n.d.d | 13.8 ± 4.5 | n.d.d | -11.6 ± 2.5 | n.d.d | -17.2 ± 5.2 | n.d.d | n.a.d | 1.6 ± 7.3 |
| 7d | n.d.d | 7.1 ± 4.5 | n.d.d | 3.2 ± 4.9 | n.d.d | -7.5 ± 2.2 | n.d.d | n.a.d | -4.4 ± 3.8 |
| 9a | n.d.d | 12.2 ± 1.6 | 488 ± 59 | 61.4 ± 2.0 | n.d.d | 24.4 ± 6.5 | n.d.d | 92.6 ± 5.4 | 1.4 ± 5.5 |
| 9b | n.d.d | 19.3 ± 3.6 | 496 ± 18 | 62.7 ± 1.2 | n.d.d | 27.6 ± 3.2 | n.d.d | 101 ± 5 | 16.6 ± 3.7 |
| 9d | n.d.d | 13.3 ± 3.1 | 752 ± 34 | 55.8 ± 1.8 | n.d.d | 25.4 ± 4.0 | n.d.d | 91.0 ± 8.3 | 9.4 ± 5.4 |
| 10c | 4870 ± 1360 | 19.9 ± 4.8 | 151 ± 36 | 81.2 ± 1.5 | 1130 ± 310 | 29.0 ± 12.0 | 11.2 ± 4.9 | 90.0 ± 6.7 | 2.8 ± 1.4 |
| 10d | 4230 ± 1310 | 19.8 ± 2.6 | 215 ± 12 | 75.5 ± 3.1 | 1320e | 24.4 ± 8.0 | 60.6 ± 12.8 | 97.0 ± 7.1 | 3.3 ± 3.0 |
Values are presented as the mean ± standard error of three or more independent experiments. The hA1, hA2B, and hA3 ARs were expressed in CHO cells and the hA2A AR in HEK-293 cells for binding and in CHO cells for the functional assay.
Ki values were determined through the radioligand binding, and the percent inhibition refers to the inhibition of binding at 1 μM.
EC50 values were determined by measuring the degree of stimulation of adenylate cyclase in cells stably expressing hA2A or hA2B AR. Percent activation refers to the activation of adenylate cyclase in comparison to the effects exerted by the full agonist NECA (100%) at 10 μM.
n.d. = not determined, i.e., binding/functional experiment was not conducted, or n.a. = no binding/activation was detected under the given assay conditions.
Performed only once.
Overall, the inhibition constants (Ki) of the dendritic ligands at all tested AR subtypes (hA1, hA2A, and hA3) were higher than those of the monovalent ligands 1–3. Interestingly, this reduced affinity of dendritic ligands was more prominent at hA1 and hA3 compared to hA2A AR. The selectivity for hA2A compared to hA3 AR was nearly an order of magnitude for the dendritic ligand 10c (hA2A, Ki/ hA3, Ki = 0.13; 0.16 for 10d), whereas the monomeric mimic of the dendrimer-attached nucleoside 3 showed selectivity (3.62-fold) for the hA3. Indeed, high-loaded dendrimer conjugates 10 successfully achieved similar Ki values at hA2A AR as that of 3. In contrast, dendrimers 9 with low-loading of CGS21680 showed a reduction in binding affinity compared to monovalent ligand 3, where 9d bearing longer PEG2000 chains was the weakest. This may suggest an increased degree of steric inhibition (by neighboring PEG chains or the PAMAM backbone) experienced by a single ligand moiety in a macromolecular carrier with fewer ligands, significantly impairing ligand accessibility.
A similar trend was observed when comparing the inhibition at a fixed concentration of 1 μM (Figure 3a, Table 2). The dendritic controls 7 displayed essentially no inhibitory effect (<15%) at all subtypes of ARs. Inhibition of radioligand binding at the hA2A AR was more pronounced for dendrimers 10 with high-loading of CGS21680 compared to 9 with low-loading, and similar to the effects by CGS21680 (1) and monomeric control 3, but somewhat lower than inhibition by APEC (2). The differences in Ki values at hA2A AR between dendrimers 9 and dendrimers 10 are statistically significant (p<0.01). The relative inhibitory effect at the hA2A versus the hA3 AR was more than twice as high (hA2A, %inhibition/ hA3, %inhibition = 2.20–3.09) for all PAMAM-PEG-CGS21680 dendrimer conjugates and nearly three times as high for dendrimers 10 with high-loading of CGS21680. This represented an improvement in hA2A selectivity considering the low selectivity in comparison to the hA3 AR (0.96–1.42) of monomers 1–3. Furthermore, selectivity for hA2A compared to hA1 has also increased (3.25–5.03 by 9 and 10; 1.77–3.40 by 1–3).
Figure 3.
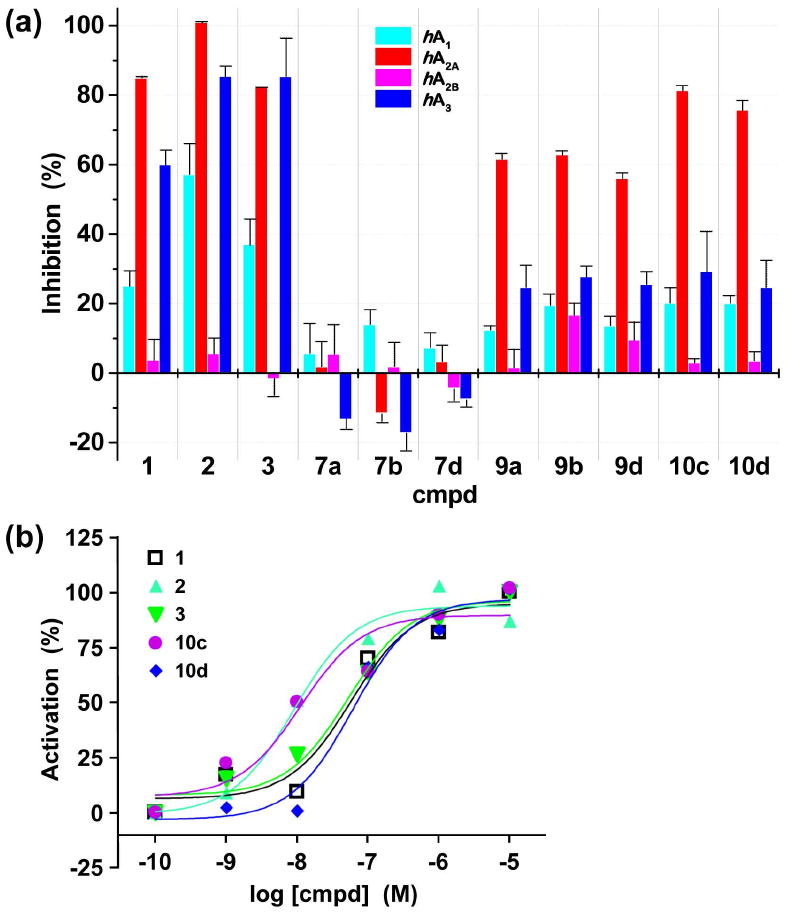
In vitro assay results of monomers (1–3) and selected dendrimers (7–10). (a) Interaction with four subtypes of hARs (percent inhibition of specific radioligand binding at hA1, hA2A, and hA3 ARs at a fixed concentration of 1 μM, and percent activation of adenylate cyclase at hA2B AR at 10 μM); (b) agonistic effect at hA2A AR as determined by measuring the degree of stimulation of adenylate cyclase in cells stably expressing the hA2A AR. The hA1, hA2B, and hA3 ARs were expressed in CHO cells and the hA2A AR in HEK-293 cells for binding and in CHO cells for the functional assay.
Cyclic AMP Accumulation Assay
The potency (i.e., agonistic effects) of monomers and dendrimers at hA2A AR was determined by measuring the stimulation of adenylate cyclase–to produce intracellular adenosine 3′,5′-cyclic monophosphate (cAMP)–in CHO cells (Figure 3b, Table 2) (12). Dendrimer 10c (EC50 = 11.2 ± 4.9 nM, with PEG750) was more potent than dendrimer 10d (EC50 = 60.6 ± 12.8 nM, with PEG2000) in activating the hA2A AR (p<0.05). Considering the numbers of CGS21680 ligands (ca. 14 for 10c; ca. 21 for 10d) and PEG chains attached to these dendrimers, the longer PEG2000 chains appeared to significantly impede the ligand-receptor interaction, whereas the shorter PEG750 chains negligibly affected receptor binding and activation.
The EC50 values at the hA2A AR of dendrimer 10c (11.2 ± 4.9 nM, with PEG750) and its corresponding monomer 3 (51.0 ± 8.0 nM) are significantly different (p<0.05), although the hA2A AR binding affinity of 10c (Ki = 151 ± 36 nM) was not significantly different from that of monomer 3 (210 ± 19 nM). Unlike the potency determined by measuring cAMP concentration that is directly associated with the intracellular signaling cascade initiated through AR activation, binding affinity was measured in isolated membranes of mammalian cells heterologously expressing specific ARs (see Experimental Section).
Recent reports described the dynamic behavior of GPCRs including receptor reorganization and oligomerization (7, 29, 30), which can be potentially regulated allosterically upon binding of a ligand. In such case, usage of clustered multivalent ligands would be particularly advantageous to amplify the desired cellular response. Therefore, this substantial enhancement in EC50 of a high-loaded dendrimer 10c might reflect the involvement of more than one receptor-ligand interaction simultaneously. Indeed, a recent computer modeling study showed the possibility to bridge both primary binding sites at the transmembrane domain of a GPCR homodimer by two separate CGS21680 units covalently attached to a single G3 PAMAM dendrimer (31). Nevertheless, although enhancement of potency and/or selectivity was observed here, it was not sufficient to prove receptor clustering or oligomerization in this system.
Platelet Aggregation Studies
The effective inhibition of ADP-induced platelet aggregation was previously demonstrated using PAMAM dendrimers with multiple copies of an A2A AR agonist (added as a DMSO solution) (10, 11). Here, the antiaggregatory effect of water-soluble PAMAM-PEG-CGS21680 dendrimer conjugates 9 and 10 was measured (Figure 4, Table S1, Supporting Information). A submicromolar inhibitory effect on platelet aggregation of 10c (IC50 = 151 ± 53 nM) was validated, which was better than that of another high-loaded dendrimer 10d (495 ± 145 nM) with longer PEG chains. Unlike the cAMP assay, here the potency of 10c was similar to those of CGS21680 1 (192 ± 90 nM) and the dendrimer-attached mimic 3 (173 ± 72 nM), but lower than that of APEC 2 (35.9 ± 17.0 nM). Dendrimer 9a bearing eight units of short PEG chains but with only one ligand on average (8PEG550-1CGS21680), exhibited a similar or slightly lower IC50 value (337 ± 77 nM) than 10d carrying ca. 21 CGS21680 units. Accordingly, it seems apparent that the intracellular activity triggered by ligand-receptor interactions is more influenced by the length of the PEG chains (i.e., sterics) than the total number of ligands available on the dendrimer surface for a potential multivalent binding. Obviously, when the PEG size and the number of ligands were similar (8PEG550-1CGS21680 (9a) vs. 15PEG550-2CGS21680 (9b)), the dendrimer with a smaller number of PEG chains showed higher activity. Likewise, when the PEG size and its number were more or less the same (6PEG2000-21CGS21680 (10d) vs. 5PEG2000-1CGS21680 (9d); 11PEG750-14CGS21680 (10c) vs. 8PEG550-1CGS21680 (9a)), the dendrimer carrying more ligands exhibited higher agonistic potency. The weakest activity was shown by 9b and 9d, either highly substituted with PEG or carrying long PEG chains, and both having only ca. 1–2 ligand attachments. Also, APEC 2 was found most potent in this study as in the cAMP assay, suggesting the importance of the terminal amino group in enhancing the agonistic effect. In fact, the number of unreacted amines in PAMAM-PEG-CGS21680 conjugates 9 and 10 estimated from the 1H NMR integration (Table 1) was in a similar range (ca. 5–7), and thus, the modulation of the agonistic effects among dendrimers by the PAMAM amino groups is somewhat unlikely. It is important to note the moderate inhibitory effects in platelet aggregation exerted by two of the dendrimers without any ligands, 7d (71% at 15 μM) and 8d (40% at 7.5 μM) (Figure S9, Supporting Information), suggesting the involvement of longer PEG2000 chains in inhibiting platelet aggregation possibly through a mechanism unrelated to the A2A AR.
Figure 4.
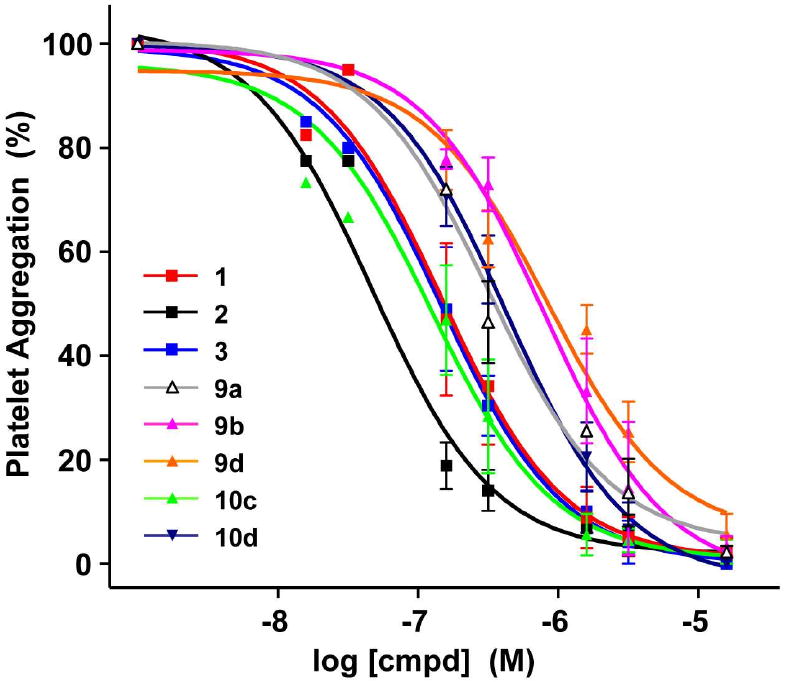
Platelet aggregation induced by ADP (5 μM) in the presence of various monomeric or dendritic A2A agonists at 37 °C. The degree of platelet aggregation was normalized against that induced by ADP at 5 μM (100%). The extent of aggregation was estimated quantitatively by measuring the maximum curve height (i.e., percent light transmission) above the baseline level.
Flow Cytometry Analysis
Our earlier platelet uptake studies using PAMAM-CGS21680 conjugates (no PEG) showed a sustained antiaggregatory effect, which might have resulted from the slow release of internalized conjugates as monitored by fluorescent confocal microscopy (10). Despite its known benefits, the presence of PEG groups on the surface of nanocarriers generally hampers the internalization process (4). Here, we investigated by flow cytometry3 the potential for internalization of the PEGylated PAMAM dendrimers into human washed platelets (Figures 5, S10, and S11, Supporting Information). The best internalization efficiency at 3 μM (30 min incubation) was achieved by dendrimers 8c and 8d, which did not carry any ligand and had a relatively poor water-solubility. Interestingly, the water-solubility of 4c and 4d dropped significantly upon conjugation of 1 equiv of AF488, whereas that of partially acetylated 5a, 5b, and 5d was not affected. A minor degree of uptake was detected from 9b (15PEG550-2CGS21680) and its precursor 7b. Neither of the high-loaded dendrimers 10c and 10d showed any indication of internalization at 3 μM. When the dendrimer concentration was increased to 50 μM (30 min incubation), some internalization was detected from 10c and 10d (Figure S10, Supporting Information), with 10c being slightly higher in its efficiency. Similar observations were made in the fluorescent confocal microscopy studies (data not shown) to confirm these results. It is difficult to conclude whether the low water-solubility (as demonstrated by the fully conjugated water-insoluble PAMAM-CGS21680 dendrimer (10)) or the presence of many cationic amino groups (11)–or the combination of both–aided in the effective internalization of PEGylated PAMAM dendrimers 8c and 8d. In any case, the internalization of dendritic controls 8c and 8d conceivably did not involve ligand interactions with the A2A AR. This suggests the involvement of uptake pathways other than GPCR-mediated endocytosis (16, 32, 33) for PEGylated dendrimers into the platelets.
Figure 5.
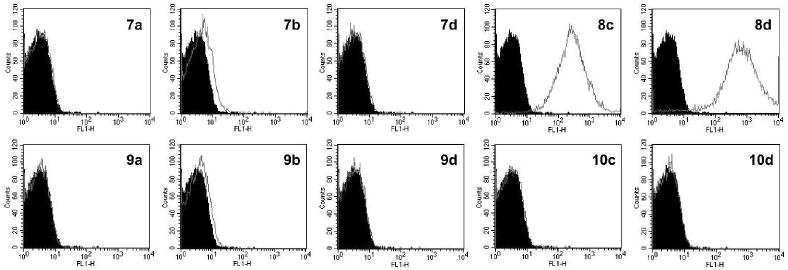
Flow cytometry histograms (AF488, control: black area) obtained from human washed platelets incubated with 3 μM of each G3 PAMAM dendrimer conjugate for 30 min at 37 °C.
In conclusion, an approach to the design and synthesis of potent and water-soluble macromolecular ligands of GPCRs was demonstrated through our systematic studies on dendritic carriers. A PAMAM dendrimer conjugate bearing multiple copies of a nucleoside ligand and relatively short PEG chains manifested enhanced receptor subtype-selectivity and highly potent agonistic effects, without experiencing any disadvantageous steric effects. Longer PEG chains reduced affinity at the A2A AR. With growing evidence to support the oligomerization of GPCRs and the potential intervention of ligands in GPCR clustering (7, 29, 30), acquiring synergistic cooperativity among ligands through multivalent binding may be desirable for GPCR therapeutics based on ligand-receptor interactions. Dendrimers of higher generations are envisioned for use in exploring multiple interactions with higher order GPCR aggregates. In addition, the potential of PEGylated PAMAM dendrimers as carriers to deliver genes across the blood-brain barrier by derivatizing their periphery with the brain-targeting ligands has been demonstrated recently (34, 35). Consequently, our PEGylation strategy described in this communication may find versatile utility in various nanomedical applications aimed at multivalent ligand-receptor interactions.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, NIDDK. We thank Dr. Haijun Yao at the Mass Spectrometry Laboratory of the University of Illinois for collecting MALDI spectra of our dendrimer samples, and Dr. Herman Yeh at the NIDDK for the helpful advice on the NMR experiments. Y.K. thanks the Can-Fite Biopharma for financial support.
Footnotes
Supporting Information Available: Experimental procedures and characterization, 1H NMR, NOESY, and MALDI MS spectra, results of platelet aggregation assays and flow cytometry histograms. This material is available free of charge via the Internet at http://pubs.acs.org.
All of our in vitro assay results (including platelet studies) are reported in dendrimer concentrations, and not in ligand concentrations. This was intended because of potential limitations in simultaneously achieving full accessibility to receptors of all ligands covalently bound to a single carrier such as in our dendrimer conjugates. We maintain that our system is different from those carriers loaded with drugs as either physically entrapped or covalently bound through cleavable linkers for their release for activation as monomeric forms.
The illustration of dendrimer conjugates shows the average number of attached pendant groups in each statistical mixture as determined by 1H NMR integrals, and reflects the approximate PEG length (550/750/2000) relative to the size of the CGS21680 ligand. The geometry of each component as drawn, particularly of the PEG chains, does not represent the actual solution conformation.
Strictly, fluorescent confocal microscopy analysis detects fluorescence only from the internalized dendrimers, while flow cytometry measures fluorescence from both internalized and surface-bound dendrimers. Accordingly, the estimation of internalization efficiency by flow cytometry analysis may be limited.
Literature Cited
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotech. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Duncan R. The dawning era of polymer therapeutics. Nature Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 4.Tomalia DA, Reyna LA, Svenson S. Dendrimers as multi-purpose nanodevices for oncology drug delivery and diagnostic imaging. Biochem Soc Trans. 2007;35:61–67. doi: 10.1042/BST0350061. [DOI] [PubMed] [Google Scholar]
- 5.Boas U, Christensen JB, Heegaard PMH. Dendrimers in medicine and biotechnology. The Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 6.Lee CC, MacKay JA, Fréchet JMJ, Szoka FC. Designing dendrimers for biological applications. Nature Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 7.Kiessling LA, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badjić JD, Nelson A, Cantrill SJ, Turnbull WB, Stoddart JF. Multivalency and cooperativity in supramolecular chemistry. Acc Chem Res. 2005;38:723–732. doi: 10.1021/ar040223k. [DOI] [PubMed] [Google Scholar]
- 9.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Kim Y, Hechler B, Klutz AM, Gachet C, Jacobson KA. Toward multivalent signaling across G protein-coupled receptors from poly(amidoamine) dendrimers. Bioconjugate Chem. 2008;19:406–411. doi: 10.1021/bc700327u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Klutz AM, Hechler B, Gao ZG, Gachet C, Jacobson KA. Application of the functionalized congener approach to dendrimer-based signaling agents acting through A2A adenosine receptors. Purinergic Signalling. 2009;5:39–50. doi: 10.1007/s11302-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Discovery. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landry Y, Niederhoffer N, Sick E, Gies JP. Heptahelical and other G-protein-coupled receptors (GPCRs) signaling. Curr Med Chem. 2006;13:51–63. [PubMed] [Google Scholar]
- 15.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nature Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 16.Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev. 2005;57:2215–2237. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Kim Y, Klutz AM, Jacobson KA. Systematic investigation of polyamidoamine dendrimers surface-modified with poly(ethylene glycol) for drug delivery applications: synthesis, characterization, and evaluation of cytotoxicity. Bioconjugate Chem. 2008;19:1660–1672. doi: 10.1021/bc700483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao ZG, Mamedova LK, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Schwabe U, Trost T. Characterization of adenosine receptors in rat brain by (−)[3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg's Arch Pharmacol. 1980;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 21.Perreira M, Jiang JK, Klutz AM, Gao ZG, Shainberg A, Lu C, Thomas CJ, Jacobson KA. “Reversine” and its 2-substituted adenine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem. 2005;48:4910–4918. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 23.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-Aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng YC, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 25.Cazenave JP, Ohlmann P, Cassel D, Eckly A, Hechler B, Gachet C. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol Biol. 2004;272:13–28. doi: 10.1385/1-59259-782-3:013. [DOI] [PubMed] [Google Scholar]
- 26.Hunter CA. Quantifying intermolecular interactions: guidelines for the molecular recognition toolbox. Angew Chem Int Ed. 2004;43:5310–5324. doi: 10.1002/anie.200301739. [DOI] [PubMed] [Google Scholar]
- 27.Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, Christopoulos A. Critical Role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem. 2007;282:25677–25686. doi: 10.1074/jbc.M702311200. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson KA. Functionalized congener approach to the design of ligands for G protein-coupled receptors (GPCRs) Bioconjugate Chem. 2009 doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gandia J, Galino J, Amaral OB, Soriano A, Lluís C, Franco R, Ciruela F. Detection of higher-order G protein-coupled receptor oligomers by a combined BRET-BiFC technique. FEBS Lett. 2008;582:2979–2984. doi: 10.1016/j.febslet.2008.07.045. [DOI] [PubMed] [Google Scholar]
- 30.Szidonya L, Cserző M, Hunyady L. Dimerization and oligomerization of G-protein-coupled receptors: debated structures with established and emerging functions. J Endocrinol. 2008;196:435–453. doi: 10.1677/JOE-07-0573. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov AA, Jacobson KA. Molecular modeling of a PAMAM- CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorg Med Chem Lett. 2008;18:4312–4315. doi: 10.1016/j.bmcl.2008.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 33.Watson P, Jones AT, Stephens DJ. Intracellular trafficking pathways and drug delivery: fluorescence imaging of fixed and living cells. Adv Drug Deliv Rev. 2005;57:43–61. doi: 10.1016/j.addr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Huang RQ, Qu YH, Ke WL, Zhu JH, Pei YY, Jiang C. Efficient gene delivery targeted to the brain using a transferring-conjugated polyethyleneglycol-modified polyamidoamie dendrimer. FASEB J. 2007;21:1117–1125. doi: 10.1096/fj.06-7380com. [DOI] [PubMed] [Google Scholar]
- 35.Huang R, Ke W, Liu Y, Jiang C, Pei Y. The use of lactoferrin as a ligand for targeting the polyamidoamine-based gene delivery system to the brain. Biomaterials. 2008;29:238–246. doi: 10.1016/j.biomaterials.2007.09.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.