

Abstract
Dark azido push–pull chromophores have the ability to be photoactivated to produce bright fluorescent labels suitable for single-molecule imaging. Upon illumination, the aryl azide functionality in the fluorogens participates in a photochemical conversion to an aryl amine, thus restoring charge-transfer absorption and fluorescence. Previously, we reported that one compound, DCDHF-V-P-azide, was photoactivatable. Here, we demonstrate that the azide-to-amine photoactivation process is generally applicable to a variety of push–pull chromophores, and we characterize the photophysical parameters including photoconversion quantum yield, photostability, and turn-on ratio. Azido push–pull fluorogens provide a new class of photoactivatable single-molecule probes for fluorescent labeling and super-resolution microscopy. Lastly, we demonstrate that photoactivated push–pull dyes can insert into bonds of nearby biomolecules, simultaneously forming a covalent bond and becoming fluorescent (fluorogenic photoaffinity labeling).
Introduction
Advances in super-resolution fluorescence imaging by controllable photoactivation of single-molecule emitters (e.g. PALM, FPALM, STORM)1–3 require new and optimized activatable fluorophores. If one single emitter is switched on at a time in a diffraction-limited region (~250 nm), its location can be determined well below the diffraction limit by fitting the point-spread function (i.e. image of the single molecule). A super-resolution image of a labeled complex structure can then be reconstructed from many successive rounds of weak photoactivation and fitting.4 Several groups have been developing photoswitchable fluorescent proteins,5–7 organic fluorophores,8–12 and quantum dots13 in order to build the toolbox of controllable emitters.14 Recently, we reported a photoactivatable azido version of a push-pull fluorophore that contains a 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (DCDHF) moiety as a very strong electron-accepting group.15 In addition to super-resolution imaging, the ability to photochemically control the fraction of emitting molecules has additional applications in pulse-chase experiments, single-molecule tracking, or in situations where the number of emitting molecules at a given time must be kept low.
Push–pull chromophores containing an electron donor, a conjugated network (π), and an electron acceptor have been explored for many years for nonlinear optics,16 photoinduced electron transfer,17 and photorefractivity;18 some molecules in this class were even found to be good single-molecule labels.19–23 In our approach, a nonfluorescent, blue-shifted azide–π–acceptor fluorogen precursor is photoconverted to a fluorescent, red-shifted amine–π–acceptor fluorophore. In the fluorogen, the donor is absent, but the product fluorophore contains all three necessary components of the complete donor–π–acceptor push–pull chromophore (Scheme 1). Because the azido fluorogens do not exhibit the red-shifted charge-transfer band typical of push–pull chromophores,24, 25 they are not resonant with the wavelengths used to excite the amino version of the fluorophore (Figure 1 and Table 1), and are therefore dark. In related work, Bouffard et al.26 designed a chemically caged DCDHF fluorophore in an effort to detect cysteine: an electron-withdrawing sulfonyl group was added to the nitrogen and thus interfered with its donor capability until it was cleaved off by any cysteine, producing an amine capable of donating electrons. Budyka et al.27 have previously reported that an azido hemicyanine dye (similar to the azido stilbazolium reported below) undergoes conversion to an amine upon irradiation with visible light; however, the fluorescence properties of this dye were not reported.
Scheme 1.

Photoconversion of dark azide-substituted fluorogens produce fluorescent amine-substituted fluorophores, which may involve insertion into C-H or C-C bonds. A dark photoproduct is also possible.
Figure 1.

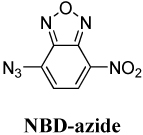
(left) Spectra of NBD-azide photoactivation. (See the SI for spectra of the other fluorogens.) Because the azide cannot participate as a donor in a charge transfer, the fluorogen exhibits blue-shifted absorption (λabs = 384 nm) with respect to its amine-donor sister (λabs = 456 nm). Upon irradiation with UV light, the azido fluorogen (solid black curve) converts to the amino fluorophore (solid grey curves). Fluorescence (dotted red lines) excited at 440 nm increases significantly after photoconversion (right) Photoactivation of DCDHF-P-azide using a 385-nm flashlight (1.1 mW cm−2). The short-wavelength azido absorption peak (triangles) disappears with time and the long-wavelength absorption peak (circles) corresponding to the fluorescent amino fluorophore grows in. The time constant (τP = 85 s) from the exponential fit of the disappearance of the azido fluorogen is used to calculate the photoconversion quantum yield (ΦP) in Table 1.
Table 1.
Photophysical and photochemical parameters of various azido fluorogens (in ethanol unless otherwise stated)
| λabs,azido (nm)[a] | λabs,amino (nm)[b] | λfl,amino[c] (nm) |
Yield[d] | ΦF[e] | ΦP[f] | ΦB (10−6)[h] | |
|---|---|---|---|---|---|---|---|
| {εmax (M−1 cm−1)} | {εmax (M−1 cm−1)} | {λP}[g] | in PVA | ||||
 |
379 {19,300}[i] |
475 {44,900}[i] |
496 | 24% | 0.003 | ~0.08 {385 nm} |
4.6 |
 |
424 {29,100} |
570 {54,100} |
613 | 65% | 0.025 | 0.0059 {407 nm} |
4.1[k] |
 |
415 {19,100} |
479 | 591 | ~50% | – | ~0.09 {385 nm} |
– |
 |
420 {18,100}[i] |
517 | 611 | – | – | – | 3.4 |
 |
407 {26,700} |
463 {20,000} |
578 | 87% | 0.0062 | 0.017 {385 nm} |
9.2 |
 |
403 {31,300} |
456 {31,100} |
599 | < 30% | 0.18 | 0.085 {385 nm} |
6.2 |
 |
375 {41,000}[j] |
449 | 580 | ~25% | – | ~0.2 {365 nm} |
– |
 |
384 {14,400}[i] |
456 | 539 | ~50% | 0.20 | ~0.2 {365 nm} |
100[l] |
Peak absorbance and molar absorption for azido fluorogen, and
amino fluorophore.
Fluorescence peak of amino fluorophore.
Overall chemical reaction yield to the fluorescent amino product (see Methods).
Fluorescence quantum yield of amino fluorophore. DCDHFs become much brighter in rigid environments.20
Photoconversion quantum yield of azido fluorogens to any product.
Wavelength used to photoactivate azido fluorogens.
Photobleaching quantum yield of amino fluorophore. Fluorescein in gelatin is 64×10−6.
In dichloromethane.
In acetonitrile.
In gelatin.
In PMMA.
While amines are strong electron-donating substituents, azides are weakly electron-withdrawing.28 Recovering fluorescence from aryl azides is possible because they are known to be photolabile. The photochemistry of aryl azides has been studied extensively;29 the photoreaction most often reported involves the loss of dinitrogen and rearrangement to a seven-membered azepine heterocycle. However, electron-withdrawing substituents on the aromatic ring can stabilize the nitrene intermediate and promote formation of the amino functionality.30 Because push–pull chromophores inherently contain a strong electron-withdrawing substituent, an azido push–pull molecule should be more prone to photoconvert to the fluorescent amino version upon irradiation with activating light that is resonant with the blue-shifted absorption of the azido fluorogen. Initially, we demonstrated this photoactivation of fluorescence with only one member of the DCDHF class of fluorophores.15 Here, we expand the concept to a broad range of push–pull dyes: DCM, stilbazolium, NBD, and other DCDHF fluorophores (for structures see Table 1). We also demonstrate fluorogenic photoaffinity labeling of biomolecules using the photochemical azide-to-amine process.
Methods
Chemical Analysis of Photoproducts
For most of the azido fluorogens, we relied on UV–vis and fluorescence spectroscopy in order to identify the amine products, comparing the spectra of the photoconverted samples to the spectra of independently synthesized versions of the amino fluorophores. The following full chemical analysis of the photoconversion of DCDHF-V-P-azide (bolded to signify fluorogen) was originally reported in the SI of reference 15. Samples for bulk chemical studies were photoconverted in ethanol, both with and without removing dissolved oxygen by bubbling N2, and analyzed using NMR and HPLC–MS.
Column chromatography and NMR
A solution of photoconverted DCDHF-V-P-azide in ethanol was separated on a TLC plate (1:3 acetone:dichloromethane) into two bands: a red band with lower Rf that was fluorescent under UV light (365 nm) and a yellow band with higher Rf that was nonemissive; the yellow band was not present when the solution of DCDHF-V-P-azide was deoxygenated by bubbling N2 before and during photoconversion. (Adequate separation was not achievable using dichloromethane and hexanes or dichloromethane alone; therefore, we resorted to acetone in the mobile-phase solvent mixture.)
For chromatography, the photoproducts were separated on a column using silica gel as the stationary phase and 2:1 hexanes:acetone as the mobile-phase solvent. Two bands were well separated: a yellow band of DCDHF-V-P-nitro eluted first, then a red band of DCDHF-V-P-amine eluted later (see Figure S1 for structures). NMR spectra of column-separated photoproducts confirm these assignments, as compared to pure, synthesized samples (although the yellow band was contaminated with some other minor photoproducts).30, 31 DCDHF-V-P-azide: 1H NMR (400 MHz, CDCl3, δ): 7.65 (d, J = 8.4 Hz, Ar, 2H), 7.61 (d, J = 16 Hz, vinyl, 1H), 7.13 (d, J = 8.4 Hz, Ar, 2H), 6.97 (d, J = 16 Hz, vinyl, 1H), 1.80 (s, CH3, 6H). DCDHF-V-P-amine (photoconverted from DCDHF-V-P-azide, column separated): 1H NMR (400 MHz, CDCl3, δ): 7.58 (d, J = 16 Hz, vinyl, 1H), 7.50 (d, J = 8.4 Hz, Ar, 2H), 6.80 (d, J = 16 Hz, vinyl, 1H), 6.70 (d, J = 8.8 Hz, Ar, 2H), 4.39 (s, NH2, 2H), 1.76 (s, CH3, 6H). DCDHF-V-P-amine (pure synthesized independently): 1H NMR (500 MHz, CDCl3, δ): 7.58 (d, J = 16 Hz, vinyl, 1H), 7.50 (d, J = 8.5 Hz, Ar, 2H), 6.80 (d, J = 17 Hz, vinyl, 1H), 6.70 (d, J = 8.5 Hz, Ar, 2H), 4.39 (s, NH2, 2H), 1.76 (s, CH3, 6H). DCDHF-V-P-nitro (photoconverted from DCDHF-V-P-azide, crude, column enriched): 1H NMR (300 MHz, CDCl3, δ): 8.34 (d, J = 8.7 Hz, Ar), 7.80 (d, J = 8.4 Hz, Ar), 7.69 (d, J = 11 Hz, vinyl), 7.12 (d, J = 14 Hz, vinyl), 1.83 (s, CH3). DCDHF-V-P-nitro (pure synthesized independently): 1H NMR (400 MHz, CDCl3, δ): 8.34 (d, J = 8.8 Hz, Ar, 2H), 7.80 (d, J = 8.8 Hz, Ar, 2H), 7.68 (d, J = 16.8 Hz, vinyl, 1H), 7.12 (d, J = 16.4 Hz, vinyl, 1H), 1.83 (s, CH3, 6H).
Purification of DCDHF-V-P-amine and DCDHF-V-P-nitro by semi-prep HPLC
An ethanolic solution containing ~1 mg mL−1 of DCDHF-V-P-azide was photoconverted using a 150-W Xe lamp for 5 min under air. Photoproducts DCDHF-V-P-amine and DCDHF-V-P-nitro were separated by HPLC on a Hypersil Hyper Prep 100 BDS–C18 column (10.0×250 mm) with linear gradient elution (5–100% acetonitrile over 25 min, 5 min hold at 100% acetonitrile; balance by volume, 0.1 M tetraethylammonium acetate buffer, pH 7.5; total flow rate, 4 mL min−1). The UV–vis absorption spectrum of the column eluent was continuously monitored using a Shimadzu diode array detector (SPD-M10A). Under these conditions, compounds DCDHF-V-P-amine and DCDHF-V-P-nitro exhibited retention times of 20.9 and 22.5 min, respectively. No detectable DCDHF-V-P-azide (RT = 23.6 min) remained after photoactivation.
HPLC–MS characterization of photoproducts
Ethanolic solutions of DCDHF-V-P-azide were photoconverted using diffuse 407-nm laser light under nitrogen or air. The photoactivation products were analyzed by HPLC–MS (Waters 2795 Separations module with 2487 Dual λ Absorbance Detector; Waters Micromass ZQ mass spectrometer). Gradient elution (2–95% acetonitrile with 0.1% formic acid over 20 min, 10 min hold at 95% acetonitrile/formic acid; balance by volume, water with 0.1% formic acid) through a C18 column (2.1×40 mm) was employed for the separation. The column eluent was subjected to electrospray ionization, and positive and negative ions with m/z from 100–1000 amu were detected.
In the absence of oxygen, photoconversion of DCDHF-V-P-azide produced DCDHF-V-P-amine (RT = 11.36 min; ESI−: m/z = 301.7, [M–H]−; ESI+: m/z = 303.5, [M+H]+) as the only major photoproduct. A putative azo dimer (RT = 16.97 min; ESI−: m/z = 599.7, [M–H]−) was observed as a minor photoproduct.
In air, photoactivation of DCDHF-V-P-azide produced a mixture of DCDHF-V-P-amine (RT = 11.43 min; ESI−: m/z = 301.5, [M–H]−; ESI+: m/z = 303.4, [M+H]+) and DCDHF-V-P-nitro (RT = 12.99 min; ESI−: m/z = 331.5, [M–H]−, 315.5 [M–O–H]−, 301.5 [M–2O–H]−) as major products. After several days in air and room lights, an unidentified species believed to be generated from DCDHF-V-P-nitro formed in the solution (RT = 19.15 min; ESI−: m/z = 367.6).
Bulk Solution Spectroscopy
Bulk solution absorption and emission spectra were acquired on a Perkin-Elmer Lambda 19 UV–vis spectrometer and a Horiba Fluoromax-2 fluorimeter using standard 1-cm path length, quartz cuvettes. Fluorescence quantum yields were referenced against standards with known quantum yields, corrected for differences in optical density and solvent refractive index.32 All quantitative measurements were performed at low concentrations (absorbance values less than 0.2) to avoid any complications with dimer or aggregate formation. Molar absorption coefficients were measured from dilutions of solutions with known concentrations.
Photoconversion in ethanol was performed using one of the following light sources: a 365-nm hand-held UV lamp (0.62 mW cm−2); a 385-nm diode flashlight (1.1 mW cm−2, see Figure S5 for spectrum); the 407-nm line from a Kr-ion laser (Coherent Innova-301, 3.1 mW cm−2).
The overall chemical reaction yields to fluorescent product listed in Table 1 were measured from the absorbance values in the photoactivation spectra. Yield was defined by [amino]f/[azido]i = (Aamino/εamino)f/(Aazido/εazido)i, where i and f refer to initial and final values, respectively. In cases where other photoproducts contributed significant absorbance at the amino peak wavelength, the final absorbance value for the amino peak Aamino,f was corrected for this additional absorbance.
Sample Preparation for Microscopy
Samples for aqueous bulk photostability measurements and quantitative single-molecule measurements were prepared using 5–10% (by mass) gelatin (type A, Bloom 200, MP Biomedicals) or 1% poly(vinyl alcohol) (PVA, 72000 g mol−1, Carl Roth Chemicals) in purified water. The gelatin solution was liquefied at 37 °C; then a small volume (<0.5 µL) of dye stock solution in ethanol was mixed with 10 µL gelatin, sandwiched between two Ar-plasma-etched glass coverslips, and allowed to gel at room temperature. Single-molecule samples for the movie were made in 1% poly(methyl methacrylate) (PMMA, Tg = 105 °C, MW = 75000 g mol−1, atactic, polydispersity ~2.8, PolySciences Inc.) in toluene by mass. For polymer samples, a small volume of stock dye solution was mixed into an aliquot of PVA in water or PMMA in toluene, then the mixture was spin-coated onto an Ar-plasma-etched glass coverslip at 3000 RPM for 30 s. Thick samples of PMMA were prepared by drop casting 200–400 µL of 10% (by mass) PMMA in toluene and allowing the film to dry for several hours.
Microscopy
Samples were studied using an Olympus IX71 inverted microscope in an epifluorescence configuration33 using 488- or 514-nm illumination from an Ar-ion laser (Coherent Innova) or the 594-nm line from a HeNe laser (Meredith Instruments, 5 µW output); the irradiance at the sample was typically 0.5–1.0 kW cm−2. The emission was collected through a 100×, 1.4 N.A. oil-immersion objective, filtered using appropriate dichroic and long-pass filters to remove scattered excitation light, and imaged onto an electron-multiplying Si EMCCD camera (Andor iXon+) with integration times of 20–100 ms.
Turn-on Ratio
To measure the effective turn-on ratio, the signal on the camera from many activated fluorophores in a PMMA film was divided by the signal from the same location in the sample before activation; the background intensity level measured in an undoped film was subtracted from both signals the (see Figure S6). This measurement is experimentally relevant because it not only uses illumination intensity levels comparable to those used in actual imaging, but also considers the decrease in contrast due to a chemical reaction yield that is less than unity (see Results and Discussion).
For the bulk experiments, the fluorogens were doped into a thick PMMA polymer film at approximately 1–2 orders of magnitude higher concentration than single-molecule experiments, but otherwise imaged under similar conditions. This approach assumes that we are working in concentration regime where emitters are dense enough to get a statistical sampling of the population but separated enough to avoid self-quenching or excimer behavior. From our previous experience with bulk and single-molecule samples of fluorophores in polymer films, we are confident that we are safely in this regime.
Because in some measurements the on and off signals fell outside the dynamic range of the CCD detector, it was necessary to use different intensities or gain levels for the dark and background measurements than for the fluorescent signal. This required that the intensities and gain levels were accurately measured and accounted for in the calculation of the turn-on ratio.
Intermediate Lifetime
The lifetime of the nonfluorescent intermediate was measured immediately after the photoconversion. Imaging in the widefield epifluorescence microscope at 594 nm (~1 kW cm−2), a sample in aqueous agarose gel was repeatedly photoactivated with 0.5 s of high-intensity 405-nm light (~1 kW cm−2). These bright bursts of violet light both photobleached any existing fluorescent species and initiated the photoconversion of the azido species to fluorescent amino products, via a nonfluorescent intermediate (putatively a nitrene, but there may be additional intermediates requiring rearrangement before fluorescence appears). By fitting with a single exponential the increase of fluorescence over time after the photobleaching/photoactivating burst, the average lifetime of the intermediate was determined to be 1.85 s. Presumably, this lifetime is dependent on the environment, solvent, and temperature; however, this was measured in an aqueous environment that is not dissimilar to the cell.
Live-Cell Microscopy
For details of Chinese Hamster Ovary (CHO) cell culture, see reference 34. CHO cells were plated on fibronectin-coated borosilicate chambered coverslips overnight prior to imaging. CHO cells were treated with 1-µM dye solution (1-mM dye stock in ethanol into growth medium) at 37 °C for 1 hr, followed by extensive PBS buffer rinses to remove excess dye. Briefly, cells were imaged at 22 °C in supplemented PBS buffer. That is, imaging was performed within 45 min after removing the cell tray from the 37 °C incubator to ensure cell viability. No changes in cell morphology were observed after photoactivation. Moreover, previous studies using DCDHFs in living cells did not encounter complications with toxicity.
Photoaffinity Labeling and Gel Electrophoresis
Photoaffinity labeling (PAL) of azido fluorogens was performed on Chinese Hamster Ovary (CHO) cells. Under red lights, 20 µL of ~1-mM NBD-azide and DCDHF-P-azide stock solutions in ethanol (kept in the dark to prevent preactivation) were mixed with separate 200-µL aliquots of CHO cells suspended in PBS buffer. The mixed solution was photoactivated using a 365-nm handheld Hg UV lamp (0.4 mW cm−2) for 30 minutes. The nitrene intermediate inserted into bonds of accessible biomolecules. For a control, aliquots of the fluorogen stock solutions were preactivated before mixing with cells; the preactivated dye is unable to participate in the covalent PAL bioconjugating photoreaction because the nitrene does not survive for more than a few seconds (see above).
The CHO cells were then lysed using RIPA buffer, and passed through 200-µL pipette tip 50 times. The insoluble portion of the lysate was spun down at 4 °C, 14000 RPM and discarded. The soluble supernatant was mixed with SDS and heated to 95 °C. The lysate was then separated on a 12% polyacrylamide SDS gel to separate the PAL fluorescence from unbound fluorophores. After the electrophoresis completed, the gel was destained using a 10% acetic acid solution in water and methanol. To image the gels, a GE Healthcare Typhoon 8600 scanner was used with 488-nm excitation, a 526 short-pass filter, and 600 PMT sensitivity setting. The protein in the gel was then stained with Coomassie Brilliant Blue and destained overnight. The Coomassie bands were imaged using white light (BioRad Gel Doc scanner with Universal Hood II).
Synthesis
DCDHF-P-azide, 2-(4-(4-Azidophenyl)-3-cyano-5,5-dimethylfuran-2(5H)-ylidene)malononitrile
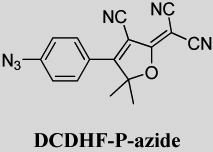
To a 200-mL round bottom flask with stirbar was added 2-[3-cyano-4-(4-fluorophenyl)-5,5-dimethyl-5H-furan-2-ylidene]-malononitrile (0.20 g, 0.716 mmol) and DMSO (8 mL). The mixture was stirred at room temperature, then sodium azide (0.08 g, 1.23 mmol) was added and the reaction continued to react at room temperature. After 2 h, the reaction mixture was completely homogeneous. The product mixture was poured into ice-water (300 mL) and stirred 0.5 h. The yellow precipitate was filtered off by suction filtration. The solid was recrystallized from 1-propanol to give the final product as a yellow solid (0.19 g, 88% yield). Mp 184 °C. IR (neat): 2924, 2228, 2113, 1567, 1533, 1366, 1279, 1188, 1111, 841 cm−1. 1H NMR (400 MHz, DMSO): δ 7.94 (ddd, J = 9.2, 2.8, 2.0 Hz, 2H), 7.40 (ddd, J = 9.2, 2.8, 2.0 Hz, 2H), 1.78 (s, 6H); 13C NMR (100 MHz, DMSO): δ 177.1, 176.6, 145.0, 130.6, 123.4, 120.3, 112.3, 111.4, 111.3, 101.6, 100.3, 55.2, 24.9. Anal. Calcd for C16H10N6O: C, 63.57; H, 3.33; N, 27.80%; found: C, 63.51; H, 3.44; N, 27.40%. UV–vis (CH2Cl2): λmax = 384 nm, ε = 2.9×104 L mol−1 cm−1.
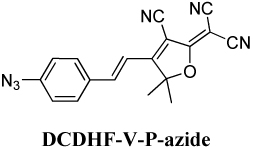
DCDHF-V-P-azide, (E)-2-(4-(4-Azidostyryl)-3-cyano-5,5-dimethylfuran-2(5H)-ylidene)malononitrile
Synthesis previously reported in supplemental material of reference 15. Literature procedures were followed for the synthesis of the precursors 4-azidobenzaldehyde35 and 3-cyano-2-dicyanomethylene-4,5,5-trimethyl-2,5-dihydrofuran.36 The 4-azidobenzaldehyde was isolated in 78% yield. Other reagents were commercially available and were used as received.
To a 100-mL round-bottom flask with stirbar was added 4-azidobenzaldehyde (0.30 g, 2 mmol), 2-(3-cyano-4,5,5-trimethyl-5H-furan-2-ylidene)-malononitrile (0.40 g, 2 mmol), pyridine (5 mL) and acetic acid (several drops). The mixture was stirred at room temperature for 2.5 days. TLC showed the desired azido product had been formed as the main product. The reaction was stopped and poured into ice-water (1 L). After stirring for 2 h, the precipitate was filtered off by suction filtration. The solid was recrystallized from 1-propanol to give the final product as a dark red solid (0.55 g, 84% yield). This compound decomposes at around 180 °C before melting (observed under microscope and DSC). IR (neat): 2227, 2119, 1600, 1530, 1281, 1184, 823 cm−1. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 16.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 2H), 7.0 (d, J = 16.4 Hz), 1.83 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 175.2, 173.5, 145.9, 144.8, 130.8, 130.5, 120.1, 114.3, 111.6, 110.8, 110.2, 99.9, 97.6, 51.1, 26.5. Anal. Calcd for C18H12N6O: C, 65.85; H, 3.68; N, 25.60%. Found: C, 65.58; H, 3.74; N, 25.94%. UV-Vis (CH2Cl2): λmax = 433 nm, ε = 2.7×104 L mol−1 cm−1.
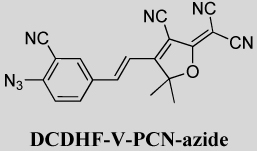
DCDHF-V-PCN-azide, 2-{4-[2[(4-Azido-3-cyano-phenyl)-vinyl-3-cyano-5,5-dimethyl-5H-furan-2-ylidene}malononitrile
3-Cyano-4-fluorobenzaldehyde
In a 50-mL single-neck flask, 3-bromo-4-fluorobenzaldehyde (2.5 g, 12.3 mmol) and CuCN (1.26 g, 14.3 mmol) was mixed with NMP (5 mL). The mixture was slowly warmed until the temperature reached 170 °C and stirred at that temperature for 40 h. TLC showed complete consumption of starting material, the temperature was gradually reduced to 80 °C, ethyl acetate (7 mL) was added dropwise followed by water (3 mL) and stirring was continued at rt for 20 min. The cold solution was filtered through a Celite pad and finally the solid was rinsed several times with ethyl acetate. The organic phase was separated, washed with saturated brine solution, dried over anhydrous MgSO4 and solvent was removed under reduced pressure. The crude product obtained was flash chromatographed on silica gel column eluted with hexane/ethyl acetate (3:1). The eluent was concentrated and the product separated as pale yellow solid. Yield: 1.2 g (65.4%). Mp: 85 °C (Lit mp: 85.5–87.5 °C). 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 8.11–8.17 (m, 2H), 7.40 (t, 8.4 Hz, 1H).
2-Azido-5-formyl-benzonitrile
In a 100-mL round-bottomed flask, sodium azide (600 mg, 9 mmol) was dissolved in DMSO (20 mL) 3-cyano-4-fluorobenzaldehyde (1.0 g, 6.7 mmol) was added and stirred at 50 °C for 6 h. The reaction mixture was cooled and poured into water (200 mL) and stirred at rt for 1 h. The precipitate formed was filtered, washed with water and air-dried. TLC and 1H NMR spectra showed the material was pure and used as such for reaction. Yield: 1.0 g (86.5%). Mp: 115 °C. IR (neat) 2869, 2141, 1696, 1680, 1579 cm−1. 1H NMR (CDCl3, 400 MHz) 9.97 (s, 1H), 8.13–8.15 (m, 2H), 7.28 (d, J= 8.8 Hz, 1H); 13CNMR (CDCl3) 188.4, 148.65, 135.73, 134.20, 132.92, 119.38, 114.34, 104.88.
2-{4-[2[(4-Azido-3-cyano-phenyl)-vinyl-3-cyano-5,5-dimethyl-5H-furan-2-ylidene}malononitrile
A mixture of 2-azido-5-formyl-benzonitrile (0.78 g, 4.5 mmol) and 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (924 mg, 4.6 mmol) was dissolved in ethanol/THF mixture (30 mL, 2:1) followed by ammonium acetate (359 mg, 4.65 mmol) was added, stirred at 0–25 °C for 16 h. TLC showed the starting material was completely consumed. The solvent was removed by rotary evaporation and the crude product was purified by silica gel column chromatography using hexane/EAC (30%) as eluent. The reddish product obtained was recrystallized from dichloromethane/1-propanol. Yield: 350 mg (22%). Mp: 178 °C IR (neat) 2920, 2228, 2134, 1622, 1586, 1553, 1381 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.85 (m, 2H), 7.54 (d, J = 16.4 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 16.4Hz, 1H), 1.78 (s, 6H); 13CNMR (100 MHz, DMSO) δ 177.00, 174.48, 145.57, 143.98, 135.29, 134.54, 131.37, 120.80, 116.53, 115.22, 112.59, 111.73, 110.56, 103.24, 100.40, 99.44, 55.50, 25.04. Anal. Calcd for C19H11N7O: C, 64.59; H, 3.14; N, 27.75; found: C, 64.25; H 3.44; N 27.50. UV–vis (EtOH): λmax = 415 nm.
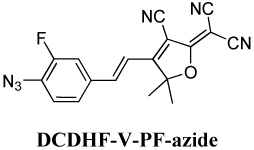
DCDHF-V-PF-azide, 2-{4-[2[(4-Azido-3-fluro-phenyl)-vinyl-3-cyano-5,5-dimethyl-5H-furan-2-ylidene}malononitrile
4-Azido-3- fluorobenzaldehyde
In a 100-mL round-bottomed flask, sodium azide (2.60 g, 40 mmol) was dissolved in DMSO (80 mL). To the mixture, 3,4-difluorobenzaldehyde (3.30 mL, 30 mmol) was added and stirred at 40 °C for 5 h. The reaction mixture was poured into water (200 mL) and stirred at rt for 2 h the precipitate formed was filtered, washed with water and dried under vacuum. Finally, the product was recrystallized from dichloromethane/1-propanol. Yield: 2.5g (50%). Mp: 43 °C. IR 2959, 2135, 1697, 1682, 1608, 1579, 1502 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 7.61–7.75 (m, 2H), 7.10–7.22 (m, 1H). 19F NMR (470 MHZ, CDCl3): −124.85 (m, 1F).
2-{4-[2[(4-Azido-3-fluro-phenyl)-vinyl-3-cyano-5,5-dimethyl-5H-furan-2-ylidene}malononitrile
A mixture of 4-azido-3-fluorobenzaldehyde (1.0 g, 6.0 mmol) and 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (1.20 g, 6.0 mmol) was dissolved in 25 mL pyridine and few drops of acetic acid was added. The mixture was stirred at room temperature for 48 h, poured into water, stirred for 2 h at room temperature, kept in the refrigerator overnight and the precipitate obtained was filtered and air dried. The crude product was purified by silica gel column chromatography using hexane/EAC (30%) as eluent and reddish brown product obtained was then recrystallized from dichloromethane/1-propanol. Yield: 250 mg (15%). Mp: 193 °C. IR (neat): 3092, 2330, 2130, 1602, 1513 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 16.4 Hz, 1H), 7.37 (m, 2H), 7.14 (t, 1H), 6.88 (d, J = 16.4 Hz, 1H), 1.78 (s, 6H). 13C NMR (CDCl3) 174.36, 172.67, 156.37, 153.86, 144.40, 144.37, 132.53, 131.55, 131.48, 126.10, 126.07, 121.89, 121.87, 116.17, 115.98, 115.26, 111.21, 110.46, 109.93, 100.50, 97.49, 26.28. 19F NMR (470 MHZ, CDCl3) –124.35 (s, 1F). HRMS (EI): Calcd for C18H11FN6O (M+) 369.08 found 369.08. UV–vis (CH2Cl2): λmax = 427 nm.
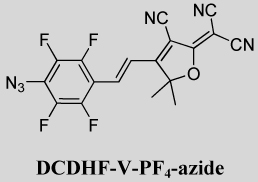
DCDHF-V-PF4-azide, (E)-2-(4-(4-Azido-2,3,5,6-tetrafluorostyryl)-3-cyano-5,5-dimethylfuran-2(5H)-ylidene)malononitrile
Previously reported in supplemental material of Pavani et al.37
4-Azido-2,3,5,6-tetrafluorobenzaldehyde: 38–41
To a 100-mL round-bottom flask with stirbar was added pentafluorobenzaldehyde (1.96 g, 0.01 mol), sodium azide (0.72 g, 0.011 mol), acetone (15 mL) and water (15 mL). The mixture was warmed to reflux under nitrogen for 10 h. TLC showed all the pentafluorobenzaldehyde was consumed and so the reaction was stopped and cooled to room temperature. The product mixture was diluted with 20 mL of water. The crude product was extracted with ether (30 mL ×5). The combined organic layer was dried over anhydrous MgSO4. The solvent was removed at room temperature under vacuum. Sublimation of the residue (50 °C/0.2 mm) gave the final product as a white solid (1.20 g, 55% yield). Mp 44 °C (lit 44–45 °C, reference 38). IR (neat): 3377, 2121, 1704, 1644, 1480, 1398, 1237, 1066, 1000, 615 cm−1. 1H NMR (400 MHz, CDCl3): δ 10.26 (m, 1H); 19F NMR (470 MHz, CDCl3): δ –149.6 (m, 2F), −155.6 (m, 2F).
(E)-2-(4-(4-Azido-2,3,5,6-tetrafluorostyryl)-3-cyano-5,5-dimethylfuran-2(5H)-ylidene)malononitrile
To a 100-mL round-bottom flask with stirbar was added 4-azido-2,3,5,6-tetrafluorobenzaldehyde (0.22 g, 1 mmol) and 2-(3-cyano-4,5,5-trimethyl-5H-furan-2-ylidene)-malononitrile (0.22 g, 1.1 mmol), 5 mL pyridine and several drops of acetic acid. The mixture was stirred at room temperature for 2.5 days. TLC showed the desired azido product had been formed as the main product. The reaction was stopped and poured into 500 mL ice water. After stirring for 2 h, the precipitate was filtered off by suction filtration. The solid was recrystallized from 1-propanol. After recrystallization, part of the azido product was converted to the corresponding amino compound. The mixture was adsorbed on silica gel, placed at the top of a silica column and eluted (CH2Cl2/EtOAc = 20:1). Fractions containing only the first product were combined and concentrated to give an orange product (40 mg, 10% yield). This is the final azido product, (E)-2-(4-(4-azido-2,3,5,6-tetrafluorostyryl)-3-cyano-5,5-dimethylfuran-2(5H)-ylidene)malononitrile. Recrystallization could not be done on this compound, since it has high photoreactivity and it readily converts to the corresponding amino compound in solvents (like propanol) in daylight. IR (neat): 2933, 2228, 2124, 1586, 1557, 1489, 1372, 1253, 998 cm−1. 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 16.8 Hz, 1H), 7.31 (d, J = 16.4 Hz, 1H), 1.82 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 174.5, 172.5, 146.9 (m), 144.4 (m), 142.0 (m), 139.4 (m), 130.7, 121.4 (t, J = 9.8 Hz), 111.1, 110.3, 109.5, 102.6, 97.8, 51.3, 26.3; 19F NMR (470 MHz, CDCl3): δ −143.5 (2F), −155.2 (2F). UV–vis (EtOH): λmax = 407 nm, ε = 2.7×104 L mol−1 cm−1.
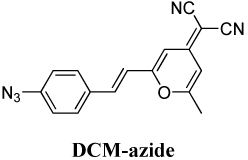
DCM-azide, 2-{2-[2-(4-Azidophenyl)-vinyl]-6-methylpyran-4-ylidene]-malononitrile
2-{2-[2-(4-Aminophenyl)-vinyl]-6-methylpyran-4-ylidene]-malononitrile
A mixture of 2,6-dimethylpyran-4-ylidene-malononitrile (1.2 g, 6.8 mmol), 4-aminobenzaldehyde (1.0 g, 8.2 mmol) and piperidine (0.68 mL, 6.9 mmol) was dissolved in 1-propanol (150 mL) and refluxed for 48 h. The reaction mixture was cooled, poured into water (500 mL), stirred for 5 h, the precipitate formed was filtered and air-dried. The crude product was purified by silica gel column chromatography using hexane/EAC 30–50% as eluent. The product was finally recrystallized from dichloromethane/1propanol. Yield: 1.50g (80%). Mp: 249 °C. IR (neat): 3478, 2957, 2200, 1647 cm−1. 1H NMR (400 MHz, DMSO) δ 7.36 (d, J = 8.4 Hz, 2H ), 7.31 (s, 1H), 6.91 (d, J = 16 Hz, 1H), 6.69 (bs, 1H), 6.57 (d, J = 16 Hz, 1H), 6.55 (d, J = 8.4Hz, 2H), 5.86 (s, 2H), 2.40 (s, 3H); 13C NMR (DMSO) δ 163.12, 160.71, 156.05, 151.12, 138.47, 129.61, 121.57, 115.31, 113.18, 111.53, 104.88, 104.22, 18.80. Anal. Calcd for C17H13N3O: C, 74.17, H, 4.76, N, 15.26; found: C, 73.91, H 4.54, N 15.38. UV–vis (CH2Cl2): λmax = 436 nm, ε = 2.73×104 L mol−1 cm−1.
2-{2-[2-(4-Azidophenyl)-vinyl]-6-methylpyran-4-ylidene]-malononitrile
A solution of NaNO2 (552 mg, 8.0 mmol) in 8 mL water was added drop wise to a solution of the 2-{2-[2-(4-aminophenyl)-vinyl]-6-methylpyran-4-ylidene]-malononitrile (1.10 g, 4.0 mmol) in 4M HCl (46 mL) at 0–5 °C. After stirring the mixture at this temperature for 45 min, a solution of NaN3 (520 mg, 8.0 m mol) in water (8 mL) was slowly added to the mixture at the same temperature. Stirring was continued for 1 h below 5 °C and then at room temperature for overnight. The precipitate obtained was filtered and air-dried. The crude product was purified by a silica gel column chromatography using hexane/EAC (7:3) as eluent and finally recrystallized from dichloromethane/1-propanol. Yield: 550 mg (45%) mp: 195 °C. IR (neat) 3078, 2208, 2118, 1655 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 16 Hz, 1H), 7.03 (d, J = 8.4 Hz, 2H), 6.64 (bs, 1H), 6.63 (d, J = 16 Hz, 1H), 6.50 (bs, 1H), 2.36 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.70, 158.63, 155.94, 141.83, 136.52, 131.02, 128.90, 119.47, 117.40, 114.68, 107.01, 106.22, 19.71. Anal. Calcd for C17H11N5O: C, 67.77, H, 3.68, N, 23.24. Found: C, 67.51, H, 3.64, N 22.97. UV–vis (CH2Cl2): λ max = 405 nm.
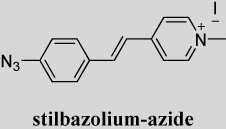
Stilbazolium-azide, 4-[2-(4-Azido-phenyl)-vinyl]-1-methyl-pyridinium iodide (4'-Azido-4-stilbazolium methiodide)
4-(2-Pyridin-4-yl-vinyl)-phenylamine (4'-amino-4-stilbazole)
Adapted from a literature method for preparing 4'-amino-4-stilbazoles as described by Loew, et al.42 To an oven-dried flask charged with nitrogen was added 4-iodoaniline (5.59 g, 25.5 mmol), 4-vinylpyridine (3.68 g, 35 mmol), palladium (II) acetate (14.4 mg, 0.064 mmol), tri-o-tolylphosphine (39.0 mg, 0.128 mmol), triethylamine (7.08 g, 70 mmol), and acetonitrile (25 mL). The flask was fitted with a reflux condenser and then charged again with nitrogen. A bubbler was quickly attached to the top of the condenser and then the flask was heated in an oil bath. The mixture was stirred at reflux for 48 hrs, after which the flask was removed from heat and cooled to room temperature. The mixture was poured into cold water and then the precipitate was collected via vacuum filtration. The product was placed in the vacuum oven and dried overnight at 55°C at approximately 20 mmHg vacuum until it reached a constant mass. The product was collected as a yellow powder in the amount of 4.26 g (85% yield). mp 270–274°C. 1H NMR (400MHz, DMSO-d6) δ 8.45 (dd, J = 1.6, 6.1 Hz, 2H), δ 7.44 (dd, J = 1.6, 6.1 Hz, 2H), δ 7.37 – 7.32 (m, 3H), δ 6.87 (d, J = 16.4 Hz, 1H), δ 6.58 (ddd, J = 1.9, 2.6, 8.6 Hz, 2H), δ 5.49 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 150.3, 145.7, 134.2, 129.0, 124.1, 120.6, 120.3, 114.2. λmax (CH3CN) = 348 nm (ε = 2.96×104 M−1 cm−1).
4-[2-(4-Azido-phenyl)-vinyl]-pyridine (4'-azido-4-stilbazole)
A solution of sodium nitrite (1.76 g, 25.5 mmol) in 10 mL water was added dropwise to a solution of 4-(2-pyridin-4-yl-vinyl)-phenylamine (2.0 g, 10.2 mmol) in 102 mL 4M HCl at 2–3°C. After stirring the mixture at this temperature for 1 hr, a solution of sodium azide (1.32 g, 20.4 mmol) in 10 mL water was slowly added dropwise, maintaining the temperature at 2–3°C. Stirring was continued for 30 min at this temperature and then the ice bath was allowed to come to ambient temperature overnight while stirring continued. Saturated aqueous sodium bicarbonate was added carefully to the mixture with stirring until the evolution of gases subsided. The precipitate was filtered out and dried overnight in the funnel to yield 1.875 g of the product as a beige powder (83% yield). mp 88.2 – 89.4°C. 1H NMR (400MHz, CDCl3) δ 8.51 (dd, J = 1.4, 6.1 Hz, 2H), δ 7.46 (ddd, J = 1.9, 2.7, 8.5 Hz, 2H), δ 7.29 (dd, J = 1.4, 6.1 Hz, 2H), δ 7.18 (d, J = 16.4 Hz, 1H), δ 6.98 (ddd, J = 2.0, 2.7, 8.5 Hz, 2H), δ 6.90 (d, J = 16.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 150.3, 144.4, 140.3, 133.1, 132.0, 128.4, 125.7, 120.8, 119.5. λmax (CH3CN) = 323 nm (ε = 3.92×104 M−1 cm−1). IR νmax (cm−1) = 2122 (N3).
4-[2-(4-Azido-phenyl)-vinyl]-1-methyl-pyridinium iodide (4'-Azido-4-stilbazolium methiodide)
In a 100 mL round bottom flask equipped with a stirbar and covered with foil, 4-[2-(4-azido-phenyl)-vinyl]-pyridine (0.57 g, 2.56 mmol) was mixed with iodomethane (0.95 g, 6.7 mmol) in acetonitrile (20 mL) and stirred at room temperature for 36 hrs. A precipitate had formed and TLC showed a single spot, so the solution was placed in the refrigerator for 30 minutes and then filtered, rinsing with diethyl ether. The product was collected as a yellow powder (0.482 g). Diethyl ether was added to the filtrates and then placed in the refrigerator for 2 hrs. Orange/brown crystals formed which were collected via filtration (0.15 g). The total amount of product collected was 0.63 g (68% yield). mp 166.2 – 168.0°C. 1H NMR (400MHz, DMSO-d6) δ 8.86 (d, J = 6.9 Hz, 2H), δ 8.20 (d, J = 6.9 Hz, 2H), δ 8.01 (d, J = 16.4 Hz, 1H), δ 7.80 (ddd, J = 1.8, 2.6, 8.5 Hz, 2H), δ 7.49 (d, J = 16.4 Hz, 1H), δ 7.26 (ddd, J = 1.8, 2.6, 8.5 Hz, 2H), δ 4.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 152.89, 145.56, 141.80, 140.08, 132.54, 130.31, 123.90, 123.28, 120.37, 47.39. λmax (CH3CN) = 373 nm (ε = 4.07×104 M−1 cm−1). IR νmax (cm−1) = 2118 (N3). Anal. Calcd for C14H13IN4: C, 46.17; H, 3.60; N, 15.38; found: C, 46.24; H, 3.69; N, 15.39.
NBD-azide, 7-Azido-4-nitrobenzoxadiazole
In a 50 mL round bottom flask equipped with a stirbar, 7-chloro-4-nitrobenzofurazan (1.5 g, 7.52 mmol) and sodium azide (0.54 g, 8.27 mmol) were stirred in ethanol (15 mL) for 6 hrs at 35°C. TLC indicated complete consumption of the starting material. The solution was poured into ice water, forming a precipitate, which was filtered and washed with water. The product was collected as a yellow powder in the amount of 1.45 g (94% yield). mp 101.1 – 101.9°C. 1H NMR (400MHz, CDCl3) δ 8.53 (d, J = 8.2 Hz, 1H), δ 7.08 (d, J = 8.2 Hz, 1H). 13C NMR (100MHz, CDCl3) δ 145.9, 143.6, 138.1, 132.2, 128.6, 114.9. λmax (CH2Cl2) = 388 nm (ε = 1.44×104 M−1 cm−1). IR νmax (cm−1) = 2114 (N3).
Results and Discussion
The various azido push–pull chromophores in Table 1 are all photoactivatable: upon pumping the blue-shifted absorption, the nonfluorescent azido fluorogens photoconvert to longer-wavelength amino versions that are fluorescent (e.g. see spectra in Figure 1 for NBD-azide; the other fluorogens follow the same trend and spectra may be found in the SI). Although there are several photoproducts, we confirmed in an earlier study that the fluorescent amino product of the DCDHF-V-P-azide photoreaction is dominant (via mass spectrometry and NMR analyses of the column- and HPLC-separated photoproducts described in Methods; see chemical reaction yields in Table 1). For the other fluorogens, the presence of an amino fluorophore in the photoreaction mixture is corroborated by recovery of the long-wavelength push–pull absorption and emission spectra. (In the case of DCM-azide, the absorption spectra of the photoreaction mixture include peaks that obscure the amino species; nevertheless, fluorescence excitation spectra reveal only one fluorescing species, which overlaps with the absorption curve of the independently synthesized DCM-amine. See SI.)
Taking into account both the localization precision43, 44 and the Nyquist–Shannon sampling,45, 46 the best emitters for photoactivation super-resolution imaging maximize the number of localized unique molecules per area per time.47 In other words, the probe must be easily photoactivated to avoid cell damage from short-wavelength illumination; must be bright; must emit many photons, then disappear; must densely label the sample; and must have a high contrast between on and off states. (For static structures, reversible switching is not ideal, because reactivation of an emitter that has already been localized is superfluous and only adds complexity to any image reconstruction.) The azido push–pull chromophores we present in this paper meet many of these critical requirements: several are photoconverted with high efficiency without a catalyst, emit millions of photons before irreversibly photobleaching, exhibit high turn-on ratios, and possess moderate molar absorption coefficients and quantum yields.
Photostability
One of the most important parameters for single-molecule fluorophores is photostability, one measure of which is the photobleaching quantum yield (ΦB), or the probability of bleaching with each photon absorbed (see Equation 1). A very low value of ΦB corresponds to not only a bright, long-lived fluorophore, but also higher localization precision for photoactivation imaging.43, 44
We have reported previously that the DCDHF molecules emit millions of photons and therefore have low values for ΦB,20, 23 and this is also true of some of the new activated fluorophores reported here. These photostable emitters enable precise localization and sophisticated imaging schemes: we demonstrated previously that single molecules of DCDHF-V-PF4-azide in PMMA can be photoactivated and localized to less than 20-nm standard deviation in all three dimensions; two molecules separated by 36 nm were resolved in three dimensions (see Figure 2B for an example of activation and imaging of single molecules).37
Figure 2.

(A) Two living Chinese Hamster Ovary (CHO) cells incubated with DCDHF-V-P-azide before activation and after a five-second flash of diffuse, moderate-irradiance (13 W cm−2) 407-nm light. The 594-nm light (500 W cm−2) for imaging was illuminating the sample the entire time, except for the brief period of 407-nm activation. False color: gray is the white-light transmission image and red the thresholded fluorescence images. Bar, 15 µm. (B) Snapshots from a movie of single molecules of DCDHF-V-PF4-amine embedded in a PMMA film immediately after photoactivation of the corresponding azide (see SI for the entire movie). The inset shows the same frame immediately before activation. Preactivated molecules were first prebleached using high-intensity 514-nm light, and then the sample was imaged at lower intensities (2 kW cm−2). To activate, a 100-ms flash of low-intensity 407-nm light (0.2 kW cm−2) was applied. Pseudocolor scale: the number of counts in this 100-ms frame ranges from 459 at the minimum pixel to 6644 at the brightest pixel (which corresponds to approximately 40–578 photons detected per pixel per 100 ms). Bars, 2 µm.
The photobleaching quantum yield is defined as the probability of photobleaching after absorbing a photon, or the ratio of the bleaching rate RB to the rate of absorbing photons Rabs:
| (1) |
where τB(P) is the decay constant in the exponential fit, the absorption cross-section is related to the molar absorption coefficient by the equation σλ = (1000)2.303ελ/NA = 2.1×10−16 cm2 for DCDHF-V-Pamine, Iλ is the irradiance at the sample, λ is the excitation wavelength, h is Planck’s constant, and c is the speed of light. (This definition will also be used for the photoconversion efficiency ΦP, see below.)
The average decay constant for a two-exponential fitting function, , is given by:
| (2) |
where fi = αiτi/∑jαjτj is the fractional area under the multi-exponential curve.32 Photobleaching quantum yield scales with the inverse of total number of photons emitted, and a lower value for ΦB indicates better photostability.
As shown in Table 1, adding electron-withdrawing fluorines to the aromatic group (DCDHF-V-PF-amine and DCDHF-V-PF4-amine) had little effect on the photostability parameter ΦB. DCM-amine is also a strong single-molecule emitter, with a ΦB comparable to most DCDHFs. DCM-amine has a higher fluorescence quantum yield in solution, and thus is more likely to be bright throughout a sample (not just in rigid environments, which is a feature of DCDHFs20, 23). While NBD-amine is brightly fluorescent in all environments, it is many times less photostable than DCDHFs. The stilbazolium-amine fluorophore is also not as photostable as the DCDHFs or even DCM-amine (a precise value was not determined).
Photoconversion Efficiency
The probability of photoconverting an azido fluorogen to any product per photon absorbed is the photoconversion quantum yield (ΦP), listed in Table 1 for the various push–pull fluorogens and demonstrated in Figure 1 right panel for DCDHF-P-azide. The higher the value of ΦP, the more the sensitive the fluorogen is to the activating light, so less potentially cell-damaging blue or UV irradiation is required to activate fluorescence.
Photoconversion was measured by monitoring changes over time in absorbance values of the reactant and photoproduct of interest in ethanol. The photoconversion quantum yield ΦP is defined in Equation 1 above, with τP as the average decay constant from the exponential fit of the decaying absorption values for the starting material (see Figure 1). Note that ΦP is the probability that the starting material will photoconvert for each photon absorbed; a fraction of those photoconverted molecules become fluorescent, because the photoreaction yield is less than unity.
Figure 2 shows that DCDHF-V-P-azide was readily activated in living CHO cells without obvious photodamage. By adding four electron-withdrawing fluorines to the benzene ring, we were able to further stabilize the nitrene and increase ΦP by more than a factor of two (compare DCDHF-V-P-azide and DCDHF-V-PF4-azide). Other acceptor groups and structures resulted in even higher ΦP values; for instance, NBD-azide needs to absorb only a few photons on average before photoreacting. These high photoconversion efficiencies enable activation at lower intensities; thus, super-resolution imaging by photoactivation should achieve higher cycling rates without causing photodamage to living cells. A drawback of experiments taking advantage of these very high ΦP values is that they require near complete darkness (or red lights) during sample preparation to prevent preactivation of the fluorogens. Stock fluorogen solutions in ethanol in the dark were resistant to thermal activation for several days or weeks (data not shown).
Turn-On Ratio
Photostability is not the only important parameter determining the ultimate resolution of a photoactivation image. Nyquist–Shannon sampling requires that there be at least two emitters per final resolution element, adding further restriction on the emitters: not only must the labeling be very dense, but also the turn-on ratio (i.e. the contrast between the bright and dark states of the molecule) must be very high, lest many weakly emitting “off” molecules in a diffraction-limited spot drown out the signal from the single “on” molecule.45–49 In other words, the limit of interest occurs when the background-subtracted intensity from one bright molecule Ion equals that of noff dark fluorogens (i.e. when Ion = noffIoff):
| (3) |
where data from DCDHF-V-P-azide in PMMA provide the numerical value. But the assumption that every dark molecule becomes fluorescent is not correct (i.e. non > noff). One could measure R by averaging over many single molecules; however, this would select only the fluorogens that become fluorescent, and would be artificially inflated. In other words, the simple ratio R is not an experimentally relevant parameter.
Alternatively, we measured an effective turn-on ratio that better corresponds to how many localizations we can get for a given region. In a bulk experiment in a PMMA film, we integrated the background-subtracted intensities over a large region before Soff and after Son activation (see Figure S6). Not all copies of the fluorogen convert to the fluorescent species, as the simple ratio R assumes above; the overall chemical reaction yield p can be 90% or lower (see Table 1). Therefore, the total emitters that will turn on is the reaction yield times the number of precursor molecules: non = pnoff. The ratio of the background-subtracted signals in a bulk experiment gives the effective turn-on ratio Reff, which is the experimentally relevant parameter:
| (4) |
The value Reff corresponds directly to the maximum number of localizations non in a diffraction-limited spot before the aggregate signal (noffIoff) of all the dark fluorogens required for that number of localizations equals the signal from one on molecule Ion. In the case of DCDHF-V-P-azide, we measured (multiple times using different samples, concentrations, and imaging parameters) the Reff to be 325 ± 15, which is over three times larger than that in ethanol.15 Moreover, the value of Reff is approximately a quarter of that of the R value measured on the single-molecule level; this discrepancy could imply that the reaction yield is p = 25%, or may result from preactivation (see below), or simply from different experimental conditions between the two experiments.
Approximating the diffraction limit to be 250 nm in diameter, the area of the diffraction-limited spot is about 50,000 nm2. With the maximum of 325 localizations in each diffraction-limited spot, the average distance between each localization is approximated by . The Nyquist–Shannon criterion45, 46 requires a sampling at least of twice the desired resolution, limiting the resolution to about 25 nm (in two dimensions). Our calculations here are similar to those in the supplemental material of Shroff et al.47
As a side note, this measured value of Reff should be considered a lower limit, because any preactivated molecules contribute to the signal in the frames before activation, thus reducing the measured value. The fraction q of preactivated molecules was kept low by protecting the fluorophore stock solution and samples from ambient room light; regardless, some preactivation does inevitably occur. The effect preactivation has on measuring Reff by including signal from preactivated molecules in the dark measurement is calculated by including the fraction q:
| (5) |
Thus, the multiplicative correction factor to convert from measured to true effective turn-on ratio is (1 + qR). Even 0.1% preactivation could artificially deflate the measured value by more than half (assuming the measured R of one isolated molecule is 1270). Therefore, minimizing preactivation (or, alternatively, maximizing prebleach as done for Figure 2B) before measuring the effective turn-on ratio can increase the value and bring it closer to the true ratio. However, prebleaching is not always an option—if the sample is highly light-sensitive—so Reff remains a practical measure of the lower limit for the turn-on ratio.
Photoaffinity Labeling
Besides super-resolution imaging,37 the azido push–pull fluorogens can be applied to other biological imaging schemes. For instance, the well-known photoaffinity labeling (PAL) requires a chemically inert binding molecule that becomes reactive upon illumination; the reactive photoproduct forms a covalent bond to a biomolecule to which it is bound or near. This technique has been used to study small-molecule binding, binding-pocket chemistries, and protein-protein interactions.50–53 Aryl azides are common PAL tags, because the nitrene intermediate is reactive and long-lived (we measured a lifetime of nearly 2 s, see Methods). Here, we demonstrate fluorogenic PAL: a dark precursor that can become both fluorescent and bioconjugated in one illumination step. Figure 3 shows proteins from CHO cells labeled with an azido push–pull fluorogen by PAL and purified by denaturing gel electrophoresis (see Methods). This demonstrates that nonspecific fluorogenic PAL is possible; moreover, it should be possible to engineer a binding pocket for one of these azido push–pull fluorogens, increasing the PAL reaction yield and producing a targeted fluorogenic bioconjugation system. Additional targeting strategies to place the fluorogen at a position of interest can also be envisioned.
Figure 3.

(A) A schematic of nonspecific fluorogenic photoaffinity labeling (PAL) of whole cells. The nitrene intermediate resulting from the photoconversion of an azide to an amine is reactive enough to insert into bonds of nearby biomolecules. The reaction simultaneously turns-on fluorescence and covalently links the probe to the biomolecule. (B) Gel electrophoresis of CHO-cell lysate demonstrating fluorogenic PAL using NBD-azide. (Similar results were observed with DCDHF-P-azide, data not shown.) The left panel shows the stained protein, imaged with white light; the right panel is fluorescence from NBD-amine photochemically cross-linked to proteins, imaged using 488 nm. The left lane in the gel (+PAL) is protein covalently labeled with NBD-amine by PAL. The right lane (−PAL) is a control performed by mixing into the cell solution preactivated NBD-amine, which is fluorescent but cannot participate in the covalent PAL bioconjugation reaction. The fluorescence signal in the control lane was significantly lower. The blurry fluorescence on the bottom of the gel is from the unbound dye at the front edge; equal brightness in both lanes indicates equal dye concentration in the PAL and control.
Some azide-based fluorogenic PAL ligands have been reported previously.54–58 In these earlier studies, the light required to excite fluorescence is in the high-energy ultraviolet (e.g. 280–350 nm), which prohibits ultrasensitive detection in living cells, because these short wavelengths pump cellular autofluorescence and cause cell damage. Moreover, photoconversion in these previous cases did not produce fluorophores photostable enough to be applied to single-molecule imaging. Our azido push–pull fluorogens can be activated and imaged using visible light, and several produce fluorophores that can be easily imaged on the single-molecule level.
Conclusion
A wide variety of azido fluorogens are photoactivatable to produce bright push–pull fluorophores, some of which emit millions of photons before photobleaching. For instance, the various DCDHF derivatives as well as DCM should prove to be high-quality single-molecule emitters given their photostability and brightness (especially in rigid environments). In addition, the azido push–pull molecules can participate in fluorogenic PAL reactions to label biomolecules covalently in situ. For cell imaging, these fluorogens will ultimately need to be targeted to biomolecules of interest, but in any case do not require the addition of other chemicals (e.g. catalysts, thiols, or oxygen scavengers). Azido push-pull fluorogens thus provide a useful new class of photoactivatable probes for a variety of applications in biological labeling and super-resolution imaging.
Supplementary Material
Acknowledgments
We thank Marissa K. Lee and Nathan R. Hobbs for assistance. This work was supported in part by Award Number R01GM086196 from the National Institute of General Medical Sciences.
Footnotes
Supporting Information Available: Spectra of all fluorogens, live-cell images, activation flashlight spectrum, images from effective turn-on ratio measurements, PAL results with pure BSA protein, general schemes, and a single-molecule movie. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 2.Hess ST, Girirajan TPK, Mason MD. Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rust MJ, Bates M, Zhuang X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moerner WE. New Directions in Single-Molecule Imaging and Analysis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/Off Blinking and Switching Behavior of Single Green Fluorescent Protein Molecules. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 6.Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Photoactivatable Fluorescent Proteins. Nat. Rev. Mol. Cell Biol. 2005;6:885–891. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- 7.Lippincott-Schwartz J, Patterson GH. Fluorescent Proteins for Photoactivation Experiments. Methods Cell Biol. 2008;85:45–61. doi: 10.1016/S0091-679X(08)85003-0. [DOI] [PubMed] [Google Scholar]
- 8.Bates M, Blosser TR, Zhuang X. Short-Range Spectroscopic Ruler Based on a Single-Molecule Switch. Phys. Rev. Lett. 2005;94:108101-1–108101-4. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu M, Zhu L, Han JJ, Wu W, Hurst JK, Li ADQ. Spiropyran-Based Photochromic Polymer Nanoparticles with Optically Switchable Luminescence. J. Am. Chem. Soc. 2006;128:4303–4309. doi: 10.1021/ja0567642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukaminato T, Umemoto T, Iwata Y, Yokojima S, Yoneyama M, Nakamura S, Irie M. Photochromism of Diarylethene Single Molecules in Polymer Matrices. J. Am. Chem. Soc. 2007;129:5932–5938. doi: 10.1021/ja069131b. [DOI] [PubMed] [Google Scholar]
- 11.Fölling J, Belov V, Kunetsky R, Medda R, Schönle A, Egner A, Eggeling C, Bossi M, Hell SW. Photochromic Rhodamines Provide Nanoscopy with Optical Sectioning. Angew. Chem. Int. Ed. 2007;46:6266–6270. doi: 10.1002/anie.200702167. [DOI] [PubMed] [Google Scholar]
- 12.Conley NR, Biteen JS, Moerner WE. Cy3-Cy5 Covalent Heterodimers for Single-Molecule Photoswitching. J. Phys. Chem. B. 2008;112:11878–11880. doi: 10.1021/jp806698p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han G, Mokari T, Ajo-Franklin C, Cohen BE. Caged Quantum Dots. J. Am. Chem. Soc. 2008;130:15811–15813. doi: 10.1021/ja804948s. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Suarez M, Ting AY. Fluorescent Probes for Super-Resolution Imaging in Living Cells. Nat. Rev. Mol. Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 15.Lord SJ, Conley NR, Lee HD, Samuel R, Liu N, Twieg RJ, Moerner WEA. Photoactivatable Push–Pull Fluorophore for Single-Molecule Imaging in Live Cells. J. Am. Chem. Soc. 2008;130:9204–9205. doi: 10.1021/ja802883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicoud JF, Twieg RJ. Nonlinear Optical Properties of Organic Molecules and Crystals. In: Chemla DS, Zyss J, editors. Design and Synthesis of Organic Molecular Compounds for Efficient Second Harmonic Generation. Vol. 1. New York: Academic Press; 1987. pp. 227–296. [Google Scholar]
- 17.Debreczeny MP, Svec WA, Wasielewski MR. Optical Control of Photogenerated Ion Pair Lifetimes: An Approach to a Molecular Switch. Science. 1996;274:584–587. [Google Scholar]
- 18.Ostroverkhova O, Moerner WE. Organic Photorefractives: Mechanisms, Materials, and Applications. Chem. Revs. 2004;104:3267–3314. doi: 10.1021/cr960055c. [DOI] [PubMed] [Google Scholar]
- 19.Willets KA, Ostroverkhova O, He M, Twieg RJ, Moerner WE. New Fluorophores for Single-Molecule Spectroscopy. J. Am. Chem. Soc. 2003;125:1174–1175. doi: 10.1021/ja029100q. [DOI] [PubMed] [Google Scholar]
- 20.Willets KA, Nishimura SY, Schuck PJ, Twieg RJ, Moerner WE. Nonlinear Optical Chromophores as Nanoscale Emitters for Single-Molecule Spectroscopy. Acc. Chem. Res. 2005;38:549–556. doi: 10.1021/ar0401294. [DOI] [PubMed] [Google Scholar]
- 21.Lord SJ, Lu Z, Wang H, Willets KA, Schuck PJ, Lee HD, Nishimura SY, Twieg RJ, Moerner WE. Photophysical Properties of Acene DCDHF Fluorophores: Long-Wavelength Single-Molecule Emitters Designed for Cellular Imaging. J. Phys. Chem. A. 2007;111:8934–8941. doi: 10.1021/jp0712598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Z, Liu N, Lord SJ, Bunge SD, Moerner WE, Twieg RJ. Bright, Red Single-Molecule Emitters: Synthesis and Properties of Environmentally Sensitive Dicyanomethylenedihydrofuran (DCDHF) Fluorophores with Bisaromatic Conjugation. Chem. Mater. 2009;21:797–810. doi: 10.1021/cm801783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lord SJ, Conley NR, Lee HD, Nishimura SY, Pomerantz AK, Willets KA, Lu Z, Wang H, Liu N, Samuel R, Weber R, Semyonov AN, He M, Twieg RJ, Moerner WE. DCDHF Fluorophores for Single-Molecule Imaging in Cells. ChemPhysChem. 2009;10:55–65. doi: 10.1002/cphc.200800581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doub L, Vandenbelt JM. The Ultraviolet Absorption Spectra of Simple Unsaturated Compounds. I. Mono- and p-Disubstituted Benzene Derivatives. J. Am. Chem. Soc. 1947;69:2714–2723. [Google Scholar]
- 25.Stevenson PE. Effects of Chemical Substitution on the Electronic Spectra of Aromatic Compounds: Part I. the Effects of Strongly Perturbing Substituents on Benzene. J. Mol. Spectrosc. 1965;15:220–256. [Google Scholar]
- 26.Bouffard J, Kim Y, Swager TM, Weissleder R, Hilderbrand SA. A Highly Selective Fluorescent Probe for Thiol Bioimaging. Org. Lett. 2008;10:37–40. doi: 10.1021/ol702539v. [DOI] [PubMed] [Google Scholar]
- 27.Budyka MF, Biktimirova NV, Gavrishova TN, Kozlovskii VI. Azido Derivative of a Hemicyanine Dye with High Sensitivity to Visible Light. Mendeleev Commun. 2007;17:159–160. [Google Scholar]
- 28.Hansch C, Leo A, Taft RW. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 29.Schriven EFV, editor. Azides and Nitrenes: Reactivity and Utility. Orlando, FL: Academic Press; 1984. [Google Scholar]
- 30.Soundararajan N, Platz MS. Descriptive Photochemistry of Polyfluorinated Azide Derivatives of Methyl Benzoate. J. Org. Chem. 1990;55:2034–2044. [Google Scholar]
- 31.Cline MR, Mandel SM, Platz MS. Identification of the Reactive Intermediates Produced upon Photolysis of p-Azidoacetophenone and its Tetrafluoro Analogue in Aqueous and Organic Solvents: Implications for Photoaffinity Labeling. Biochemistry. 2007;46:1981–1987. doi: 10.1021/bi061269j. [DOI] [PubMed] [Google Scholar]
- 32.Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Springer Science; 2006. [Google Scholar]
- 33.Moerner WE, Fromm DP. Methods of Single-Molecule Fluorescence Spectroscopy and Microscopy. Rev. Sci. Instrum. 2003;74:3597–3619. [Google Scholar]
- 34.Vrljic M, Nishimura SY, Moerner WE. McConnell, H. M. Cholesterol Depletion Suppresses the Translational Diffusion of Class II Major Histocompatibility Complex Proteins in the Plasma Membrane. Biophys. J. 2005;88:334–347. doi: 10.1529/biophysj.104.045989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartoszewska B, Szczerek I. Synthesis and use of 2,6-Bis(p-Azidobenzylidene)Cyclohexanone. Organika. 1979:31–35. [Google Scholar]
- 36.Moerner WE, Twieg RJ, Kline DW, He M. Fluorophore Compounds and Their Use in Biological Systems. 10/604,282. US Patent Application. 2005 Application number.
- 37.Pavani SRP, Thompson MA, Biteen JS, Lord SJ, Liu N, Twieg RJ, Piestun R, Moerner WE. Three-Dimensional, Single-Molecule Fluorescence Imaging Beyond the Diffraction Limit by using a Double-Helix Point Spread Function. Proc. Nat. Acad. Sci. U. S. A. 2009;106:2995–2999. doi: 10.1073/pnas.0900245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keana JFW, Cai SX. New Reagents for Photoaffinity Labeling: Synthesis and Photolysis of Functionalized Perfluorophenyl Azides. J. Org. Chem. 1990;55:3640–3647. [Google Scholar]
- 39.Keana JFW, Cai SX. Functionalized Perfluorophenyl Azides: New Reagents for Photoaffinity Labeling. J. Fluorine Chem. 1989;43:151–154. [Google Scholar]
- 40.Stromgaard K, Saito DR, Shindou H, Ishii S, Shimizu T, Nakanishi K. Ginkgolide Derivatives for Photolabeling Studies: Preparation and Pharmacological Evaluation. J. Med. Chem. 2002;45:4038–4046. doi: 10.1021/jm020147w. [DOI] [PubMed] [Google Scholar]
- 41.Wiegand M, Lindhorst TK. Synthesis of Photoactive Alpha-Mannosides and Mannosyl Peptides and their Evaluation for Lectin Labeling. Euro. J. Org. Chem. 2006;2006:4841–4851. [Google Scholar]
- 42.Hassner A, Birnbaum D, Loew LM. Charge-Shift Probes of Membrane Potential. Synthesis. J. Org. Chem. 1984;49:2546–2551. [Google Scholar]
- 43.Thompson RE, Larson DR, Webb WW. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ober RJ, Ram S, Ward ES. Localization Accuracy in Single-Molecule Microscopy. Biophys. J. 2004;86:1185–1200. doi: 10.1016/S0006-3495(04)74193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nyquist H. Certain Topics in Telegraph Transmission Theory. Trans. AIEE. 1928;47:617–644. [Google Scholar]
- 46.Shannon CE. Communication in the Presence of Noise. Proc. IRE. 1949;37:10–21. [Google Scholar]
- 47.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-Cell Photoactivated Localization Microscopy of Nanoscale Adhesion Dynamics. Nat. Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-Color Superresolution Imaging of Genetically Expressed Probes within Individual Adhesion Complexes. Proc. Nat. Acad. Sci. U. S. A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biteen JS, Thompson MA, Tselentis NK, Bowman GR, Shapiro L, Moerner WE. Super-Resolution Imaging in Live Caulobacter Crescentus Cells using Photoswitchable EYFP. Nat. Methods. 2008;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotzyba-Hibert F, Kapfer I, Goeldner M. Recent Trends in Photoaffinity Labeling. Angew. Chem. Int. Ed. 1995;34:1296–1312. [Google Scholar]
- 51.Vodovozova E. Photoaffinity Labeling and its Application in Structural Biology. Biochemistry (Moscow) 2007;72:1–20. doi: 10.1134/s0006297907010014. [DOI] [PubMed] [Google Scholar]
- 52.Dormán G, Prestwich GD. Using Photolabile Ligands in Drug Discovery and Development. Trends Biotechnol. 2000;18:64–77. doi: 10.1016/s0167-7799(99)01402-x. [DOI] [PubMed] [Google Scholar]
- 53.Baruah H, Puthenveetil S, Choi Y, Shah S, Ting AY. An Engineered Aryl Azide Ligase for Site-Specific Mapping of Protein-Protein Interactions through Photo-Cross-Linking. Angew. Chem. Int. Ed. 2008;47:7018–7021. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dockter ME. Fluorescent Photochemical Surface Labeling of Intact Human Erythrocytes. J. Biol. Chem. 1979;254:2161–2164. [PubMed] [Google Scholar]
- 55.Dreyfuss G, Schwartz K, Blout ER, Barrio JR, Liu F, Leonard NJ. Fluorescent Photoaffinity Labeling: Adenosine 3',5'-Cyclic Monophosphate Receptor Sites. Proc. Natl. Acad. Sci. U. S. A. 1978;75:1199–1203. doi: 10.1073/pnas.75.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thevenin B, Shahrokh Z, Williard R, Fujimoto E, Kang J, Ikemoto N, Shohet S. A Novel Photoactivatable Cross-Linker for the Functionally-Directed Region-Specific Fluorescent Labeling of Proteins. Eur. J. Biochem. 1992;206:471–477. doi: 10.1111/j.1432-1033.1992.tb16949.x. [DOI] [PubMed] [Google Scholar]
- 57.Moreland RB, Dockter ME. Preparation and Characterization of 3-Azido-2,7-Naphthalene Disulfonate: A Photolabile Fluorescent Precursor Useful as a Hydrophilic Surface Probe. Anal. Biochem. 1980;103:26–32. doi: 10.1016/0003-2697(80)90230-4. [DOI] [PubMed] [Google Scholar]
- 58.Angelides KJ. Application of Photoactivatable Fluorescent Active-Site Directed Probes to Serine-Containing Enzymes. Biochim. Biophys. Acta Protein Struct. 1981;669:149–156. doi: 10.1016/0005-2795(81)90236-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.