

Abstract
Cytoskeletal protein phosphorylation is frequently altered in neuropathologic states but little is known about changes during normal aging. Here we report that declining protein phosphatase activity, rather than activation of kinases, underlies aging-related neurofilament hyperphosphorylation. Purified PP2A or PP2B dephosphorylated the heavy neurofilament (NFH) subunit or its extensively phorphorylated carboxyl-terminal domain in vitro. In cultured primary hippocampal neurons, inhibiting either phosphatase induced NFH phosphorylation without activating known neurofilament kinases. Neurofilament phosphorylation in the mouse CNS, as reflected by levels of the RT-97 phosphoepitope associated with late axon maturation, more than doubled during the 12 month period after NFH expression plateaued at p21. This was accompanied by declines in levels and activity of PP2A but not PP2B, and no rise in activities of neurofilament kinases (Erk1,2, cdk5 and JNK1,2). Inhibiting PP2A in mice in vivo restored brain RT-97 to levels seen in young mice. Declining PP2A activity, therefore, can account for rising neurofilament phosphorylation in maturing brain, potentially compounding similar changes associated with adult-onset neurodegenerative diseases.
Keywords: neurofilament, phosphorylation, dephosphorylation, kinases, phosphatases, maturation, aging, RT-97 epitope, immunoreactivity
1. Introduction
Neurofilaments (NFs) are the major cytoskeletal components of large myelinated axons and their subunits are among the most highly phosphorylated brain proteins. NFs in the central nervous system (CNS) consist of the four subunits, α-internexin, NFH (High), NFM (Medium), and NFL (Low) (Chin and Liem 1990; Yuan, et al. 2009), each containing a short head domain, an α helical rod domain, and a carboxyl-terminal tail domain that varies in length depending on the subunit. The exceptionally long tail domains of NFM and NFH contain 5 and 52 KS/TP repeats, respectively, which are endogenously phosphorylated by proline directed kinases (Nixon and Sihag 1991; Pant, et al. 2000; Sihag et al., 2007) (Fig. 1).
Figure 1.

A cartoon of NF-H shows its polypeptide domains (A) and sequences of the KSPXXXK and KSPXK type of recombinant proteins derived from the rat and human NF-H tail domains respectively (B).
Phosphorylation causes the tail domains to project perpendicularly from the neurofilament and act as “sidearms” that regulate filament spacing (Nixon and Sihag 1991) ( Elder, et al. 1998; Mukhopadhyay, et al. 2004; Sihag, et al. 2007). These events stabilize a large cross-linked NF network that is a structural framework enabling the marked expansion of axon caliber during maturation required for proper impulse conduction (Gasser and Grundfest 1939a; Gasser and Grundfest 1939b;. Nixon and Lewis 1986; Nixon and Logvinenko 1986) and the optimal topographical organization of vesicular organelles and receptors within axons and synaptic terminals (Ehlers, et al. 1998; Kim, et al. 2002; Perrot and Julien 2009; Rao, et al. 2009). The appearance of the phosphorylation-dependent epitope RT-97 on NFH and NFM sidearms is an important marker of the establishment of a metabolically stable stationary cytoskeleton and the axon caliber growth initiated after axons establish synaptic connections and acquire myelin. Importantly, increased levels of the RT-97 phosphoepitope on NF proteins and tau, especially in neuronal perikarya, identifies affected neurons in certain neurodegenerative diseases, including Alzheimer’s disease (AD), where cytoskeletal protein hyperphosphorylation is believed to contribute to disease pathogenesis (Anderton, et al. 1982; Kesavapany, et al. 2007; Sternberger, et al. 1985; Veeranna, et al. 2004).
The RT-97 phosphoepitope has been shown to be regulated by MAPK family members, such as Erks and JNKs, that phosphorylate KSPXK and KSPXXXK motifs along NFH and NFM tail domains (Giasson and Mushynski 1997; Veeranna, et al. 1998) and also by cdk5, which phosphorylates only KSPXK sites (Shetty, et al. 1993). Evidence that phosphate groups turn over on NF proteins in vivo (Nixon and Lewis 1986) suggests that phosphatases may also be important in regulating phosphate topography along cytoskeletal polypeptides, as underscored by studies of cytoskeletal proteins in neurodegenerative diseases (Gong, et al. 1993; Gong, et al. 1995; Gong, et al. 2000; Kesavapany, et al. 2007; Veeranna, et al. 2004). In AD brain, for example, abnormal hyperphosphorylation of NF and tau is accompanied by significantly decreased mRNA expression, protein levels (Vogelsberg-Ragaglia, et al. 2001), and methylation (Sontag, et al. 2004) of protein phosphatase 2A (PP2A) and lowered protein levels of protein phosphatase 1 (PP1) (Gong, et al. 1993; Gong, et al. 1995; Gong, et al. 2000). These changes are believed to compound effects of additional disease-related activation of certain protein kinases that can phosphorylate NF proteins and tau (Ferrer, et al. 2005; Pei, et al. 2002; Veeranna, et al. 2004; Wang, et al. 2001; Webber, et al. 2005).
Despite the intense interest in the abnormal phosphorylation of the cytoskeleton in relation to disease pathogenesis, changes in the state of phosphorylation of cytoskeletal proteins during the course of normal brain maturation and aging are not well characterized or understood in terms of underlying molecular mechanisms. In the present study, we investigated these issues in hippocampal neurons and the normal mouse CNS. These studies strongly implicate declining activity of PP2A in aging brain. This decline would be expected to compound similar changes in phosphatase activities and protein hyperphosphorylation in major aging-related neurodegenerative diseases, thereby, providing further understanding of how brain aging may contribute to late-onset neurological disease.
2. Materials and Methods
Mice, antibodies and other reagents
Aged C57BL6 mice (Rodent Resource Facility of the National Institute of Aging, Charles River Laboratories, Wilmington, MA) acclimatized for one week in the Nathan S. Kline Institute for Psychiatric Research (NKI) animal facility. All animal experiments were performed according to “Principles of Animal Care” (NIH 1985) and approved by the Institutional Animal Care and Use Committee at the NKI. We obtained the following antibodies commercially, monoclonal antibody SMI- 33 dephospho-epitope on NFH and NFM (Sternberger Monoclonal Inc. Baltimore, MD); polyclonal antibodies against p35 and cdk5 (Santa Cruz Biotechnology Inc., Santa Cruz, CA); phospho-Erk1,2, phospho-independent Erk1,2, phospho JNK 1,2, JNK 1,2 and antibodies to catalytic subunits of PP2A and PP2Bα (Cell Signaling, Boston, MA) and PP1 assay kit ( New England Biolabs Inc, Ipswich, MA). A rabbit polyclonal antibody against NFL was made in this laboratory. The RT-97 monoclonal antibody clone was a kind gift from Brian Anderton (Institute of Psychiatry, London). Anti-mouse and anti-rabbit secondary antibodies conjugated to alkaline phosphatase (Promega, Madison, WI); or horseradish peroxidase conjugated anti-mouse and anti-rabbit secondary antibodies (Jackson Immunochemical Laboratories, Baltimore, MD); or Alexa 488 and Alexa 568 tagged secondary antibodies and all cell culture reagents (Invitrogen, Carlsbad, CA). Recombinant Erk2 and MEK1 proteins were kind gifts from Dr. N.G. Ahn (University of Colorado, Boulder, CO). Additional commercial reagents, included microcystin LR (Calbiochem, San Diego, CA); okadaic acid (OA) and cyclosporine A (Biomol, Plymouth Meeting, PA); purified bovine brain PP2B (Sigma, St. Louis, MO); purified PP2A, monoclonal antibody to catalytic subunit of PP2A clone 1D6 and a polyclonal antibody to catalytic subunit of PP1 α (Millipore, Bedford, MA); γ−32P ATP (Perkin Elmer Life Sciences, Boston, MA); ECL kit (GE Health care, Piscataway, NJ); P81 phosphocellulose paper (Whatman, Maidstone, UK); and dialysis tubing (Spectrum, Los Angeles, CA).
Preparation and expression of neurofilament proteins
The NF protein pellet was prepared from mouse spinal cords as described earlier (Veeranna, et al. 1995; Veeranna, et al. 1998). A rat NFH tail fragment with 24 KSPXXXK repeats and another fragment derived from human NFH with 14 KSPXK repeats, both tagged with GST fusion protein, were expressed and purified as described previously ( Pant, et al. 1997; Veeranna, et al. 1998). Full length NFH was expressed as described earlier (Takahashi, et al. 1995).
Phosphorylation and dephosphorylation of phosphatase substrates
Phosphorylation of the bacterially expressed NFH by cdk5 and Erk,2 was carried out essentially as described previously (Shetty, et al. 1993; Veeranna, et al. 1995). Phosphorylation of KSPXK fusion protein by cdk5 and KSPXXXK GST fusion protein by Erk2 was performed as described earlier (Veeranna, et al. 1995; Veeranna, et al. 1998). We performed dephosphorylation of 32P-labeled substrates using PP2A and PP2B as described earlier (Mitsuhashi, et al. 2000; Veeranna, et al. 1995), although transfer of the 32P labeled substrate was performed without further purification after phosphorylation, the phosphatase reaction was carried out in the presence of olomoucine (1 mM) and U-0126 ( 50 µM ) or roscovitine (50 µM). Purified phosphatases, PP2A and PP2B obtained commercially, were used to dephosphorylate recombinant NFH and KSPXXXK fusion proteins that had been phosphorylated by recombinant cdk5 and Erk2 respectively. After dephosphorylation, they were subjected to SDS PAGE and the gels were silver stained and dried before autoradiography to monitor loss of 32P labeling. Alternatively, the gels were subjected to electrotransfer and Western blot analysis to visualize loss of phospho-dependent immunoreactivity (IR) after dephosphorylation.
Immunoprecipitation and activity measurement of PP2A
Each spinal cord was homogenized (1:5 W/V) in 50 mM Hepes buffer (pH 7.5) containing EDTA (0.1 mM), NaCl (120 mM), AEBSF (0.1 mM), 0.5% NP-40, and 25 µg/ml each of leupeptin, aprotinin and pepstatin. Homegenates were centrifuged for 30 min at 15000 × g using a table top refrigerated centrifuge. Protein (1.25 mg) from the supernatant was mixed with 20 µl of A plus G agarose beads pre-coupled to 5 µg of PP2Ac primary antibody clone 1D6 (Millipore Corporation, Bedford, MA) and incubated for 2 hrs at 4°C. Beads were washed twice with 10 volumes of homogenization buffer with a final wash of 50 mM Tris (pH 7.5) phosphatase assay buffer containing MgCl2 (10 mM), MnCl2 (1 mM), 0.02% Brij-35, and BSA (1 mg/ml). PP2A activity was monitored as described earlier (Veeranna, et al. 1995) in the presence and absence of 10 nM OA, using an aliquot of the immunoprecipitate as enzyme source and 32P labeled histone 1 phosphorylated by baculovirus expressed cdk5/p25 complex (Millipore Corporation, Bedford, MA) as substrate.
Measurement of activity of protein phosphatase 1 (PP1)
The supernatants obtained after the immunoprecipitation of PP2Ac from spinal cords were used for the measurement of PP1 activity with an assay kit as described (Cohen et al 1989). Myelin basic protein phosphorylated by PKA catalytic subunit was used as a substrate to assay the PP1 activity at 25° C in triplicate in three separate sets of samples. In order to obtain the activity specific to PP1, the assays were carried out in the presence and absence of 2nM and 1 µM Okadaic acid which inhibits PP2A and PP1 activity at respective concentrations and the data were calculated per g protein and represented as percent dephosphorylation
Primary neuronal cultures and treatment with phosphatase inhibitors
Primary hippocampal neurons were established from embryonic day-19 Sprague Dawley rat embryos (Charles River Laboratories, Wilmington, MA). Rat pups were decapitated and hippocampal regions were dissected from cerebral cortices in Hibernate-E media (Brain Bits, Springfield, IL). Dissociated hippocampal neurons were obtained by incubating the hippocampi in Hibernate-E containing 15 units/ml of papain (Worthington Biochemicals, NJ) for 15 mins at 37°C before triturating in Neurobasal medium containing 20% fetal bovine serum (Hyclone, Logan, UT), DNAse (0.2 mg/ml) and 0.1M MgSO4. Undissociated neurons were removed from the cell suspension by passing the cell suspension through a 40 µm cell strainer (Fisher Scientific, NY). Neurons were centrifuged at 200 × g for 3 mins at 20°C and the pellet was resuspended in Neurobasal medium supplemented with B27, penicillin (100 U/ml), streptomycin (100 U/ml) and L-glutamine (0.5 mM) (Invitrogen, Carlsbad, CA). Neurons were then plated at a density of 150,000 cells/ml on circular glass coverslips and 6-well tissue culture dishes, coated with poly-D-lysine (50 µg/ml, Sigma, St. Louis, MO), and incubated in a humidified atmosphere containing 5% CO2: 95% O2 at 37°C. The following drugs were investigated: OA (10 nM) to specifically inhibit PP2A; anisomycin to stimulate JNKs; veratridine (1 and 5 µM) and cyclosporine A (1 and 5 µM) or both combined to inhibit calcineurin (PP2B). Each inhibitor was added to the media 7 days after cell plating and after 24 hrs, neurons were harvested and analyzed for RT-97 IR.
Immunofluorescence labeling of hippocampal neurons
Twenty four hrs after treatments, hippocampal neurons were fixed in 4% paraformaldehyde in PBS (pH 7.4), permeabilized for 20 mins in 0.2% Triton-X-100, blocked in 4% normal goat serum (NGS) in PBS for 1 hr and incubated with primary antibodies against NFH (RT-97) diluted in 4% NGS in phosphate buffered saline containing 0.2% Triton X 100 for 1 hr at room temperature (RT). After three washes in blocking solution, the neurons were incubated with anti-rabbit and anti-mouse Alexa 488 or Alexa 568. Secondary antibodies were diluted in the same buffer as the primary antibodies and incubated for 1 hr at RT. Cells were washed and mounted on the cover slips and analyzed by laser confocal microscopy (Leica, Wetzlar, Germany) using a TCS software program.
Intracerebral injection of Okadaic acid
Mice were anesthetized and placed on a stereotaxic apparatus with a mouse adapter (David Kopf Instruments), and the head was leveled in the x, y, and z planes using the sagittal suture, lambda and the bregma as landmarks (Franklin and Paxinos 1997). The body temperature of the mouse was monitored with a heating pad. The scalp was shaved and a midline incision made starting slightly behind the eyes, exposing the skull area. A hole was drilled in the skull and a 32-gauge needle of 0.5µl syringe (Hamilton, Reno, NV) was inserted into the striatum −0.1 mm anterior-posterior (AP), −2 mm medial-lateral (ML), and 3 mm dorsal-ventral (DV), using the bregma as a reference for AP and ML coordinates and the skull as a reference for DV coordinates. One min after the needle was inserted, 0.3 µl of OA solution (100 µM) or PBS was injected at a constant flow rate over 90 secs. To prevent reflux of the injected solution along the needle track, the needle remained stationary in this configuration for an additional 2 mins, was then raised 0.5 mm and remained in this position for one min, and again was raised 0.5 mm and kept for another min before being completely removed. The animals were sacrificed after 12 hrs.
Immunohistochemistry
Mice aged 4 months were anesthetized with a mixture (0.01 ml/g body weight, intraperitonial) of ketamine (10 mg/ml) and xylazine (1 mg/ml) and transcardially perfused with 30 ml of saline to wash out the blood. The spinal cords and the brains were removed and immersion-fixed in 4% paraformaldehyde (PFA) in 0.1 M sodium cacodylate buffer for 2 days at 4°C. Forty micron thick sagittal brain sections and longitudinal spinal sections were cut with a Vibratome and processed for immunocytochemistry. Briefly, sections were first washed with TBS and endogenous peroxidases were blocked with 1% H2O2 (diluted in TBS containing 10% methanol). After washing with the dilution buffer (1% BSA, 0.4% Triton X-100 and 1% normal goat serum in TBS), the sections were blocked with 20% normal goat serum (diluted in TBS) for 1 hr at room temperature. After briefly rinsing with the dilution buffer, the sections were incubated in the primary antibody RT-97 (1:3000 dilution) overnight at 4°C. After 30-mins washing, the sections were incubated in a biotinylated goat anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA) for 1 hr and then incubated in a Vector Standard ABC solution (Vector Laboratories, Burlingame, CA) for 1 hr. Signal was detected with 3,3’-diaminobenzidine tetra hydrochloride, (Vector Laboratories Inc., Burlingame, CA). The sections were then mounted onto glass slides, air dried overnight, and dehydrated in an ascending ethanol series. Images were taken with an Axioskop microscope (Zeiss) equipped with a digital camera aided by AxioVision 4 software (Zeiss).
SDS-PAGE and Western blot analysis
After mice were euthanized with isoflurane 99.9% (Baxter, Guayama, Puerto Rico) and decapitated, spinal cords and sciatic nerves of C57BL6 mice were removed surgically, frozen on dry ice, and stored at −80°C. Frozen tissues were homogenized on ice in a buffer containing 50 mM Tris (pH 7.5); 0.5 mM EDTA (pH 8); beta glycero-phosphate (20 mM), sodium fluoride (10 mM); sodium orthovanadate (1 mM); and PMSF (1mM); and 5mg/ ml each of leupeptin, aprotinin, and pepstatin. An equal volume of a solution containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 2% SDS was added. Homogenates were sonicated for 20 secs, boiled for 10 min, and clarified by centrifugation at 16,000 × g in a table top refrigerated centrifuge for 5 min.
Protein concentration in each fraction was assayed by the BCA method (Pierce, IL), and protein aliquots (10–30 µg) were loaded for each lane on a 7% or 10% gel for electrophoresis unless otherwise indicated. Proteins were electro-transferred from gels to nitrocellulose membranes using a Genie blotter (Idea Scientific, Minneapolis, MN) and blocked using 3% nonfat dry milk (Biorad, Hercules, CA) in Tris-buffered saline containing Tween −20 (20mM Tris-HCl, pH 7.4;150 mM NaCl; 0.2% Tween 20) for 2 hrs. The membranes were incubated overnight at 4 °C with primary antibodies at appropriate dilutions. Blots were developed with alkaline phosphatase-conjugated secondary antibodies using the chromogenic substrate BCIP/NBT (Promega, Madison, WI) or with chemiluminescence based CDP star (Tropix, Bedford, MA) or peroxidase conjugated secondary antibodies using the ECL kit (GE Healthcare, Piscataway, NJ) or ABC kit (Vector Laboratories, Burlingame, CA).
3. Results
PP2A and PP2B eliminate the RT-97 phosphoepitope on NFH C-terminal domains
To determine which protein phosphatase regulates phosphorylation of the RT-97 epitope, we performed Western blot analysis with antibodies to PP2Ac and PP2Bα, which confirmed earlier findings (Veeranna, et al. 1995) that small proportions (0.26 and 1.6%, respectively) of the total tissue contents of these phosphatases are tightly associated with neurofilaments isolated from mouse spinal cord (Fig. 2A) after two washes in a tris HCl buffer pH 6.8, containing 100 mM NaCl, 1 mM each of EDTA and EGTA and 1% Triton X 100. To investigate the activity of phosphatases toward the RT-97 phosphoepitope, we incubated purified PP2A or PP2B with recombinant NFH subunits or a NFH tail domain sequence, each of which were 32P labeled with both recombinant cdk5 and Erk2. Western blot analysis of the substrates with RT-97 monoclonal antibody (Mab) after SDS-PAGE and autoradiography indicated that both PP2A and PP2B dephosphorylated KSPXK sites that were phosphorylated by cdk5 or Erk2 (Fig. 2B,C) and KSPXXXK sites that were phosphorylated by Erk2. Both phosphatases also partially reversed MAPK-mediated phosphorylation of a KSPXXXK GST fusion protein derived from the NFH tail sequence (Fig. 2D). Similarly, phosphorylation sites on native NFH identified by the RT-97 phosphoepitope were dephosphorylated by PP2A (data not shown) and PP2B (Fig. 2E).
Figure 2. Identification of NF protein phosphatases.

(A) Isolated neurofilaments contain tightly associated PP2A and PP2B: Neurofilaments isolated from mouse spinal cord (10 µg protein) were subjected to SDS PAGE on a 4–20% gradient gel. One lane of the gel was stained with Coomassie Brilliant Blue. (lane 1) The remaining gel was subjected to immunoblot analysis with antibodies to PP2A (Lane 3) or PP2B (Lane 2). (B–D) Dephosphorylation of NF proteins by purified PP2A and PP2B in vitro. Recombinant NFH was phosphorylated by purified cdk5 (B) or Erk2 (C) and a recombinant KSPXXXK 24 repeat fusion protein derived from NFH tail sequence was phosphorylated by Erk2 (D). In each case, the autoradiograph (upper panel), and the Western blot analysis (lower panel) immunolabeled with RT-97 monoclonal antibody are presented. Shown are (E) western blot analyses of PP2B mediated dephosphorylation of native NFH in the NF pellet from spinal cord probed with antibodies to RT-97 (left panel) or SMI-33 (right panel)( representative data are presented)
Regulation of RT-97 phosphoepitope levels by phosphatases in primary hippocampal neurons and brains in vivo
To analyze the turnover of phosphate groups on NF tail domains in neurons, we treated primary mouse hippocampal neurons (7 days in vitro) with specific phosphatase inhibitors. OA treatment raised levels of RT-97 immunoreactivity, especially in neuritic processes shown by immunocytochemical labeling (Fig. 3A,B), which mimicked the pattern of distribution seen in more mature axons in vivo (.Nixon, et al. 1994; Vidal-Sanz, et al., 1987). By Western blot analysis, we observed an increase (50%, p<0.05, Fig. 3C) in RT-97 immunoreactivity compared to untreated control neurons. Because OA treatment can potentially elevate RT-97 immunoreactivity by activating JNKs under conditions of cellular stress (Veeranna, et al. 2008), we measured levels of activated JNKs and Erks in lysates of OA- treated and untreated control neurons by Western blot analysis using antibodies to the total and phosphorylated forms of these protein kinases. The ratios between the pErks and total Erks, as well as pJNKs and total JNKs, were not significantly altered in OA-treated neurons (Fig. 3 C, D).
Figure 3. Okadaic acid inhibition of PP2A and PP2B elevate RT-97 IR levels in hippocampal neurons.

Hippocampal neurons in culture (7DIV) untreated (A) or treated (B) with 10 nM Okadaic acid overnight were fixed in 4% paraformaldehyde and immune stained with RT-97 monoclonal antibody. that specifically stains neurofilaments phosphorylated at KSP repeats. Western blot analysis of lysates of hippocampal neurons treated with and without 10 nM okadaic acid overnight and lysed in MPER (Thermo Scientific Rockford, IL) in the presence of protease and phosphatase inhibitors (panel C). A bar graph in panel D indicates the relative band intensities of pJNK1,2, pErk 1,2 and RT-97 on gels (n=3,*p<0.05)
To confirm effects of acute phosphatase inhibition on NFH phosphorylation in vivo, we injected OA stereotaxically into the striatum of anesthetized mice and analyzed these mice after 12 hrs. Immunocytochemical analyses showed that RT-97 IR in nerve fiber bundles coursing through the striatum was markedly increased only on the injected side of the brain (Fig. 4 A, B). These fibers represent axonal trajectories between the somato-sensory thalamus and cortex and also include myelinated striatopallidonigral bundles. By Western blot analyses, RT-97 IR increased significantly (95% p<0.05, Fig. 4C) in striata from OA-treated mice compared to those from saline-injected controls, consistent with the predicted action of OA on PP2A. In contrast to the select phosphatase changes seen in primary hippocampal neurons, however, the levels of activated JNKs and Erks were also increased in OA-injected brain compared to those in control brains (Fig.4 C, D), which may also have contributed to the increased phosphorylation (Veeranna, et al. 2008).
Figure 4. Okadaic acid injection to mouse brain enhances NF phosphorylation.

Vibratome sections of mouse brain, injected with PBS (A) or okadaic acid (B) in the striatun, were immunostained with monoclonal antibody RT-97 that specifically stains neurofilaments phosphorylated at KSP repeats. Images were taken from the striatum showing increased RT-97 immunostaining of nerve fiber bundles in okadaic acid-injected mouse brain. (Scale bar = 100 µM) (C) RT-97 IR rises in Okadaic acid-injected mice striatum: Erk1,2, pErk1,2, JNK1,2, pJNK1,2, and RT-97 immunoreactivities were assessed by Western blot analyses of whole homogenates from three sets of striata dissected from three months old mice injected with 100 nM Okadaic acid. Blots were developed using a HRP-based chemiluminescence probe (D) Graphic representation of pJNK1,2, pErk1,2 and RT-97 immunoreactivities. The band intensities were quantified using NIH image software (n=3 *p<0.05).
To investigate the role of PP2B in dephosphorylation of RT-97 epitope, we next treated hippocampal neuronal cultures with veratridine, a depolarizing agent that enhances intracellular calcium, thereby activating PP2B. This treatment showed marked reduction (86%, p<0.01, Fig. 5A, B) in the levels of RT-97 immunoreactivity. Simultaneous treatment with veratridine and cyclosporine A, a specific inhibitor of calcineurin (PP2B), partly reversed veratridine-induced dephosphorylation of RT-97 sites to levels that were significantly higher than veratridine alone treated levels (Fig. 5A, B). Cyclosporine A treatment alone yielded levels of RT-97 IR comparable to control levels indicating that basal PP2B activity is negligible in the absence of calcium (Fig. 5A, B). Consistent with our results on in vitro dephosphorylation of NFs by purified PP2B (Fig. 2B–D), these data indicate that phosphorylation of RT-97 epitopes can be modulated in intact neurons by PP2B, particularly when calcium homeostasis is altered. Immunocytochemical analyses of comparable neuronal cultures yielded the same result as the Western blot analyses (Fig. 5 C–F), showing that RT-97 IR signal in veratridine plus cyclosporine treated neurons was consistently more intense than in control and veratridine-treated neurons.
Figure 5. Cyclosporine A blocks veratridine-induced dephosphorylation of the RT-97 epitope mediated through PP2B in hippocampal neurons.
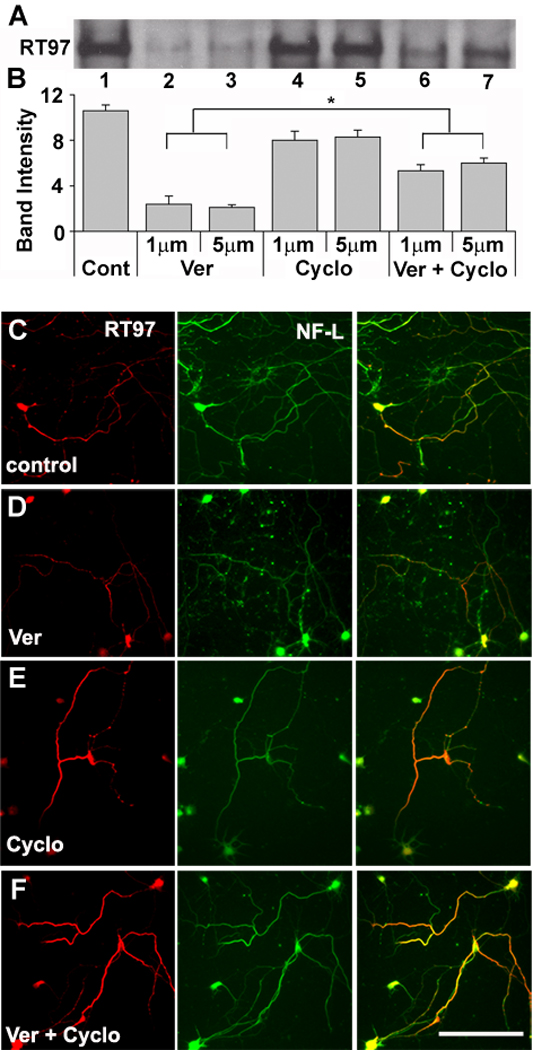
(A) Western blot analysis of whole homogenates of hippocampal neurons treated with vehicle (Cont;1) veratridine ( Ver ; 2,3), Cyclosporine A (Cyclo; 4,5) and both together (6,7) at 1 and 5 µM. Blots were developed using a HRP based chemiluminescence method (B) Graphic representation of RT-97 immunoreactivities in figure 5A. The band intensities were quantified using NIH image software (n=3*p<0.05). (C–F) Hippocampal neurons in culture (7 DIV) following treatment with veratridine, (E) cyclosporine A, (D) and both together (F) at 1 and 5 µM concentration for 24 hours were fixed in 4% paraformaldehyde along with untreated control neurons (C) and then subjected to double immunostaining using monoclonal antibody RT-97 together with polyclonal antibody raised against NFL. A mixture of anti-mouse and anti-rabbit secondary antibodies tagged to fluorophores Alexa 568 red and Alexa 488 green, respectively were used to develop and visualize the neurons.
Phosphatase modulation of the RT-97 epitope on NFH C-terminal domains during brain maturation and aging
In order to understand the impact of brain maturation and aging on NF phosphorylation, we examined the phosphorylation state of NFH and NFM by immuno-cytochemical analysis of RT-97 immunoreactivity in the brains of mice aged 5, 30 and 120 days (Fig. 6A) and by Western blot analyses of neurofilament proteins from homogenates of spinal cord (Fig. 6 B–F) and sciatic nerve (Fig. 7 A–D) from mice at ages ranging from 3 days to 2 years of age. The increase in RT-97 IR was evident from immunocytochemical analyses of fiber bundles coursing through the striatum, as in Fig. 4A and B, which displayed marked age-related increases in phosphorylation at RT-97 sites (Fig. 6 A). Western blot analyses of RT-97 phosphoepitope levels and of total NFH, reflected by levels of SMI-33 IR, in homogenates of mouse spinal cord revealed an age-dependent increase in RT-97 immunoreactivity that was 6 fold higher at 2 years than at 3 days. By contrast, SMI-33 IR, reflecting total levels of NFH independently of phosphorylation state, rose less than 3 fold, all during the period between postnatal day 3 and 21 (Fig. 6 B–F). The net rise in RT-97 IR after total NFH expression had plateaued at p21 indicated that the phosphorylation of NFH continues to rise during adulthood for 12 months. Similarly, in sciatic nerve, RT-97 IR increased 4 fold from p7 to p180 and plateaued at 2 years of age. By contrast, during the same period, SMI-33 IR rose minimally, establishing that the NF phosphorylation state markedly increased during maturation and aging (Fig. 7 A–D).
Figure 6. RT-97 IR increases during brain maturation and aging.

Vibratome sections of brain from mice aged 5 days, 30 days and 4 months were immunostained with monoclonal antibody RT-97 that specifically stains neurofilaments phosphorylated at KSP repeats. Images taken from the striatum of mice at 5 days, 30 days and 4 months of age show changes of RT-97 immunostaining during brain maturation. Scale bar = 20 µm. (B) Western blot analysis were performed on whole homogenates of of spinal cords from mice at postnatal ages of 3, 7, 12, 21, 30, 180, 365 and 660 days using antibodies for neurofilaments, RT-97, SMI-33, neurofilament kinases and phosphatases, loaded in respective lanes 1–8. Blots were developed using HRP-based chemiluminescence or a DAB kit from Vector laboratories. Quantification of band intensities was done using Multi gauge V2.3 software. (C–G) The graphic representations of RT-97, and SMI-33 along with NF kinases or phosphatases are shown. Band densities were quantified using Multi gauge V2.3 software and graphed using Microsoft Excel. (H) The graphic representations of RT-97 and SMI-33 along with PP2A and PP1 activities in spinal cords are shown. Phosphatase activities were monitored by radiometric assays as described in the text and are expressed as % dephosphorylation. (The data represent mean ± SEM, n=3)
Figure 7. The expression of phosphatases declines in sciatic nerve during maturity and aging.

(A) Western blot analysis performed on whole homogenates of sciatic nerves from mice at postnatal ages of 7, 12, 21, 30, 180, 365 and 660 days using antibodies for neurofilaments, RT-97, SMI-33, neurofilament kinases and phosphatases, loaded in respective lanes 1–7. Blots were developed using HRP-based chemiluminescence or a DAB kit from Vector laboratories. Quantification of band intensities was done using Multi gauge V2.3 software. (B–D) The graphic representations of RT-97, and SMI-33 along with NF phosphatases are shown. Band densities were quantified using Multi gauge V2.3 software and graphed using Microsoft Excel (The data represent mean +/− SEM, n=3)
In order to examine the basis for the continued NFH phosphorylation and rises in RT-97 epitope levels during brain maturation and aging (p21-2yr) after the leveling off of NFH expression, we performed Western blot analyses on the kinases and phosphatases that have been shown to regulate RT-97 epitope levels. The levels of catalytic subunits of PP2Ac immunoreactivity declined 5-fold between p12 and 2yr (p<0.0001, Fig. 6, B, C). PP1 immunoreactivity declined 2.8 fold (p< 0.05, Fig. 6 F), however, PP2B levels were not significantly altered during this period (Fig. 6B, C). To verify whether PP2A activity declines during maturation and aging, we monitored its activity. These data revealed that PP2A activity rises significantly (28%, p<0. 05) from p3 to p12 but then declines 38% by between p21 and 2 years of age (p<0.05) (Fig. 6F). PP1 activity, which was five fold less than that of PP2A, did not decrease significantly during maturity and aging. Similar patterns of phosphatase alterations were observed in sciatic nerves from these mice (Fig. 7A–D), where the catalytic subunit expression of PP2A declined 3.5 fold between p12 and 2yr (p<0.0001). Similarly, catalytic subunit expression of PP1 declined 7 fold between p12 and 2yr (p<0.0001). However, the expression levels of the PP2Bα subunit were not significantly altered during this period.
Protein kinase activation decreased during brain maturation. p35, the major activator of cdk5, significantly decreased with age, while its catalytic subunit, cdk5 decreased marginally (Fig. 6B, D). Similarly, during the period of NFH hyperphosphorylation (p21-2yr) p-Erk1, 2 levels decreased 28% relative to their levels during early postnatal development (p3–21) (Fig. 6A, E). The levels of P-JNK1, 2 also declined 5.6 fold from p21 to 2yr and another 3.7 fold from p3 to p21 (Fig. 6 B, E). Similar changes in expression of these kinases were observed in sciatic nerve during maturation and aging (data not shown).
4. Discussion
NF phosphorylation state involves a complex balance between activities of multiple kinases and phosphatases (Nixon 1993; Pant, et al. 2000) (Fig. 8). We previously identified the molecular determinants of the RT-97 epitope, which is the major repeated phosphorylation motif along the long C-terminal domains of NFH and NFM, and showed that it can be generated by multiple protein kinases, including Erk1,2, JNK1,2, and cdk5 (Veeranna, et al. 2008). Here we have shown that PP2A and PP2B modulate turnover of phosphate groups at the RT-97 phosphoepitope and, thus, the phosphorylation state of NFH C-terminal domains in intact neurons. Previous in vitro studies (Giasson, et al. 1996; Mata, et al. 1997; Sacher, et al. 1992; Sacher, et al. 1994; Shetty, et al. 1992; Strack, et al. 1997; Veeranna, et al. 1995; Wang, et al. 2001) have suggested a role of PP1, PP2A and PP2B in the dephosphorylation of NFs. Further, it has been shown that PP2A removes phosphates on KSPXK motifs on the NF tail domain added by cdk5, although this motif represents only 20% of the total KSPs (Veeranna, et al. 1995). We showed in this study that either PP2A or PP2B dephosphorylate the KSPXK sites and, in addition, also dephosphorylate KSPXXXK motifs that constitute 80% of the KSPs on the NFH tail domain. Moreover, OA, an inhibitor of PP1 and PP2A (Bialojan and Takai 1988; Ishihara, et al. 1989; Veeranna, et al. 1995), elevated levels of RT-97 IR when administered to hippocampal neurons in culture or intracerebrally in mice. These findings, coupled with in vitro analyses showing direct actions of the purified phosphatases in reducing RT-97 levels on neurofilament proteins, strongly suggest that PP2A contributes to regulation of the phosphorylation state of the NFH C-terminus and to RT-97 levels. PP1, which has been reported to play a very minor role in NF dephosphorylation (Strack, et al. 1997) because of its low abundance was confirmed to be 5-fold less abundant in spinal cord than PP2a and decreased non-significantly during maturity and aging, suggesting a minor role in the NF hyperphosphorylation during brain maturation and aging.
Figure 8. Both kinases and phosphatases regulate NF phosphorylation.

Phosphorylation at the RT-97 epitope in NFH based on this study (italicised) and Veeranna et al., 2008, is affected by kinases and phosphatases shown in the diagram‥ Also note that various upstream factors and cross talk among kinases influence NF phosphorylation.
In addition to PP2A-mediated modulation of NF phosphorylation state, a specific inhibitor of PP2B, cyclosporine A, partially prevented veratridine-induced dephosphorylation at RT-97 sites suggesting that this phosphatase may also regulate the RT-97 phosphoepitope, consistent with an earlier report that it may regulate phosphorylation at the SMI-31 epitope (Mata, et al. 1997). The enhanced phosphorylation to generate RT-97 IR in hippocampal neurons could be attributed directly to inhibition of OA-sensitive phosphatases. Rises in NFH phosphorylation occurred despite unaltered activation of known NF kinases. Increased NF phosphorylation after OA administration in vivo, however, could reflect an additional contribution of NF kinases, JNKs and Erk1,2 which were activated under these conditions. This difference in the activation of kinases between in vitro neuronal cultures and mouse brains in vivo in response to treatment with OA reflects the many differences between these two systems, including the contribution of dominant glial cells to the contents of brain tissue. Collectively, these data implicate PP2A as the major phosphatase contributing to the regulation of proline-directed NFH dephosphorylation, consistent with its role in regulating the phosphorylation of tau (Goedert, et al. 1995).
Elevated NFH tail domain phosphorylation has been observed with aging in rat CNS (Gou, et al. 1995), although the basis for these changes were not identified (Julien and Mushynski 1982; Lee, et al. 1987). Our data show that NF phosphorylation during early postnatal development rises in proportion to the increase in NF expression. NF phosphorylation during this developmental period has been shown to be due to increased activities of NF kinases, including Erk1,2 cdk5, and possibly JNK1,2 (Veeranna, et al. 1997; Veeranna, et al. 2008), which outbalance the relatively high phosphatase levels present during the same developmental period. By p21, most large axons have established synaptic connections, acquired myelin, and achieved maximal or near maximal caliber, which is largely dependent on the local accumulation of NF within axons (Sanchez, et al. 2000). At this stage of late postnatal development, levels of NF protein expression have plateaued; however, the state of phosphorylation of NFH and NFM continued to increase, which is attributable principally to declining PP2A levels and its activity. Expression of PP1 also declined during the same period, but has been considered less active than PP2A toward NF (Strack, et al. 1997). Decreased PP2A gene expression was reported earlier in the hypothalamus and cortex of aged mice (Jiang, et al. 2001). In whole rat brain, PP2A levels were recently reported as being unchanged during maturation although a decline in PP2A expression is not inconsistent with the immunoblot presented in this report (Yu, et al. 2009). It has also been suggested that PP2B activity is elevated in aged rats based on evoked normal LTD in response to the PP2B inhibitor FK 506 (Jouvenceau and Dutar 2006). Because PP2B requires Ca2+ for activity, its regulation is complex and its protein levels and in vitro activity measurements are difficult to interpret in terms of in vivo activity.
Our data provide strong support for a mechanism of aging-related shifts in equilibrium between the activities of kinases and phosphatases (Kesavapany, et al. 2007; Lin and Lu 2005; Pant, et al. 2000; Veeranna, et al. 2004) as the basis for elevated levels of hyperphosphorylated NF in older mice. Although no evidence is currently available, a decrease in O-GlcNAcylation at potential phosphorylation sites (Deng, et al. 2008; Ludemann, et al. 2005) could conceivably contribute to these aging effects since O-GlcNAcylation of NFs occurs on the same serine and threonine residues as phosphorylation. In addition, heavily phosphorylated neurofilaments ( Pant 1988) are more resistant to calpain proteolysis (Elhanany, et al. 1994; Pant 1988) and conformation induced by phosphorylation or integration of NF into the cytoskeletal network could conceivably reduce accessibility of certain sites to phosphatases. These additional theoretical possibilities, however, would compound the demonstrated effects of phosphatase declines in promoting aging-related NF hyperphosphorylation.
Higher states of NF phosphorylation during aging may increase the stability and alignment of neurofilaments within the cytoskeleton (Goldstein, et al. 1987; Gotow 2000; Gou, et al. 1995). Novel functions for neurofilaments, specific NF subunits, and specific NF polypeptide domains have emerged, including roles as a scaffold for vesicular organelles (Rao, et al. 2009) and receptors (Ehlers, et al. 1998; Kim, et al. 2002; Mueller, et al. 2004; Ratnam and Teichberg 2005). In some cases, these functions are mediated by the extensively phosphorylated C-terminal domains of NF protein (Kim, et al. 2002). How hyperphosphorylation of NF during normal brain maturation and aging may alter those functions of NF remains to be investigated.
Hyperphosphorylation of NF and tau in age-related neurodegenerative disorders has been attributed to activation of multiple protein kinases and reduced activity of protein phosphatases (Liu and Wang 2009;. Nixon 1993; Sontag, et al. 2004; Veeranna, et al. 2004; Wang, et al. 2001; Webber, et al. 2005). These enzymatic changes have been implicated in promoting abnormal perikaryal NF accumulation, tau aggregation, and defective axonal transport leading to neuronal cell death (Hanger, et al. 2009; Julien and Mushynski 1998; Julien 1999; Noble, et al. 2003). Levels of PP2A I1 and PP2A I2, the endogenous inhibitors of PP2A, are also elevated in AD brain (Tanimukai, et al. 2005). These changes, increased demethylation of Leu 309, and increased Tyr 307 phosphorylation on the PP2AC subunit contribute to the lowering PP2A activity in AD (Liu and Wang 2009).
Our study demonstrates declining phosphatase activities during aging leading to NF hyperphosphorylation, suggesting that, therefore, raising PP2A activity might reduce the hyperphosphorylation of cytoskeleton in aging and age-related neurodegenerative disorders. In this context, PP2A activation by silencing the endogenous protein inhibitors of PP2A, PP2A I1 and PP2A I2 could be one rational approach (Tanimukai, et al. 2005). Other pharmacologic approaches that elevate PP2A activity have been shown to have potential therapeutic effects in neurological disease models (Vitek, et al. 2009).
Acknowledgements
The authors thank Corrinne Peterhoff for assistance with figures, and Nicole Piorkowski for assistance with manuscript preparation. This work was supported by NIA AG05604 (R.A.N.) and the NIH intramural program (H.C.P).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
None of the authors have any actual or potential conflicts of interest to disclose.
References
- Anderton BH, Breinburg D, Downes MJ, Green PJ, Tomlinson BE, Ulrich J, Wood JN, Kahn J. Monoclonal antibodies show that neurofibrillary tangles and neurofilaments share antigenic determinants. Nature. 1982;298:84–86. doi: 10.1038/298084a0. [DOI] [PubMed] [Google Scholar]
- Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SS, Liem RK. Transfected rat high-molecular-weight neurofilament (NF-H) coassembles with vimentin in a predominantly nonphosphorylated form. J Neurosci. 1990;10:3714–3726. doi: 10.1523/JNEUROSCI.10-11-03714.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and ‥ quantitating protein phosphatases in mammalian tissues. FEBS lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- Deng Y, Li B, Liu F, Iqbal K, Grundke-Iqbal I, Brandt R, Gong CX. Regulation between O-GlcNAcylation and phosphorylation of neurofilament-M and their dysregulation in Alzheimer disease. FASEB J. 2008;22:138–145. doi: 10.1096/fj.07-8309com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Fung ET, O'Brien RJ, Huganir RL. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J Neurosci. 1998;18:720–730. doi: 10.1523/JNEUROSCI.18-02-00720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder GA, Friedrich VL, Jr, Kang C, Bosco P, Gourov A, Tu PH, Zhang B, Lee VM, Lazzarini RA. Requirement of heavy neurofilament subunit in the development of axons with large calibers. J Cell Biol. 1998;143:195–205. doi: 10.1083/jcb.143.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhanany E, Jaffe H, Link WT, Sheeley DM, Gainer H, Pant HC. Identification of endogenously phosphorylated KSP sites in the high-molecular-weight rat neurofilament protein. J Neurochem. 1994;63:2324–2335. doi: 10.1046/j.1471-4159.1994.63062324.x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in steriotaxic coordinates. San Diego, California: Academic Press; 1997. [Google Scholar]
- Gasser HS, Grundfest H. Axondiameters in relation to the spike dimensions and conduction velocity in mammalian A fibers. Am J Physiol. 1939a;127:393–414. [Google Scholar]
- Gasser HS, Grundfest H. Axondiameters in relation to the spike dimensions and conduction velocity in mammalian A fibers. Am J Physiol. 1939b;127:393–414. [Google Scholar]
- Giasson BI, Cromlish JA, Athlan ES, Mushynski WE. Activation of cyclic AMP-dependent protein kinase in okadaic acid-treated neurons potentiates neurofilament fragmentation and stimulates phosphorylation of Ser2 in the low-molecular-mass neurofilament subunit. J Neurochem. 1996;66:1207–1213. doi: 10.1046/j.1471-4159.1996.66031207.x. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Mushynski WE. Study of proline-directed protein kinases involved in phosphorylation of the heavy neurofilament subunit. J Neurosci. 1997;17:9466–9472. doi: 10.1523/JNEUROSCI.17-24-09466.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Qi Z, Wang JH, Cohen P. Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. J Neurochem. 1995;65:2804–2807. doi: 10.1046/j.1471-4159.1995.65062804.x. [DOI] [PubMed] [Google Scholar]
- Goldstein ME, Sternberger NH, Sternberger LA. Phosphorylation protects neurofilaments against proteolysis. J Neuroimmunol. 1987;14:149–160. doi: 10.1016/0165-5728(87)90049-x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in alzheimer's disease [In Process Citation] J Biol Chem. 2000;275:5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- Gotow T. Neurofilaments in health and disease. Med Electron Microsc. 2000;33:173–199. doi: 10.1007/s007950000019. [DOI] [PubMed] [Google Scholar]
- Gou JP, Eyer J, Leterrier JF. Progressive hyperphosphorylation of neurofilament heavy subunits with aging: possible involvement in the mechanism of neurofilament accumulation. Biochem Biophys Res Commun. 1995;215:368–376. doi: 10.1006/bbrc.1995.2475. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- Jiang CH, Tsien JZ, Schultz PG, Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci U S A. 2001;98:1930–1934. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau A, Dutar P. A role for the protein phosphatase 2B in altered hippocampal synaptic plasticity in the aged rat. J Physiol Paris. 2006;99:154–161. doi: 10.1016/j.jphysparis.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE. Multiple phosphorylation sites in mammalian neurofilament polypeptides. J Biol Chem. 1982;257:10467–10470. [PubMed] [Google Scholar]
- Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol. 1998;61:1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- Julien JP. Neurofilament functions in health and disease. Curr Opin Neurobiol. 1999;9:554–560. doi: 10.1016/S0959-4388(99)00004-5. [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Patel V, Zheng YL, Pareek TK, Bjelogrlic M, Albers W, Amin N, Jaffe H, Gutkind JS, Strong MJ, Grant P, Pant HC. Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol Biol Cell. 2007a;18:3645–3655. doi: 10.1091/mbc.E07-03-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim OJ, Ariano MA, Lazzarini RA, Levine MS, Sibley DR. Neurofilament-M interacts with the D1 dopamine receptor to regulate cell surface expression and desensitization. J Neurosci. 2002;22:5920–5930. doi: 10.1523/JNEUROSCI.22-14-05920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Carden MJ, Schlaepfer WW, Trojanowski JQ. Monoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF-H and NF-M) and demonstrate their existence in the normal nervous system of adult rats. J Neurosci. 1987;7:3474–3488. doi: 10.1523/JNEUROSCI.07-11-03474.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta. 2005;1739:311–322. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Liu R, Wang JZ. Protein phosphatase 2A in Alzheimer's disease. Pathophysiology. 2009;16:273–277. doi: 10.1016/j.pathophys.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Ludemann N, Clement A, Hans VH, Leschik J, Behl C, Brandt R. O-glycosylation of the tail domain of neurofilament protein M in human neurons and in spinal cord tissue of a rat model of amyotrophic lateral sclerosis (ALS) J Biol Chem. 2005;280:31648–31658. doi: 10.1074/jbc.M504395200. [DOI] [PubMed] [Google Scholar]
- Mata M, Honegger P, Fink DJ. Modulation of phosphorylation of neuronal cytoskeletal proteins by neuronal depolarization. Cell Mol Neurobiol. 1997;17:129–140. doi: 10.1023/A:1026337322916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi S, Shima H, Kikuchi K, Igarashi K, Hatsuse R, Maeda K, Yazawa M, Murayama T, Okuma Y, Nomura Y. Development of an assay method for activities of serine/threonine protein phosphatase type 2B (calcineurin) in crude extracts. Anal Biochem. 2000;278:192–197. doi: 10.1006/abio.1999.4422. [DOI] [PubMed] [Google Scholar]
- Mueller HT, Haroutunian V, Davis KL, Meador-Woodruff JH. Expression of the ionotropic glutamate receptor subunits and NMDA receptor-associated intracellular proteins in the substantia nigra in schizophrenia. Brain Res Mol Brain Res. 2004;121:60–69. doi: 10.1016/j.molbrainres.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Kumar S, Hoh JH. Molecular mechanisms for organizing the neuronal cytoskeleton. Bioessays. 2004;26:1017–1025. doi: 10.1002/bies.20088. [DOI] [PubMed] [Google Scholar]
- NIH. Laboratory animal welfare; U.S. government principles for the utilization and care of vertebrate animals used in testing, research and training; notice. Fed Regist. 1985;50:20864–20865. [PubMed] [Google Scholar]
- Nixon RA, Lewis SE. Differential turnover of phosphate groups on neurofilament subunits in mammalian neurons in vivo. J Biol Chem. 1986;261:16298–16301. [PubMed] [Google Scholar]
- Nixon RA, Logvinenko KB. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J Cell Biol. 1986;102:647–659. doi: 10.1083/jcb.102.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Sihag RK. Neurofilament phosphorylation: a new look at regulation and function. Trends Neurosci. 1991;14:501–506. doi: 10.1016/0166-2236(91)90062-y. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The regulation of neurofilament protein dynamics by phosphorylation: clues to neurofibrillary pathobiology. Brain Pathol. 1993;3:29–38. doi: 10.1111/j.1750-3639.1993.tb00723.x. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Paskevich PA, Sihag RK, Thayer CY. Phosphorylation on carboxyl terminus domains of neurofilament proteins in retinal ganglion cell neurons in vivo: influences on regional neurofilament accumulation, interneurofilament spacing, and axon caliber. J Cell Biol. 1994;126:1031–1046. doi: 10.1083/jcb.126.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Pant AC, Veeranna, Pant HC, Amin N. Phosphorylation of human high molecular weight neurofilament protein (hNF-H) by neuronal cyclin-dependent kinase 5 (cdk5) Brain Res. 1997;765:259–266. doi: 10.1016/s0006-8993(97)00561-1. [DOI] [PubMed] [Google Scholar]
- Pant HC. Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem J. 1988;256:665–668. doi: 10.1042/bj2560665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant HC, Veeranna, Grant P. Regulation of axonal neurofilament phosphorylation. Curr Top Cell Regul. 2000;36:133–150. doi: 10.1016/s0070-2137(01)80006-6. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer's disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/s0169-328x(02)00488-6. [DOI] [PubMed] [Google Scholar]
- Perrot R, Julien JP. Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 2009;23:3213–3225. doi: 10.1096/fj.09-129585. [DOI] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Kumar A, Montagna L, Campbell J, Yuan A, Espreafico EM, Julien JP, Nixon RA. The myosin va head domain binds to the neurofilament-L rod and modulates local organelle content and distribution within axons. FASEB J. 2009 doi: 10.1371/journal.pone.0017087. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnam J, Teichberg VI. Neurofilament-light increases the cell surface expression of the N-methyl-D-aspartate receptor and prevents its ubiquitination. J Neurochem. 2005;92:878–885. doi: 10.1111/j.1471-4159.2004.02936.x. [DOI] [PubMed] [Google Scholar]
- Sacher MG, Athlan ES, Mushynski WE. Okadaic acid induces the rapid and reversible disruption of the neurofilament network in rat dorsal root ganglion neurons. Biochem Biophys Res Commun. 1992;186:524–530. doi: 10.1016/s0006-291x(05)80839-3. [DOI] [PubMed] [Google Scholar]
- Sacher MG, Athlan ES, Mushynski WE. Increased phosphorylation of the amino-terminal domain of the low molecular weight neurofilament subunit in okadaic acid-treated neurons. J Biol Chem. 1994;269:18480–18484. [PubMed] [Google Scholar]
- Sanchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty KT, Veeranna, Guru SC. Phosphatase activity against neurofilament proteins from bovine spinal cord: effect of aluminium and neuropsychoactive drugs. Neurosci Lett. 1992;137:83–86. doi: 10.1016/0304-3940(92)90304-p. [DOI] [PubMed] [Google Scholar]
- Shetty KT, Link WT, Pant HC. cdc2-like kinase from rat spinal cord specifically phosphorylates KSPXK motifs in neurofilament proteins: isolation and characterization. Proc Natl Acad Sci U S A. 1993;90:6844–6848. doi: 10.1073/pnas.90.14.6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313:2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL., 3rd Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004a;63:287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- Sternberger NH, Sternberger LA, Ulrich J. Aberrant neurofilament phosphorylation in Alzheimer disease. Proc Natl Acad Sci U S A. 1985;82:4274–4276. doi: 10.1073/pnas.82.12.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. 1997;49:15–28. doi: 10.1016/s0169-328x(97)00117-4. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Amin N, Grant P, Pant HC. P13suc1 associates with a cdc2-like kinase in a multimeric cytoskeletal complex in squid axoplasm. J Neurosci. 1995;15:6222–6229. doi: 10.1523/JNEUROSCI.15-09-06222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005;166:1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna, Shetty KT, Link WT, Jaffe H, Wang J, Pant HC. Neuronal cyclin-dependent kinase-5 phosphorylation sites in neurofilament protein (NF-H) are dephosphorylated by protein phosphatase 2A. J Neurochem. 1995;64:2681–2690. doi: 10.1046/j.1471-4159.1995.64062681.x. [DOI] [PubMed] [Google Scholar]
- Veeranna, Grant P, Pant HC. Expression of p67 (Munc-18), Cdk5, P-NFH and syntaxin during development of the rat cerebellum. Dev Neurosci. 1997;19:172–183. doi: 10.1159/000111203. [DOI] [PubMed] [Google Scholar]
- Veeranna, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC. Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci. 1998;18:4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna, Lee JH, Pareek TK, Jaffee H, Boland B, Vinod KY, Amin N, Kulkarni AB, Pant HC, Nixon RA. Neurofilament tail phosphorylation: identity of the RT-97 phosphoepitope and regulation in neurons by cross-talk among proline-directed kinases. J Neurochem. 2008;107:35–49. doi: 10.1111/j.1471-4159.2008.05547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Sanz M, Bray GM, Villegas-Perez MP, Thanos S, Aguayo AJ. Axonal regeneration and synapse formation in the superior colliculus by retinal ganglion cells in the adult rat. J Neurosci. 1987;7:2894–2909. doi: 10.1523/JNEUROSCI.07-09-02894.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek MP, Christensen DJ, Wilcock D, Oddo J, Gharkholonarehe N, Ohkubo N, Toku K, Van Nostrand WE, Li F, Colton CA. Novel mechanism where ApoE mimetics activate PP2A activity and reduce Alzheimer's pathology in three different transgenic models. Alzheimer's & Dementia: The Journal of the Alzheimer's Association. 2009;5:P111. [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol. 2001;168:402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- Wang J, Tung YC, Wang Y, Li XT, Iqbal K, Grundke-Iqbal I. Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett. 2001;507:81–87. doi: 10.1016/s0014-5793(01)02944-1. [DOI] [PubMed] [Google Scholar]
- Webber KM, Smith MA, Lee HG, Harris PL, Moreira P, Perry G, Zhu X. Mitogen- and stress-activated protein kinase 1: convergence of the ERK and p38 pathways in Alzheimer's disease. J Neurosci Res. 2005;79:554–560. doi: 10.1002/jnr.20380. [DOI] [PubMed] [Google Scholar]
- Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108:1480–1494. doi: 10.1111/j.1471-4159.2009.05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sasaki T, Rao MV, Liem RK, Nixon RA. Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J Neurosci. 2009 doi: 10.1523/JNEUROSCI.1942-09.2009. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]