

Abstract

A Kiyooka aldol condensation of an aldehyde with a trimethylsilyl ketene acetal and the oxazaborolidinone prepared from N-Ts-(S)-valine gives two of the four possible aldol adducts which were oxidized and deprotected to complete the synthesis of (−)-berkelic acid and (−)-22-epi-berkelic acid. This synthesis establishes the absolute stereochemistry and assigns the stereochemistry at C-22. A biosynthetic pathway is proposed that is consistent with the known absolute stereochemistry at the quaternary carbon of spiciferone A, spicifernin, and berkelic acid and provides a simple explanation for the differing stereochemistry at C-18 and C-19 of spicifernin and berkelic acid.
Introduction
Berkelic acid (1) and its congener, spiciferone A (2), were recently isolated by Stierle and coworkers from a Penicillium species obtained from the surface water of Berkeley Pit Lake, an abandoned open-pit copper mine (see Chart 1).1 The acidic water (pH 2.5) is contaminated with metal sulfates at high concentrations so that only extremophiles, which may produce novel natural products, can survive. The structure of berkelic acid (1) was determined to be a novel spiroketal from the analysis of the NMR and mass spectral data of 1 and the analogous methyl ester. The relative stereochemistry of the side chain stereocenter (C-22) and the absolute stereochemistry were not assigned.
CHART 1.
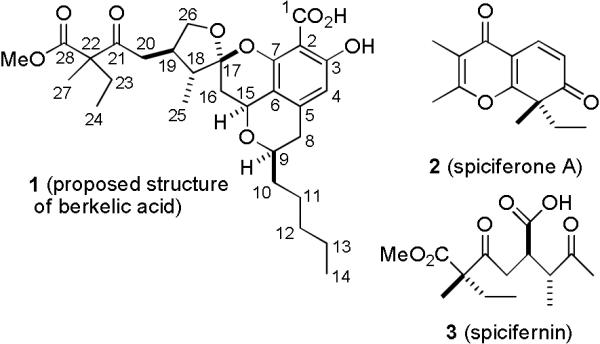
Structures of Berkelic Acid (1), Spiciferone A (2), and Spicifernin (3)
Berkelic acid (1) inhibits MMP-3 in the micromolar range (GI50 = 1.87 μM) and caspase-1 in the millimolar range (GI50 = 0.098 mM). Inhibitors that act by blocking the activity of proteolytic enzymes (MMPs) used by tumor cells to promote metastatic spread provide a new approach for cancer treatment. Berkelic acid was tested in the National Cancer Institute antitumor screen against 60 human cell lines and showed selective activity toward ovarian cancer OVCAR-3 with a GI50 of 91 nM.1
We hypothesized that 1 could be prepared by a short sequence starting from ketal aldehyde 4 and 2,6-dihydroxybenzoic acid 5 (see Scheme 1). Benzoic acid 5, a synthetic, and possibly biosynthetic, precursor to pulvilloric acid (6), has been synthesized in both racemic2 and optically pure form.3 Berkelic acid (1) might be biosynthesized by an analogous coupling of 5 and a ketal aldehyde related to 4. Alternatively, it is possible that berkelic acid (1) is biosynthesized by the nucleophilic addition of ketone 7 to pulvilloric acid (6). Ketal aldehyde 4 and ketone 7 are related to spicifernin (3) which occurs with spiciferone A (2) in Cochliobolus spicifier.4
SCHEME 1.
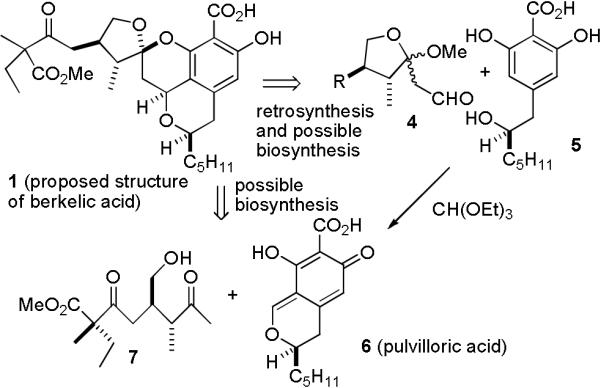
Retrosynthesis and Possible Biosynthesis of (−)-Berkelic Acid (1)
In a model study,5,6 we condensed 4a with (±)-5 in MeOH containing Dowex 50WX8-400-H+ for 12 h at 25 °C to provide a mixture of the tetracyclic acids that was treated with diazomethane to give 8a (24%) and 9a (24%) and minor amounts of the other two diastereomers (see Scheme 2). The desired isomer 8a was calculated to be 2.4 kcal/mol more stable than 9a using MMX with conformational searching.7 As expected, treatment of the mixture of four methyl esters with TFA in CDCl3 afforded a mixture containing mainly 8a, which was isolated in 50% yield from 5.
SCHEME 2.
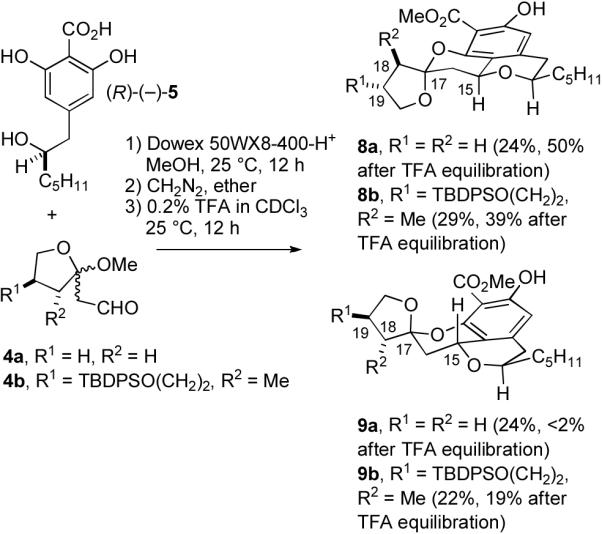
Preparation of Tetracycles 8a and 8b
For the synthesis of (−)-berkelic acid,8 we prepared (R)-(−)-5 and 4b and condensed them in MeOH containing Dowex 50WX8-400-H+ for 12 h at 25 °C to provide a mixture of the tetracyclic acids that was treated with diazomethane to give 8b (29%) and 9b (22%) and minor amounts of a third diastereomer. Equilibration with TFA in CDCl3 had little effect, giving 8b (39%) and 9b (19%). As a result of the substituents on the tetrahydrofuran, the desired isomer 8b was calculated7,9 to be only 0.8 kcal/mol more stable than 9b, which is consistent with the formation of a 2:1 mixture of 8b and 9b after equilibration.
At first this was of great concern, but the 1H NMR spectral data of the desired product 8b did not fit well with the data for berkelic acid methyl ester suggesting that the stereochemistry of berkelic acid had been assigned incorrectly. The trans stereochemistry at C-18 and C-19 appeared to be correctly assigned. The relative stereochemistry of these two centers relative to the anomeric center C-17 was assigned solely on the basis of an NOE from the methyl group (C-25) to H-16β and an H-20 (see Figure 1).1 However, our MMX calculations indicated that this NOE is more consistent with structure 10, which has the opposite stereochemistry to 1 at both C-18 and C-19, because structure 1 should have NOEs to both H-16β and H-16α. As this work was being completed, Fürstner reported a synthesis of the enantiomer of berkelic acid methyl ester and reassigned the stereochemistry at C-18 and C-19 as shown in 10.10
FIGURE 1.
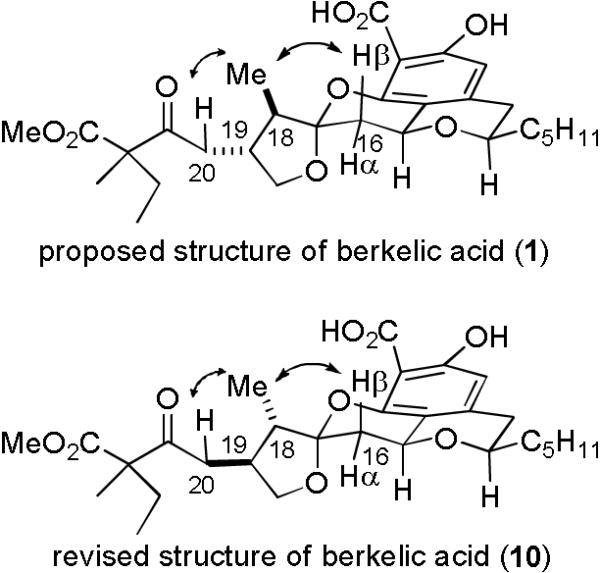
Proposed and revised structures of berkelic acid showing NOE correlations to the C-25 methyl group.
We therefore prepared ent-4b and condensed it with (R)-(−)-5 in MeOH containing Dowex 50WX8-400-H+ for 60 h at 25 °C to provide a mixture of the tetracyclic acids corresponding to esters 11 and 12 (see Scheme 3).8 Partial cleavage of the TBDPS group occurred because this reaction was slower than that with 4, requiring 60 h for complete reaction. Esterification with diazomethane provided silyl ether 11 (30%) and alcohol 12 (22%) with no diastereomers formed at C-15 and C-17 before or after TFA equilibration. The selectivity is consistent with the calculated strain energies, which indicated that 11 is 3.9 kcal/mol more stable than the isomer analogous to 9b. As we expected, the spectral data of 11 fit well with those of berkelic acid methyl ester suggesting that 10 is the structure of berkelic acid with the opposite stereochemistry at C-18 and C-19 to that originally proposed in 1 and that the stereochemistry at both C-15 and C-17 in the natural product is thermodynamically controlled.
SCHEME 3.
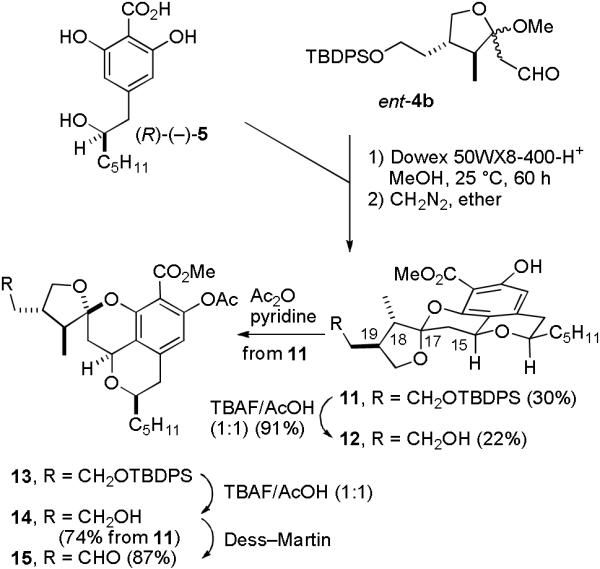
Synthesis of Tetracyclic Acetoxy Aldehyde 15
Although the structure of 12 is very close to the revised structure of berkelic acid (10), the introduction of the five-carbon side chain, the assignment of the stereochemistry at C-22 and the choice of protecting group for the aromatic acid and phenol proved to be very challenging. We describe here the full details of the completion of the synthesis,8 the assignment of stereochemistry at C-22, and an analysis of the biosynthesis of berkelic acid.
Results and Discussion
Protection of 11 with Ac2O and pyridine afforded 13, which was desilylated with TBAF/HOAc to give alcohol 14 in 74% overall yield from 11. Dess-Martin oxidation of 14 gave aldehyde 15 in 87% yield. Attempted oxidation of phenol alcohol 12 with Dess-Martin periodinane resulted in decomposition, establishing that protection of the phenol is necessary.11
Unfortunately, addition of 15 to the enolate prepared by treatment of methyl 2-methylbutanoate with LDA at −78 °C destroyed the aldehyde and cleaved the acetate group, rather than forming the desired aldol adducts. These aldol conditions are successful with 3-phenylbutanal (83%) and citronellal (70%). Acidic Mukaiyama aldol conditions were equally unsuccessful: no aldol products were obtained from the TiCl4-catalyzed reaction of 15 and trimethylsilyl ketene acetal 1912 (see Scheme 4) prepared from methyl 2-methylbutanoate, although aldol adducts (74%) were obtained from 3-phenylbutanal under these conditions. We needed to find a protecting group for the phenol that was compatible with the aldol reaction and to develop a procedure that utilized both TBDPS ether phenol 11 and hydroxy phenol 12.
SCHEME 4.

Synthesis of a Mixture of Berkelic Acid Methyl Ester and the C-22 Epimer (21)
Desilylation of 11 with TBAF/AcOH in THF afforded alcohol 12 in 91% yield. Protection of both hydroxy groups in 12 with TBSOTf and 2,6-lutidine gave bis TBS ether 16, whereas protection of 12 with TBSCl and imidazole in DMF selectively protected the primary alcohol. Treatment of bis TBS ether 16 with Ce(OTf)n•xH2O (19–23% Ce) selectively cleaved the alkyl TBS ether rather than the phenol TBS group to give alcohol 17 in 81% yield.13 Oxidation of 17 with Dess-Martin periodinane afforded aldehyde 18.
Unfortunately, addition of 18 to the enolate prepared by treatment of methyl 2-methylbutanoate with LDA at −78 °C gave no aldol products. The N-methylimidazole and LiCl-catalyzed Mukaiyama aldol reaction14 proceeds under very mild conditions and appeared to be compatible with the functionality in 18. We were pleased to find that reaction of 18 with trimethylsilyl ketene acetal 19 (10 equiv), LiCl (0.5 equiv), N-methylimidazole (0.25 equiv), and 4 Å molecular sieves in DMF afforded an inseparable mixture of all four aldol products, which was subjected to Dess-Martin oxidation to give an inseparable 1:1 mixture of keto esters 20 in 44% overall yield. Removal of the TBS group using CsF in MeOH afforded a 1:1 mixture of the methyl esters of berkelic acid and 22-epi-berkelic acid (21) in 88% yield. The 1H NMR spectral data of 21 match those of berkelic acid methyl ester in CDCl3 but there are doubled peaks for the C-20 methylene group and the methyl and ethyl groups attached to C-22 as expected, since 21 is a mixture of isomers at C-22. The correspondence of the 1H NMR spectral data of 21 and natural berkelic acid methyl ester confirmed the reassignment of the stereochemistry at C-18 and C-19.
Selective cleavage of the methyl benzoate of 21 in the presence of the β-keto ester group is not possible, as was also observed by Fürstner,10 so we needed to use a different protecting group for the carboxylic acid. The protection of the phenol by silylating both the alcohol and phenol and selective liberation of the alcohol is also cumbersome. We therefore decided to protect both the phenol and carboxylic acid with allyl groups as reported by Oikawa in his synthesis of prelasalocid.15 Condensation of ent-4b and (R)-(−)-5 with Dowex 50WX8-400-H+ in MeOH for 60 h gave crude tetracyclic salicylic acids with a partially deprotected side chain. Protection of this mixture with allyl bromide and K2CO3 in DMF gave silyl ether 22 (32%) and alcohol 23 (20%) without allylation of the primary alcohol of 23 (see Scheme 5). Additional alcohol 23 was prepared by desilylation of 22 with TBAF/HOAc (86%) so that the overall yield of 23 from ent-4b is 48%. Aldehyde 24 was prepared in 88% yield by Dess-Martin oxidation of 23.
SCHEME 5.
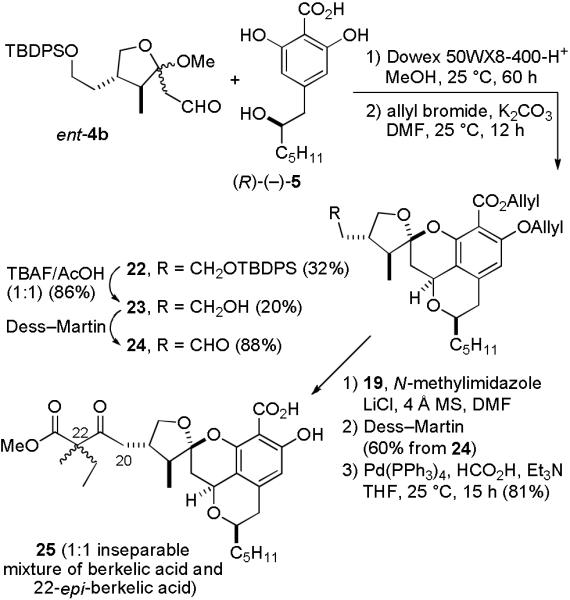
Synthesis of a Mixture of Berkelic Acid and the C-22 Epimer (25)
Treatment of aldehyde 24 with trimethylsilyl ketene acetal 19, LiCl, and N-methylimidazole afforded an inseparable mixture of all four possible aldol products, which was subjected to Dess-Martin oxidation to give a 1:1 mixture of keto esters in 60% yield from 24. Removal of both allyl groups15 with Pd(Ph3P)4, Et3N, and HCO2H afforded 25 (1:1 mixture of berkelic acid and 22-epi-berkelic acid) in 81% yield. The 1H and 13C NMR spectral data of 25 match those of natural berkelic acid both in CDCl3 and CD3OD, but there are doubled peaks for the C-20 methylene group and the methyl and ethyl groups attached to C-22 in the 1H NMR spectrum. The optical rotation of synthetic 25, [α] D22 − 104.6 (c = 0.07, MeOH), has the same sign as that of the natural berkelic acid, [α]D22 − 83.5 (c = 0.0113, MeOH),1 suggesting that the absolute stereochemistry of natural berkelic acid is most likely as shown in 25. The use of allyl protecting groups for both the phenol and carboxylic acid provides a very efficient solution of selectively protecting an acid and phenol without protecting a primary alcohol. The allyl groups also protect the phenol during the Dess-Martin oxidation of the C-21 alcohol to a ketone. In his synthesis of the methyl ester of berkelic acid, Fürstner reported that only the Swern oxidation (35%) could be used to oxidize the C-21 alcohol with a free phenol because other oxidants destroy the electron rich aromatic ring.10
We still needed to develop a procedure to either control the stereochemistry at the quaternary center16 C-22, or at least allow us to separate the aldol products. Kobayashi reported the diastereoselective alkylation of the enolate of 26 (see Scheme 6).17 Unfortunately, no aldol products were obtained from addition of the sodium enolate of 26 to the model aldehyde heptanal. TBS ketene aminal 27 undergoes a vinylogous Mukaiyama aldol reaction with hexanal at the γ-, not α-position so this reagent cannot be used.18 We prepared 27 and hydrogenated it with 5% Pd/C under 1 atm of H2 to give 28 stereospecifically as determined by an NOE between the methyl and TBS groups. We hoped that 28 would undergo a Mukaiyama aldol reaction now that a vinylogous Mukaiyama reaction was no longer possible. To our disappointment, attempted Mukaiyama aldol reactions of 28 with heptanal using either TiCl4 at various temperatures, LiCl and N-methylimidazole, or TMSOTf and i-Pr2EtN were unsuccessful.
SCHEME 6.
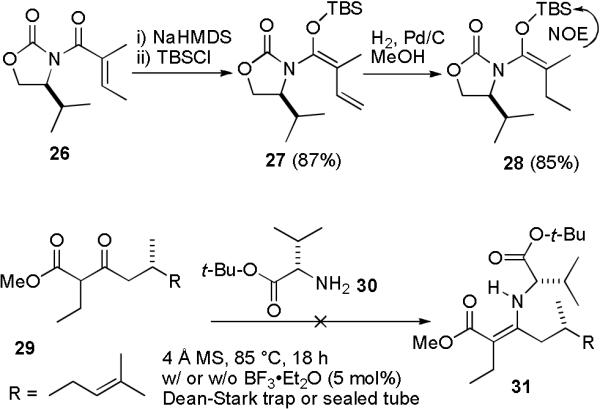
Unsuccessful Aldol Reactions
Koga reported the highly enantioselective alkylation of α-alkyl β-keto esters to give α,α-dialkyl β- keto esters.19,20 Chiral enamines were prepared from the α-alkyl β-keto esters and (S)-valine tert-butyl ester (30)21 in the presence of a catalytic amount of BF3•OEt2. Metalation of the enamines with LDA, coordination of the lithium cations with an additive (HMPA or THF), alkylation of the enamides and hydrolysis of the resulting imines affords the α,α-dialkyl β-keto esters with >90% ee. β-Keto ester 29 was synthesized by Dess-Martin oxidation of 39 (see below) to test this approach. To our surprise, 29 didn't react with (S)-valine tert-butyl ester (30) to give enamine 31 under a variety of conditions that were successful in our hands with ethyl 2-methylacetoacetate and ethyl 2-oxocyclohexanecarboxylate as reported.19,20 All of the reported examples of enamine formation from 30 use either cyclic β-keto esters or ethyl 2-methylacetoacetate, suggesting that our failure with 29 results from steric interactions between the bulky substituents of 29 and the valine moiety. This approach was therefore not pursued.
Kiyooka reported that stoichiometric oxazaborolidinone (S)-32, which is prepared in situ from N-Ts-(S)-valine22 and BH3•THF, induces aldol reactions between aldehydes and trimethylsilyl ketene acetals. Reaction occurs selectively at the Si-face of the aldehyde with excellent control of the stereochemistry at the β-carbon with the hydroxy group. Mixtures of isomers are formed at the α-carbon center.23 We needed to control the stereochemistry at the α-carbon; the stereochemistry of the β-carbon with the hydroxy group is irrelevant since it is oxidized to a ketone. Although Kiyooka's protocol won't solve our stereoselectivity problem, the mildly acidic conditions should be compatible with our substrate and we may be able to separate the aldol products if only two, rather than four, isomers are formed.
As expected, reaction of 24 and 19 as described by Kiyooka using (S)-32 afforded only 33 (40%) and 34 (40%) which were fortunately separable by preparative TLC, although at this time we didn't know which isomer was which (see Scheme 7). To our surprise, the analogous reaction using (R)-32 resulted in reduction of 24 to give primary alcohol 23. This suggests that the stereochemical preferences of 24 and (S)-32 are matched, while those of 24 and (R)-32 are mismatched leading to reduction of the aldehyde instead of the desired aldol reaction. Very different yields have been previously observed with matched and mismatched aldehydes.24
SCHEME 7.

Synthesis of Berkelic Acid (35) and 22-epi-Berkelic Acid (36)
Dess–Martin oxidation of the less polar isomer 33 afforded the keto ester in 85% yield which was deprotected with Pd(Ph3P)4, Et3N, and HCO2H to afford berkelic acid (35), although the stereochemistry at C-22 was still unassigned. The 1H and 13C NMR spectral data of 35 in both CDCl3 and CD3OD are identical to those of natural berkelic acid.8 The optical rotation of synthetic berkelic acid (35), [α]D22−115.5 (c = 0.55, MeOH), has the same sign as that of the natural product, [α]D22 − 83.5 (c = 0.0113, MeOH), indicating that the absolute stereochemistry is as shown. The differing numerical values may result from the low sample concentration used for the natural product. A similar sequence from the more polar isomer 34 afforded 22-epi-berkelic acid (36). The spectral data of 36 are similar to those of berkelic acid, but there are significant differences, most notably in the 1H NMR spectra for the C-20 methylene group and the methyl and ethyl groups attached to C-22. In CDCl3, the chemical shifts of the two protons on C-20 of 36 (δ 2.90, dd, J = 17.6, 2.9; δ 2.38, dd, J = 17.6, 10.0) are further apart from each other than those of 35 (δ 2.85, dd, J = 17.0, 2.4; δ 2.42 dd, J = 17.0, 9.8). The optical rotation of synthetic 22-epi-berkelic acid (36), [α]D22 − 107.0 (c = 0.43, MeOH), is very similar to that of berkelic acid indicating that the C-22 stereochemistry has a minor effect on the rotation as expected.
It still remained to assign the stereochemistry of 33–36 at C-22. Fráter reported the stereoselective α- alkylation of chiral β-hydroxy esters in excellent diastereoselectivity using chelation to control the stereochemistry of the alkylation.25 Fráter's alkylation might allow us to use the hydroxy group stereochemistry introduced at C-21 in the Kiyooka aldol reaction to control the stereochemistry at the quaternary center C-22. We decided to explore this approach first in a model system. Addition of (R)-citronellal (37) to the lithium enolate of methyl butyrate (38) afforded 39 as a mixture of four isomers (see Scheme 8). Treatment of 39 with 2.2 equiv of LDA gave a mixture of chelated alkoxy enolates 40 and 41, which was treated with MeI and HMPA as described by Fráter to give 16% of an inseparable 1:1 mixture of 42 and 43 containing a little 44 and 45. This sequence typically proceeds with excellent relative stereocontrol, but in poor yield.26 Therefore it isn't practical to use it with aldehyde 24 to prepare berkelic acid. However, it might be possible to prepare 42–45 with known stereochemistry and to assign the stereochemistry of 33–36 by comparison of the spectral data of hydroxy esters 33 and 34 with hydroxy esters 42–45.
SCHEME 8.

Synthesis of Aldol Adducts 42–45
Reaction of (R)-citronellal (37) and 19 using N-Ts-(S)-valine ((S)-32) gave 42 (38%) and 44 (40%). A similar sequence with the mismatched N-Ts-(R)-valine ((R)-32) provided 43 (21%) and 45 (18%). Isomers 42 and 43 are less polar than 44 and 45, so that these compounds can be easily separated. The stereochemistry can now be easily assigned. The compound produced by both Fráter's procedure, which controls the relative stereochemistry at C-2 and C-3, and Kiyooka's procedure using N-Ts-(S)-valine, which controls the absolute stereochemistry at C-3, must be 42. Therefore, the other isomer produced from Kiyooka's procedure using N-Ts-(S)-valine must be 44. The compound produced by both Fráter's procedure and Kiyooka's procedure using N-Ts-(R)-valine must be 43. Therefore, the other isomer produced from Kiyooka's procedure using N-Ts-(R)-valine must be 45.
The structure assignments of 42–45 were confirmed by Dess-Martin oxidation of 45 and 42 to keto ester 46 and of 43 and 44 to keto ester 47, and by conversion of 42 and 44 to ketals 48 and 49, respectively, whose stereochemistry was established by NOE studies (see Scheme 9).27 At this point, the structure assignments of 42–45 were secure, although they are based on the known selectivity of the Kiyooka aldol reaction for the Si face with N-Ts-(S)-valine, which has been confirmed in numerous examples.23
SCHEME 9.
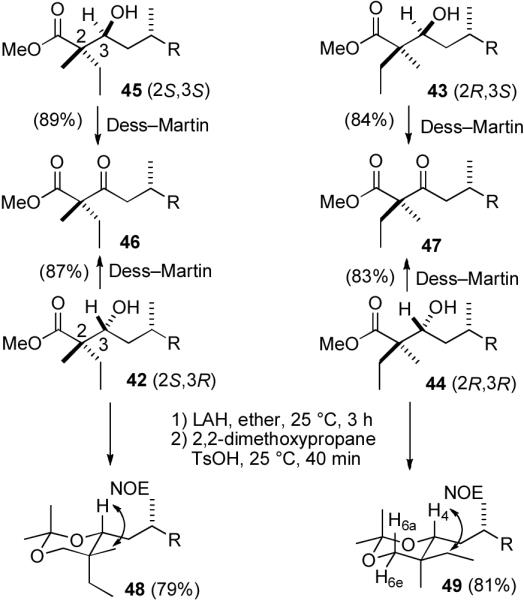
Structure Assignments of Aldol Adducts 42–45
We then compared the spectral data of 42–45 with those of 33 and 34. The subtle differences in the 1H and 13C NMR spectra of (21R,22S)-33, (2S,3R)-42, (2R,3S)-43 on the one hand and (21R,22R)-34, (2R,3R)-44, and (2S,3S)-45 on the other hand are identical in all 10 features that we can distinguish as shown in Tables S1 and S2 (see Supporting Information) suggesting that the stereochemistry of 33 and 34 can be assigned as shown by analogy to that of 42–45. The chromatographic properties also support this assignment: 33, 42, and 43 are less polar than 34, 44, and 45, respectively. Heathcock assigned the stereochemistry of aldol products by analysis of the 13C NMR spectral data.28 However, this approach was not extended to α,α-dialkyl aldol products and is not useful with our compounds.
Because berkelic acid was reported to show selective activity toward ovarian cancer OVCAR-3 with a GI50 of 91 nM,1 we submitted synthetic berkelic acid (35) and 22-epi-berkelic acid (36) to the NCI human disease-oriented 60-cell line in vitro antitumor screening protocol. Unfortunately, neither of them showed significant activity against any cell lines at 10−5 M (see Supporting Information). The racemic model acid prepared by hydrolysis of 8a inhibited lung cancer cell line HOP-92 and ovarian cancer IGROV1 very effectively at 10−5 M, but had little effect on other cell lines.
The proposed biosynthesis in Scheme 1 needs to be reconsidered in light of the revision of the stereochemistry of berkelic acid (35) at C-18 and C-19 and the assignment of the stereochemistry at C-22 and the absolute stereochemistry. The stereochemistry of berkelic acid (35) at C-9 is the same as that of pulvilloric acid (6). Berkelic acid co-occurs with spiciferone A (2), which makes it plausible that a spicifernin-like compound might be an intermediate in its biosynthesis. However, spicifernin (3) has the same stereochemistry as berkelic acid at C-22, but the opposite stereochemistry at C-18 and C-19. It is not immediately obvious how to relate the stereochemistry at C-22 of berkelic acid to that of the quaternary center of the congener spiciferone A (2). We therefore considered the biogenetic relationship between spiciferone A (2), spicifernin (3), and spiciferinone (54), which were isolated from same strain of Cochliobolus spicifer.4
The relative and absolute stereochemistry of spiciferone A (2) and spicifernin (3) have been assigned as shown.4 From feeding studies with [1,2-13C]- and [1-13C,2H3]-acetate, Nakajima established that they are biosynthesized from acetate and S-adenosylmethionine and proposed that they are biosynthesized from 51 (see Scheme 10). Because CD3 was incorporated only in the ethyl groups of spiciferone A (2) spicifernin (3), he suggested 51 was formed by a retro aldol reaction of a hydroxycyclodecatetraone.4 However, deuterium washout from the starter acetate has been observed in the biosynthesis of monensin A and other natural products, so aldehyde 51 might be derived from the reduction of polyketide 50.29 An aldol reaction of 51 through pathway A should afford 52, which could undergo dehydration to give spiciferone A (2). An aldol reaction of 51 through pathway B should afford 53, which would undergo dehydration to give spiciferinone (54). Oxidative cleavage of 54 at the indicated bond will give aldehyde ester 55. Hydrolysis of the pyran will give β-keto aldehyde 56 which can easily lose formic acid to give aldehyde 57. This could also occur by oxidation of the aldehyde to an acid and decarboxylation of the β-keto acid. Oxidation of 57 will give spicifernin (3) and reduction will give the possible berkelic acid precursor 58. The stereochemistry at the quaternary carbon center (C-22) was already set in 51. However, the stereochemistry at C-18 and C-19 is set in the further elaboration of spiciferinone (54), which proceeds through easily epimerizable intermediates. Four isomers are possible, but equilibration can occur at either C-18 or C-19 before reduction or oxidation of aldehyde 57. This scheme provides a simple biosynthetic pathway from the common intermediate 51 to spiciferone A (2), spicifernin (3), spiciferinone (54) and berkelic acid precursor 58. It is consistent with the known absolute stereochemistry at the quaternary carbon of the spiciferone A (2), spicifernin (3), and berkelic acid (35) and provides a simple explanation for the differing stereochemistry at C-18 and C-19 of spicifernin (3) and berkelic acid (35).
SCHEME 10.

Proposed Biosynthesis of Spiciferone A (2), Spicifernin (3), Spiciferinone (54) and Possible Berkelic Acid Precursor (58)
In conclusion, we have completed the first synthesis of (−)-berkelic acid (35), established the absolute stereochemistry, and assigned the stereochemistry at C-22. Although the aldol reaction to introduce the side chain is challenging, it proceeds effectively under Kiyooka's conditions using N-Ts-(S)-valine ((S)-32) to give only two of the four possible aldol products.
Experimental Section
Methyl (αS,βR,2S,3S,3'aS,4S,5'R)- and (αR,βR,2S,3S,3'aS,4S,5'R)- α,3-Dimethyl-α-ethyl- 3',3'a,4,5,5',6'-hexahydro-β-hydroxy-3-methyl-5'-pentyl-8'-(2-propenyloxy)-9'-(2- propenyloxycarbonyl)-spiro[furan-2(3H),2'-[2H]pyrano[2,3,4-de][1]benzopyran]-4-butanoate (33 and 34)
To a solution of N-Ts-(S)-valine22,23 (84 mg, 0.31 mmol) in dry CH2Cl2 (2 mL) was added BH3•THF (0.31 mL, 1 M solution in THF, 0.31 mmol) by syringe over 3 min under N2 at 0 °C. The solution was stirred for 30 min at 0 °C and additionally for 30 min at 25 °C. The solution was cooled −78 °C and a solution of aldehyde 24 (44.0 mg, 88.4 μ-mol) in CH2Cl2 (0.5 mL) was added dropwise over 3 min. After stirring for 5 min, silyl ketene acetal 19 (49 mg, 0.31 mmol) in CH2Cl2 (0.5 mL) was added dropwise over 3 min. The reaction mixture was stirred for 4 h at −78 °C and quenched with 1 M aqueous HCl (4 mL). The mixture was allowed to warm to 25 °C and the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were washed with saturated NaHCO3 (2 × 10 mL), dried (MgSO4) and concentrated to yield 71.6 mg of crude aldol product. Preparative TLC (4:1 hexanes/EtOAc, developed four times) gave 21.5 mg (40%) of 33 followed by 21.9 mg (40%) of 34.
Data for 33: [α]D22 −95.0 (c 0.22, CHCl3); 1H NMR 6.23 (s, 1, H-4), 6.07–5.92 (m, 2), 5.40 (br d, 1, J = 17.1), 5.36 (br d, 1, J = 16.5), 5.25 (br d, 1, J = 9.8), 5.22 (br d, 1, J = 9.2), 4.86–4.72 (m, 3, H-15), 4.51 (d, 2, J = 4.9), 4.28 (dd, 1, J = 8.5, 8.5, H-26), 3.86–3.78 (m, 1, H-9), 3.77–3.69 (m, 1, H-21), 3.709 (s, 3, OMe), 3.65 (dd, 1, J = 8.5, 8.5, H-26), 2.75 (dd, 1, J = 17.1, 3.4, H-8), 2.60 (dd, 1, J = 17.1, 11.0, H-8), 2.27–2.20 (m, 1, H-19), 2.22 (d, 1, J = 6.1, OH), 2.14 (dd, 1, J = 12.2, 5.5, H-16), 1.96 (dd, 1, J = 12.2, 12.2, H-16), 1.80 (dq, 1, J = 13.4, 7.3), 1.71–1.60 (m, 3), 1.59–1.46 (m, 3), 1.45–1.24 (m, 6), 1.14 (s, 3, H-27), 1.03 (d, 3, J = 6.7, H-25), 0.90 (t, 3, J = 6.7), 0.87 (t, 3, J = 7.3); 13C NMR 176.94, 165.66, 155.55, 149.09, 135.89, 132.96, 132.40, 118.17, 117.13, 114.65, 109.52, 108.52, 104.23, 76.22, 75.31, 73.87, 69.38, 68.11, 65.64, 51.79, 51.23, 49.04, 42.48, 36.32, 35.31, 34.55, 34.41, 31.79, 28.38, 25.14, 22.61, 16.75, 14.04, 11.87, 8.99; IR (neat) 3508, 2953, 2933, 2860, 1739, 1732, 1715; HRMS (EI) calcd for C35H50O9(M+) 614.3455, found 614.3451.
Data for 34: [α]D22 − 96.3 (c 0.22, CHCl3); 1H NMR 6.23 (s, 1, H-4), 6.07–5.92 (m, 2), 5.40 (br d, 1, J = 17.1), 5.36 (br d, 1, J = 16.5), 5.25 (br d, 1, J = 9.8), 5.22 (br d, 1, J = 9.2), 4.86–4.72 (m, 3, H-15), 4.51 (d, 2, J = 4.9), 4.31 (dd, 1, J = 8.5, 8.5, H-26), 3.86–3.77 (m, 1, H-9), 3.726 (s, 3, OMe), 3.74–3.68 (m, 1, H-21), 3.66 (dd, 1, J = 8.5, 8.5, H-26), 2.75 (dd, 1, J = 17.1, 3.4, H-8), 2.60 (dd, 1, J = 17.1, 11.0, H-8), 2.51 (d, 1, J = 7.3, OH), 2.30–2.21 (m, 1, H-19), 2.14 (dd, 1, J = 12.2, 4.9, H-16), 1.97 (dd, 1, J = 12.2, 12.2, H-16), 1.81–1.44 (m, 7), 1.43–1.17 (m, 6), 1.13 (s, 3, H-27), 1.04 (d, 3, J = 6.1, H-25), 0.90 (t, 3, J = 6.4), 0.85 (t, 3, J = 7.3); 13C NMR 177.58, 165.63, 155.56, 149.10, 135.87, 132.96, 132.39, 118.11, 117.12, 114.64, 109.51, 108.43, 104.22, 75.49, 75.30, 74.00, 69.37, 68.11, 65.62, 51.86, 51.45, 49.07, 42.57, 36.31, 34.57, 34.49, 34.41, 31.78, 29.60, 25.13, 22.60, 16.94, 14.03, 11.87, 8.80; IR (neat) 3522, 2955, 2933, 2860, 1737, 1732, 1715; HRMS (EI) calcd for C35H50O9 (M+) 614.3455, found 614.3458.
Methyl (αS,2S,3S,3'aS,4S,5'R)-9'-Carboxy-α,3-dimethyl-α-ethyl-3',3'a,4,5,5',6'-hexahydro-8'-hydroxy-3-methyl-β-oxo-5'-pentyl-spiro[furan-2(3H),2'-[2H]pyrano[2,3,4-de][1]benzopyran]-4-butanoate (35, berkelic acid)
To a solution of Pd(PPh3)4 (6.1 mg, 5.3 μmol) and the above allyl ether and allyl ester prepared from 33 (16.2 mg, 26.4 μmol) in dry THF (2 mL) was added NEt3 (148 μL, 1.06 mmol) and HCO2H (40 μL, 1.06 mmol) under N2 at 25 °C. The yellow solution was stirred at 25 °C for 15 h and quenched with saturated NaHCO3 (5 mL). The aqueous layer was extracted with ether (4 × 5 mL). The combined organic layers were dried (MgSO4) and concentrated. Flash chromatography on silica gel (80:1:0 to 200:1:1 CH2Cl2/MeOH/AcOH) gave 11.1 mg (78%) of berkelic acid (35): [α]D22 − 115.5 (c 0.55, MeOH); {lit.1 [α]D20 − 83.5 (c 0.0113, MeOH)}; 1H NMR (CDCl3, the residual peak of solvent is referenced as δ 7.24 rather than 7.27 to facilitate comparison with the literature data1) 11.82 (s, 1, OH), 11.13–10.92 (br, 1, OH), 6.42 (s, 1, H-4), 4.76 (dd, 1, J = 12.2, 5.4, H- 15), 4.44 (dd, 1, J = 8.5, 8.5, H-26), 3.84–3.76 (m, 1, H-9), 3.73 (s, 3, OMe), 3.59 (dd, 1, J = 8.5, 8.5, H-26), 2.85 (dd, 1, J = 17.0, 2.4, H-20), 2.78 (dd, 1, J = 17.7, 3.7, H-8), 2.60 (dd, 1, J = 17.7, 11.0, H-8), 2.54–2.45 (m, 1, H-19), 2.42 (dd, 1, J = 17.0, 9.8, H-20), 2.21 (dd, 1, J = 12.2, 5.4, H-16), 2.05 (dd, 1, J = 12.2, 12.2, H-16), 1.95 (dq, 1, J = 14.2, 7.3, H-23), 1.87 (dq, 1, J = 10.7, 6.8, H-18), 1.80 (dq, 1, J = 14.2, 7.3, H-23), 1.68–1.57 (m, 1), 1.58–1.43 (m, 2), 1.43–1.20 (m, 5), 1.32 (s, 3, H-27), 1.09 (d, 3, J = 6.8, H-25), 0.88 (t, 3, J = 6.6), 0.83 (t, 3, J = 7.6); 1H NMR (CD3OD, 500 MHz) 6.27 (s, 1, H-4), 4.72 (dd, 1, J = 12.2, 5.4, H-15), 4.30 (dd, 1, J = 8.5, 8.5, H-26), 3.84–3.77 (m, 1, H-9), 3.73 (s, 3, OMe), 3.50 (dd, 1, J = 8.5, 8.5, H-26), 2.88 (dd, 1, J = 17.6, 3.1, H-20), 2.78 (dd, 1, J = 17.3, 3.7, H-8), 2.70–2.62 (m, 1, H-19), 2.54 (dd, 1, J = 17.3, 11.2, H-8), 2.53 (dd, 1, J = 17.6, 10.5, H-20), 2.14 (dd, 1, J = 12.2, 5.4, H-16), 1.94 (dq, 1, J = 14.2, 7.3, H-23), 1.91 (dd, 1, J = 12.2, 12.2, H-16), 1.88–1.78 (m, 2, H-23, H-18), 1.63–1.48 (m, 3), 1.46–1.27 (m, 5), 1.32 (s, 3, H-27), 1.08 (d, 3, J = 6.3, H-25), 0.92 (t, 3, J = 6.8), 0.83 (t, 3, J = 7.6); 13C NMR (CDCl3) 206.0, 173.4, 170.5, 162.5, 149.8, 142.2, 112.2 (2 C), 110.5, 98.6, 75.2, 73.5, 67.2, 59.7, 52.5, 48.2, 41.6, 39.4, 36.2, 34.30, 34.29, 31.8, 27.9, 25.0, 22.6, 18.4, 14.0, 12.0, 8.7; 13C NMR (CD3OD, 400 MHz) 208.8, 174.8, 173.7, 163.4, 153.1, 142.3, 113.8, 110.7, 109.4, 101.1, 76.6, 74.2, 69.5, 61.0, 52.9, 42.6, 40.5, 37.4, 35.4, 35.0, 33.0, 28.9, 26.2, 23.7, 18.9, 14.4, 11.9, 9.0 (a peak near δ 49.2 is obscured by the solvent peak); IR (neat) 3238, 2957, 2933, 2860, 1713, 1694; HRMS (EI) calcd for C29H40O9(M+) 532.2672, found 532.2659. The 1H and 13CNMR spectral data in both CDCl3 and CD3OD are identical to those of the natural product (see Tables S2–S5).1
Methyl (αR,2S,3S,3'aS,4S,5'R)-9'-Carboxy-α,3-dimethyl-α-ethyl-3',3'a,4,5,5',6'-hexahydro-8'- hydroxy-3-methyl-β-oxo-5'-pentyl-spiro[furan-2(3H),2'-[2H]pyrano[2,3,4-de][1]benzopyran]-4-butanoate (36, 22-epi-berkelic acid)
An identical reaction with the allyl ether and allyl ester prepared from 34 (14.0 mg, 22.8 μmol) afforded 8.7 mg (72%) of 22-epi-berkelic acid (36): [α]D22 − 107.0 (c 0.43, MeOH); 1H NMR (CDCl3, the residual peak of solvent is referenced as δ 7.24 rather than 7.27 to facilitate comparison with the literature data1) 11.83 (s, 1, OH), 11.08–10.97 (br, 1, OH), 6.42 (s, 1, H-4), 4.77 (dd, 1, J = 12.2, 5.4, H-15), 4.45 (dd, 1, J = 8.5, 8.5, H-26), 3.84–3.76 (m, 1, H-9), 3.73 (s, 3, OMe), 3.59 (dd, 1, J = 8.5, 8.5, H-26), 2.90 (dd, 1, J = 17.6, 2.9, H-20), 2.79 (dd, 1, J = 17.6, 3.9, H-8), 2.60 (dd, 1, J = 17.6, 11.2, H-8), 2.54–2.44 (m, 1, H-19), 2.38 (dd, 1, J = 17.6, 10.0, H-20), 2.21 (dd, 1, J = 12.2, 5.4, H-16), 2.06 (dd, 1, J = 12.2, 12.2, H-16), 1.95 (dq, 1, J = 14.2, 7.3, H-23), 1.87 (dq, 1, J = 11.3, 6.8, H-18), 1.80 (dq, 1, J = 14.2, 7.3, H-23), 1.68–1.57 (m, 1), 1.58–1.43 (m, 2), 1.43–1.20 (m, 5), 1.33 (s, 3, H-27), 1.09 (d, 3, J = 6.8, H-25), 0.88 (t, 3, J = 6.6), 0.81 (t, 3, J = 7.3); 1H NMR (CD3OD, 500 MHz) 6.27 (s, 1, H-4), 4.73 (dd, 1, J = 12.2, 5.4, H-15), 4.30 (dd, 1, J = 8.5, 8.5, H-26), 3.84–3.77 (m, 1, H-9), 3.73 (s, 3, OMe), 3.50 (dd, 1, J = 8.5, 8.5, H-26), 2.92 (dd, 1, J = 17.6, 3.0, H-20), 2.79 (dd, 1, J = 17.3, 3.6, H-8), 2.72–2.62 (m, 1, H-19), 2.55 (dd, 1, J = 17.3, 10.3, H-8), 2.49 (dd, 1, J = 17.6, 10.5, H-20), 2.14 (dd, 1, J = 12.2, 5.4, H-16), 1.94 (dq, 1, J = 14.2, 7.3, H-23), 1.91 (dd, 1, J = 12.2, 12.2, H-16), 1.88–1.78 (m, 2, H-23, H-18), 1.64–1.49 (m, 3), 1.47–1.30 (m, 5), 1.33 (s, 3, H-27), 1.08 (d, 3, J = 6.3, H-25), 0.92 (t, 3, J = 6.8), 0.82 (t, 3, J = 7.3); 13C NMR (CDCl3) 206.0, 173.4 (tiny), 170.5, 162.5, 149.8, 142.2, 112.17, 112.15, 110.5, 98.6, 75.2, 73.5, 67.2, 59.7, 52.5, 48.2, 41.5, 39.3, 36.2, 34.31, 34.29, 31.8, 27.9, 25.0, 22.6, 18.3, 14.0, 12.0, 8.6; 13C NMR (CD3OD, 400 MHz) 208.7, 174.8, 173.6, 163.4, 153.1, 142.3, 113.8, 110.7, 109.4, 101.1, 76.6, 74.1, 69.5, 61.0, 52.9, 42.6, 40.5, 37.4, 35.4, 35.0, 33.0, 28.8, 26.2, 23.7, 18.8, 14.4, 11.9, 8.9 (a peak near δ 49.2 is obscured by the solvent peak); IR (neat) 3233, 2957, 2933, 2860, 1713, 1694; HRMS (EI) calcd for C29H40O9 (M+) 532.2672, found 532.2667.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (GM-50151) for support of this work. We thank Prof. D. Stierle for copies of spectral data of berkelic acid and berkelic acid methyl ester.
Footnotes
Supporting Information Available. Experimental procedures for reactions not in the experimental section, comparison tables S1–S2 of NMR data, and 1H and 13C NMR spectral data. Data for the acid prepared by hydrolysis of 8a, 35, and 36 in the NCI 60 cell screen. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Stierle AA, Stierle DB, Kelly K. J. Org. Chem. 2006;71:5357–5360. doi: 10.1021/jo060018d. [DOI] [PubMed] [Google Scholar]
- 2.Bullimore BK, McOmie JFW, Turner AB, Galbraith MN, Whalley WB. J. Chem. Soc. C. 1967:1289–1293. [Google Scholar]
- 3.Rödel T, Gerlach H. Liebigs Ann. Chem. 1997:213–216. [Google Scholar]
- 4.(a) Nakajima H, Hamasaki T, Maeta S, Kimura Y, Takeuchi Y. Phytochemistry. 1990;29:1739–1743. [Google Scholar]; (b) Nakajima H, Matsumoto R, Kimura Y, Hamasaki T. J. Chem. Soc., Chem. Commun. 1992:1654–1656. [Google Scholar]; (c) Nakajima H, Fujimoto H, Matsumoto R, Hamasaki T. J. Org. Chem. 1993;58:4526–4528. [Google Scholar]; (d) Nakajima H, Fukuyama K, Fujimoto H, Baba T, Hamasaki T. J. Chem. Soc., Perkin Trans. 1. 1994:1865–1869. [Google Scholar]
- 5.Zhou J, Snider BB. Org. Lett. 2007;9:2071–2074. doi: 10.1021/ol0704338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For another model study, see: Marsini MA, Huang Y, Lindsey CC, Wu K-L, Pettus TRR. Org. Lett. 2008;10:1477–1480. doi: 10.1021/ol8003244.. Huang Y, Pettus TRR. Synlett. 2008:1353–1356. doi: 10.1055/s-2008-1072750..
- 7.PCMODEL version 8.0 from Serena Software was used with MMX. Calculations were carried out on analogues with the pentyl side chain replaced by a methyl group to minimize irrelevant conformational complexity.
- 8.Wu X, Zhou J, Snider BB. Angew. Chem. Int. Ed. 2009;48:1283–1286. doi: 10.1002/anie.200805488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calculations were carried out on analogues with both the pentyl and TBDPSOCH2CH2 side chains replaced by methyl groups to minimize irrelevant conformational complexity.
- 10.Buchgraber P, Snaddon TN, Wirtz C, Mynott R, Goddard T, Fürstner A. Angew. Chem. Int. Ed. 2008;47:8450–8454. doi: 10.1002/anie.200803339. [DOI] [PubMed] [Google Scholar]
- 11.For the necessity of protection of the free phenols before Dess-Martin oxidation, see: Yang Z-Q, Danishefsky SJ. J. Am. Chem. Soc. 2003;125:9602–9603. doi: 10.1021/ja036192q.. Yang Z-Q, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348.. Wagner J, Andres H, Rohrbach S, Wagner D, Oberer L, France J. J. Org. Chem. 2005;70:9588–9590. doi: 10.1021/jo051112h..
- 12.Iida A, Nakazawa S, Okabayashi T, Horii A, Misaki T, Tanabe Y. Org. Lett. 2006;8:5215–5218. doi: 10.1021/ol0619361. [DOI] [PubMed] [Google Scholar]
- 13.(a) Crouch RD. Tetrahedron. 2004;60:5833–5871. [Google Scholar]; (b) Bartoli G, Cupone G, Dalpozzo R, De Nino A, Maiuolo L, Procopio A, Sambri L, Tagarelli A. Tetrahedron Lett. 2002;43:5945–5947. [Google Scholar]
- 14.Hagiwara H, Inoguchi H, Fukushima M, Hoshi T, Suzuki T. Tetrahedron Lett. 2006;47:5371–5373. [Google Scholar]
- 15.For the protection of a salicylic acid with two allyl groups, see: Migita A, Shichijo Y, Oguri H, Watanabe M, Tokiwano T, Oikawa H. Tetrahedron Lett. 2008;49:1021–1025..
- 16.For a review of stereoselective formation of quaternary carbon centers, see: Denissova I, Barriault L. Tetrahedron. 2003;59:10105–10146..
- 17.(a) Hosokawa S, Sekiguchi K, Enemoto M, Kobayashi S. Tetrahedron Lett. 2000;41:6429–6433. [Google Scholar]; (b) Abe T, Suzuki T, Sekiguchi K, Hosokawa S, Kobayashi S. Tetrahedron Lett. 2003;44:9303–9305. [Google Scholar]
- 18.Shirokawa S.-i., Kamiyama M, Nakamura T, Okada M, Nakazaki A, Hosokawa S, Kobayashi S. J. Am. Chem. Soc. 2004;126:13604–13605. doi: 10.1021/ja0465855. [DOI] [PubMed] [Google Scholar]
- 19.(a) Ando K, Takemasa Y, Tomioka K, Koga K. Tetrahedron. 1993;49:1579–1588. [Google Scholar]; (b) Tomioka K, Ando K, Takemasa Y, Koga K. J. Am. Chem. Soc. 1984;106:2718–2719. [Google Scholar]
- 20.For the application of Koga's chemistry, see: Cristau H-J, Marat X, Vors J-P, Virieux D, Pirat J-L. Current Org. Synth. 2005;2:113–119.. Peese KM, Gin DY. Org. Lett. 2005;7:3323–3325. doi: 10.1021/ol051184v..
- 21.Chen H, Feng Y, Xu Z, Ye T. Tetrahedron. 2005;61:11132–11140. [Google Scholar]
- 22.(a) Berry MB, Craig D. Synlett. 1992:41–44. [Google Scholar]; (b) Västilä P, Pastor IM, Adolfsson H. J. Org. Chem. 2005;70:2921–2929. doi: 10.1021/jo048146u. [DOI] [PubMed] [Google Scholar]
- 23.(a) Kiyooka S.-i., Kaneko Y, Komura M, Matsuo H, Nakano M. J. Org. Chem. 1991;56:2276–2278. [Google Scholar]; (b) Kiyooka S.-i., Shahid KA. Tetrahedron: Asymmetry. 2000;11:1537–1542. [Google Scholar]; (c) Kiyooka S.-i., Shahid KA, Goto F, Okazaki M, Shuto Y. J. Org. Chem. 2003;68:7967–7978. doi: 10.1021/jo034901c. [DOI] [PubMed] [Google Scholar]; (d) Kiyooka S.-i., Hena MA. J. Org. Chem. 1999;64:5511–5523. doi: 10.1021/jo990342r. [DOI] [PubMed] [Google Scholar]; (e) Fujiyama R, Goh K, Kiyooka S.-i. Tetrahedron Lett. 2005;46:1211–1215. [Google Scholar]; (f) Ishiwara K, Yamamoto H. In: Modern Aldol Reactions, Vol. 2: Metal Catalysis. Mahrwald R, editor. Wiley-VCH; Weinheim: 2004. pp. 25–68. [Google Scholar]
- 24.Kiyooka S.-i., Maeda H, Hena MA, Uchida M, Kim C-S, Horiike M. Tetrahedron Lett. 1998;39:8287–8290. [Google Scholar]
- 25.(a) Fráter G, Müller U, Günther W. Tetrahedron. 1984;40:1269–1277. [Google Scholar]; (b) Fráter G. Helv. Chem. Acta. 1979;62:2825–2828. [Google Scholar]
- 26.(a) Tayama E, Hashimoto R. Tetrahedron Lett. 2007;48:7950–7952. [Google Scholar]; (b) Akeboshi T, Ohtsuka Y, Sugai T, Ohta H. Tetrahedron. 1998;54:7387–7394. [Google Scholar]
- 27.Crump RANC, Fleming I, Hill JHM, Parker D, Reddy NL, Waterson D. J. Chem. Soc., Perkin Trans. 1. 1992:3277–3294. [Google Scholar]
- 28.Heathcock CH, Pirrung MC, Sohn JE. J. Org. Chem. 1979;44:4294–4299. [Google Scholar]
- 29.Sood GR, Ashworth DM, Ajaz AA, Robinson JA. J. Chem. Soc., Perkin Trans. 1. 1988:3183–3193. [Google Scholar]; Florova G, Kazanina G, Reynolds KA. Biochemistry. 2002;41:10462–10471. doi: 10.1021/bi0258804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.