

Abstract
Autophagy is an evolutionarily conserved cell survival pathway that enables cells to recoup ATP and other critical biosynthetic molecules during nutrient deprivation or exposure to hypoxia, which are hallmarks of the tumour microenvironment. Autophagy has been implicated as a potential mechanism of resistance to anticancer agents as it can promote cell survival in the face of stress induced by chemotherapeutic agents by breaking down cellular components to generate alternative sources of energy. Disruption of autophagy with chloroquine (CQ) induces the accumulation of ubiquitin-conjugated proteins in a manner similar to the proteasome inhibitor bortezomib (BZ). However, CQ-induced protein accumulation occurs at a slower rate and is localized to lysosomes in contrast to BZ, which stimulates rapid buildup of ubiquitinated proteins and aggresome formation in the cytosol. The histone deacetylase (HDAC) inhibitor vorinostat (VOR) blocked BZ-induced aggresome formation, but promoted CQ-mediated ubiquitinated protein accumulation. Disruption of autophagy with CQ strongly enhanced VOR-mediated apoptosis in colon cancer cells. Accordingly, knockdown of the essential autophagy gene Atg7 also sensitized cells to VOR-induced apoptosis. Knockdown of HDAC6 greatly enhanced BZ-induced apoptosis, but only marginally sensitized cells to CQ. Subsequent studies determined that the CQ/VOR combination promoted a large increase in superoxide generation that was required for ubiquitinated protein accumulation and cell death. Finally, treatment with the CQ/VOR combination significantly reduced tumour burden and induced apoptosis in a colon cancer xenograft model. Collectively, our results establish that inhibition of autophagy with CQ induces ubiquitinated protein accumulation and VOR potentiates CQ-mediated aggregate formation, superoxide generation and apoptosis.
Keywords: apoptosis, autophagy, bortezomib, proteasome, aggresome, chloroquine, histone deacetylase, superoxide, cancer
Introduction
A hallmark feature of aggressive cancers is a general increase in protein synthesis and protein degradation, both of which are required to increase tumour growth [1]. There are two major protein degradation systems in cells, the proteasome pathway and the autophagy pathway. The roles of these pathways in regulating cell growth, survival and metastasis of cancer cells make them attractive therapeutic targets [2–4]. Consistent with this idea, the proteasome inhibitor bortezomib (BZ; Velcade), is currently being used for the treatment of multiple myeloma and mantle cell lymphoma. Autophagy is a bulk degradation pathway that degrades unwanted proteins and defective organelles [5]. However, cancer cells can activate this pathway to recycle proteins to generate energy in an attempt to survive stressful conditions [6, 7]. Preclinical studies conducted with an Akt-driven tumour model established that autophagy is preferentially activated in tumour cells during the early stages of tumorigenesis when the vasculature is immature and nutrients and oxygen are limited [8].
Despite having diverse mechanisms of action, many frontline anticancer agents ultimately shutdown conventional metabolic pathways due to the cellular stress that they impose and thus, would be predicted to stimulate autophagy [9]. The induction of autophagy has been observed in malignant cells following treatment with a number of cancer therapeutics including arsenic trioxide, rapamycin, histone deacetylase (HDAC) inhibitors, tamoxifen, imatinib and ionizing radiation [10–15]. Although rigorous testing is required to fully elucidate the consequences of autophagy induction for the activity of individual treatments, it could significantly compromise efficacy by providing cancer cells with a mechanism to combat therapeutic stress. Therefore, inhibition of autophagy may be a promising novel strategy with broad applications in cancer therapy.
Chloroquine (CQ) is a synthetic 4-aminoquinoline that has been used for more than 60 years in human beings for the treatment of rheumatoid arthritis (RA) [16] and HIV and the prophylaxis and treatment of malaria [17, 18]. CQ functions as a weak base and is trapped in acidic cellular compartments including lysosomes [19]. Thus, deacidification of lysosomes by CQ impairs the activity of most lysosomal proteases due to their strict pH requirements. Consequently, CQ inhibits the last step in the autophagic degradation process [20]. Here we demonstrate that inhibition of autophagy with CQ stimulates superoxide generation and ubiquitin-conjugated protein accumulation. Furthermore, the combination of CQ with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA, vorinostat [VOR]) strongly enhances superoxide generation, ubiquitinated protein accumulation and apoptosis induction in colon cancer cells and has significant activity in a human colon cancer xenograft model.
Materials and methods
Animals and cell lines
Female nude mice (BALB/c background) were purchased from Harlan (Indianapolis, IN, USA). HT29 and HCT8 colon cancer cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA). Human colon cancer cell lines were maintained in minimal essential medium supplemented with 10% foetal bovine serum.
Antibodies and chemicals
Antibodies were obtained from the following commercial sources: anti-actin (Sigma, St. Louis, MO, USA), anti-HDAC6, Atg7 and LC3-II (Cell Signaling, Beverly, MA, USA), anti-ubiquitin, cathepsin D and LAMP-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Horseradish peroxidase-conjugated secondary antibodies for immunoblotting were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA). Alexa Fluor 488 goat anti-mouse and Texas Red goat anti-rabbit were obtained from Molecular Probes (Eugene, OR, USA). BZ and VOR were purchased from the CTRC pharmacy. CQ, 3-methyladenine (3-MA), propidium iodide (PI), cycloheximide (CHX) and N-acetyl-cysteine (NAC) were obtained from Sigma Chemical (St. Louis, MO, USA).
Immunocytochemistry and immunohistochemistry
Colon cancer cells were plated on chamber slides prior to drug exposure. Cells were fixed with 4% paraformaldehyde, permeabilized using 0.2% triton-X-100, and incubated overnight with indicated primary antibodies. Fluorescent secondary antibodies were used to visualize protein localization. ToPro-3 (Molecular Probes) was used to counterstain the nucleus. Images were obtained using a Zeiss LSM 510 Meta confocal microscope (Oberkochem, Germany) with a 40×/1.3 objective. Zeiss LSM 510 Version 3.2 SP2 software was used for image acquisition.
Paraffin sections were prepared as previously described [21]. Analysis of DNA fragmentation by TUNEL was performed with a commercial kit (Promega, Madison, WI, USA). PI was used to counterstain the nucleus. All slides were mounted using Prolong anti-fade reagent (Molecular Probes). Images were obtained by fluorescent microscopy. Percentages of TUNEL+ cells were determined by manual counting of five random fields per section. Images were captured using an Olympus fluorescent microscope (Center Valley, PA, USA) with a DP71 camera and a 20× objective. Image-Pro Plus software Version 6.2.1 (MediaCybernetics, Bethesda, MD, USA) was used for image acquisition.
Transmission electron microscopy
Transmission electron microscopy of cells was performed as previously described [22]. Sections were cut in an LKB Ultracut microtome (Leica, Deerfield, IL, USA), stained with uranyl acetate and lead citrate, and examined in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA, USA). Images were captured using the AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA, USA).
Immunoblotting
Colon cancer cells were incubated with 100 nM BZ or 50 μM CQ for the indicated times. Cells were collected using a cell scraper at 4°C and were then lysed as previously described [23]. Approximately 50 μg of total cellular protein from each sample were subjected to SDS-PAGE, proteins were transferred to nitrocellulose membranes and the membranes were blocked with 5% non-fat milk in a Tris-buffered saline solution containing 0.1% Tween-20 for 1 hr. The blots were then probed overnight with relevant antibodies, washed, and probed with species-specific secondary antibodies coupled to horseradish peroxidase. Immunoreactive material was detected by enhanced chemiluminescence (West Pico, Pierce, Inc., Rockville, IL, USA).
Preparation and transfection of siRNAs
HDAC6 and Atg7 SMARTpool siRNA were obtained from Dharmacon (Lafayette, CO, USA). For control, siRNA directed against firefly luciferase (non-target) was used. Cells were transfected with 100 nM of each siRNA using Oligofectamine (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Transfected cells were incubated at 37°C for 24 hrs without changing the medium. Efficiency of RNAi was measured by immunoblotting using anti-HDAC6 and Atg7 antibodies.
Quantification of drug-induced apoptosis and cytotoxicity
Apoptosis was measured by PI or active caspase-3 staining followed by flow cytometric analysis as described previously [24]. Following drug incubation, cells were harvested, pelleted by centrifugation and resuspended in phosphate-buffered saline (PBS) containing 50 μg/ml PI, 0.1% Triton-X-100 and 0.1% sodium citrate. Active caspase-3 levels were determined using a commercial kit (BD Biosciences, San Jose, CA, USA). Fluorescence was quantified by flow cytometric analysis.
Quantification of ubiquitin-conjugated protein accumulation
Aggregate formation was detected by the presence of ubiquitin-containing inclusion bodies that were observed by immunocytochemistry with an anti-ubiquitin antibody followed by a fluorescent secondary antibody and visualized by fluorescent microscopy. Approximately 200 cells were scored as aggregate-positive or -negative by confocal microscopy and repeated three times.
Clonogenic survival assays
Cells were treated for 24 hrs with 50 μM CQ, 2.5 μM VOR, or the combination for 24 hrs. Drug-treated cells were then washed twice in PBS followed by the addition of fresh media. The cells were incubated for 10 days in a humidified incubator at 37°C with 5% CO2. Colonies were washed in PBS, fixed with methanol and stained with crystal violet. Colonies were scored using an Alpha Innotech (San Leandro, CA, USA) gel documentation system.
Quantification of intracellular superoxide generation
HCT8 and HT29 colon cancer cells were treated with CQ and VOR for 24 hrs or BZ and VOR for 12 hrs. Intracellular superoxide generation was detected by staining cells with hydroethidine (Molecular Probes) as described previously [23]. Fluorescence was quantified by flow cytometry.
Implantation of tumour cells and treatment schedule
HCT8 colon cancer cells were harvested from culture flasks after brief trypsinization and transferred to serum-free HBSS. Only single-cell suspensions of >90% viability determined by trypan blue exclusion were used. Tumour cells (1×107 cells) were injected into the right flank of female nude mice and allowed to establish tumours. Following tumour formation, animals were pair-matched by tumour size and placed into groups of eight mice. Animals were then treated by intraperitoneal injection of 60 mg/kg CQ, oral gavage of 100 mg/kg VOR, or both agents for 21 days. Tumour volume and animal weight measurements were recorded. For immunohistochemistry, tumour tissue was formalin-fixed and paraffin embedded.
Statistical analyses
Statistical significance of differences observed in drug-treated and control samples were determined using the Tukey–Kramer comparison test or the Student’s t-test. Differences were considered significant in all experiments at P < 0.05.
Results
Chloroquine and bortezomib stimulate ubiquitin-conjugated protein accumulation in colon cancer cells
CQ disrupts lysosomal function and inhibits the last step in the autophagic degradation process [20]. Consistent with this mechanism of action, CQ treatment induced lipid modification of LC3-I into LC3-II and increased the expression and cytosolic localization of cathepsin D (Fig. 1A). We suggested that CQ treatment would result in an accumulation of proteins normally degraded by autophagy. Inhibition of proteasomal activity with BZ or autophagy with CQ resulted in the accumulation of ubiquitinated proteins in colon cancer cells as assessed by immunoblotting (Fig. 1B). However, inhibition of autophagy with CQ induced a much slower rate of protein accumulation as compared with proteasomal inhibition (Fig. 1C). Analysis of the subcellular distribution of these ubiquitin conjugates by immunofluorescent anti-ubiquitin staining and confocal microscopy revealed that BZ stimulated perinuclear aggresome formation, whereas CQ induced lysosomal accumulation of these aggregates (Fig. 1D).
Fig 1.
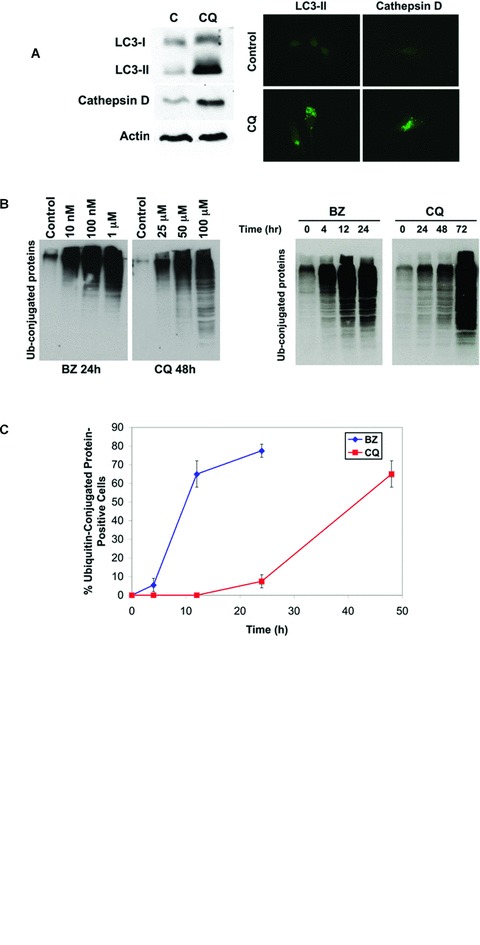
CQ treatment induces ubiquitin-conjugated protein accumulation. (A) CQ disrupts lysosomal function causing an accumulation of cathepsin D and conversion of LC3-I to LC3-II. HCT8 cells were treated with 50 μM CQ for 48 hrs. Cathepsin D and LC3-II levels were determined by immunoblotting and immunocytochemistry as described in the ‘Materials and methods’. (B) BZ and CQ stabilize ubiquitinated proteins. HCT8 cells were treated with the indicated concentrations of BZ for 24 hrs and CQ for 48 hrs (left) or incubated with 100 nM BZ or 50 μM CQ for the indicated times (right). Immunoblotting was performed with an anti-ubiquitin antibody as described in ‘Materials and methods’. (C) Kinetic analysis of ubiquitinated aggregate formation. HCT8 cells were incubated with 100 nM BZ or 50 μM CQ for the indicated times. Quantification of ubiquitinated aggregates was performed by immunofluorescent staining with an anti-ubiquitin antibody. Approximately 200 cells were counted and scored as aggregate-positive or aggregate-negative using a confocal microscope. Mean ± S.D., n= 3. (D) CQ-induced aggregates are localized within the lysosome. HCT8 cells were incubated for 24 hrs with 100 nM BZ or 50 μM CQ for 48 hrs and stained for detection of ubiquitin (green) and the lysosomal marker LAMP-2 (red). Fluorescence was visualized by confocal microscopy.
HDAC inhibition disrupts BZ-induced aggresome formation, but enhances CQ-mediated ubiquitinated protein accumulation
We previously demonstrated that BZ stimulates aggresome formation and that HDAC inhibition disrupts this process in pancreatic cancer and multiple myeloma models [3, 21]. Consistent with these results, we observed aggresome formation and disruption in HT29 and HCT8 colon cancer cell lines following 24 hrs of BZ and BZ/VOR treatment (Fig. 2A and B). A 48-hr exposure to CQ stimulated lysosomal ubiquitinated protein accumulation and this effect was strongly augmented by the addition of VOR (Fig. 2A and B). The protein aggregates were organized as electron dense particles when visualized by transmission electron microscopy (Fig. 2C).
Fig 2.
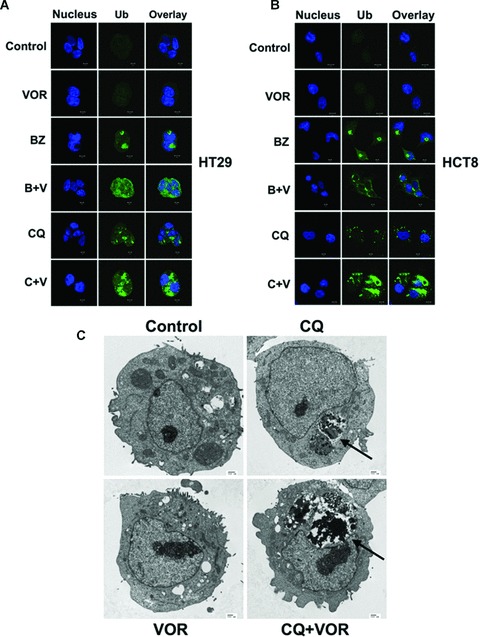
VOR disrupts BZ-induced aggresome formation and enhances CQ-mediated aggregate accumulation. (A, B) Effects of VOR on BZ- and CQ-induced ubiquitinated protein accumulation. HCT8 and HT29 cells were incubated with 100 nM BZ for 24 hrs or with 2.5 μM VOR or 50 μM CQ for 48 hrs. Ubiquitinated aggregates were visualized with an anti-ubiquitin antibody (green). The nucleus was counterstained with ToPro-3 (blue). Images were generated by confocal microscopy and are representative of the treatment group. (C) Electron microscopy of drug-exposed treated HCT8 cells. Arrows indicate electron dense material suggestive of protein aggregates in the CQ and CQ+VOR treated cells.
Ubiquitin-conjugated protein accumulation correlates with the induction of apoptosis
Kinetic analysis of aggresome formation in HCT8 colon cancer cells demonstrated that approximately 70% of cells possessed an aggresome by 24 hrs after treatment with 100 nM BZ, whereas CQ required 48 hrs to induce a similar level of protein aggregation (Fig. 1C). To determine whether protein aggregation correlated with the ability of these agents to induce apoptosis, HCT8 and HT29 cells were treated with BZ or the autophagic inhibitors (CQ and 3-MA) for 24 hrs with or without VOR. The BZ+VOR combination induced high levels of apoptosis (60–70%), whereas the CQ+VOR combination stimulated low levels of apoptosis (10%) (Fig. 3A). However, 48-hr treatment with CQ+VOR led to a strong induction in apoptosis as measured by caspase-3 activation (Fig. 3B) and DNA fragmentation (Fig. 3C). In addition, this drug combination also led to a significant reduction in clonogenic survival (Fig. 3D). We have previously demonstrated that inhibiting protein synthesis with CHX blocks BZ and BZ+VOR-mediated apoptosis in multiple myeloma and pancreatic cancer models [3, 22]. Consistent with these results, CHX also prevented ubiquitinated protein accumulation in HCT8 colon cancer cells treated with BZ and CQ (Fig. 3E) and apoptosis induced by the CQ+VOR or CQ+3-MA combinations (Fig. 3F).
Fig 3.
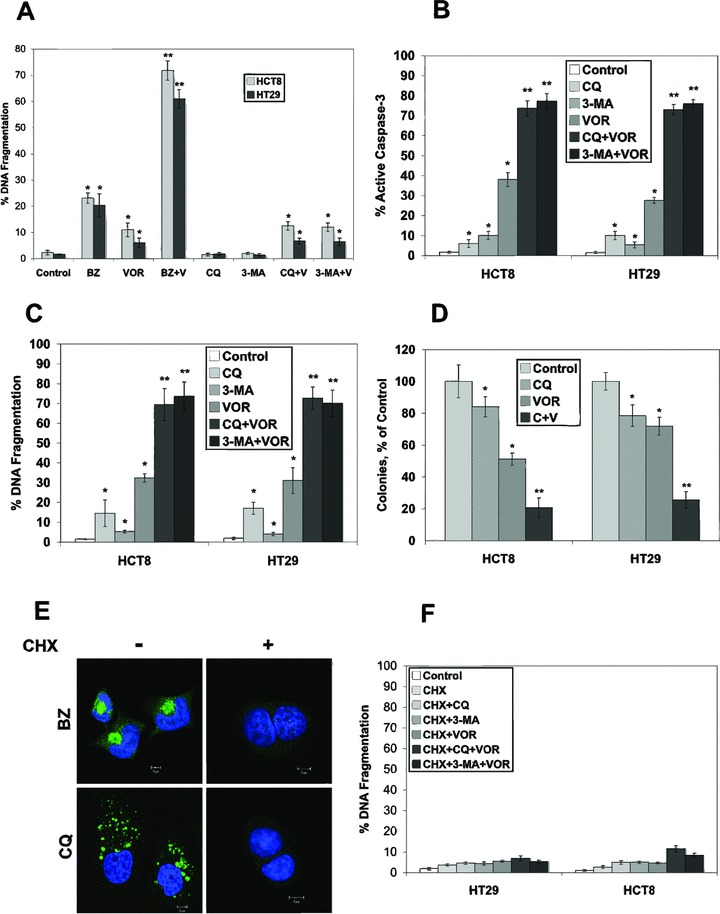
VOR sensitizes colon cancer cells to apoptosis induced by BZ, CQ and 3-MA. (A) Cells were treated with 100 nM BZ, 2.5 μM VOR, 50 μM CQ, 5 mM 3-MA or combinations for 24 hrs. Apoptosis was determined by PI staining and flow cytometry. Mean ± S.D., n= 3. *Indicates significant difference from the control group and **indicates a significant difference from BZ and VOR-single agent groups. P < 0.05. (B) and (C) VOR enhances apoptosis induced by CQ and 3-MA after 48 hrs of treatment. Cells were treated with 2.5 μM VOR, 50 μM CQ, 5 mM 3-MA or combinations for 48 hrs. Apoptosis was determined by active caspase-3 assay (B) or PI staining (C) followed by flow cytometry. Mean ± S.D., n= 3. *Indicates significant difference from the control group. **Indicates a significant difference from BZ and VOR-single agent groups. P < 0.05. (D) CQ combined with VOR strongly reduces the clonogenic survival of HCT8 and HT29 colon cancer cells. Cells were treated with 50 μM CQ, 2.5 μM VOR, or both agents for 24 hrs. Cells were then incubated in fresh media for 10 days. Colonies were stained and scored as described in ‘Materials and methods’. Mean ± S.D., n= 3. *Indicates a significant difference from control and **represents a significant difference compared to single agent treatment groups. P < 0.05. (E) CHX blocks BZ- and CQ-mediated ubiquitin-conjugated aggregate accumulation. HCT8 cells were treated with 100 nM BZ or 50 μM CQ in the presence or absence of 25 μM CHX for 24 and 48 hrs, respectively. Aggregate formation was detected by immunofluorescence using an anti-ubiquitin antibody. (F) CHX inhibits apoptosis induced by the CQ/VOR and 3-MA/VOR combinations. Cells were treated with 25 μM CHX, with 2.5 μM VOR, 50 μM CQ, 5 mM 3-MA or combinations for 48 hrs. Apoptosis was measured by PI staining and flow cytometry. Mean ± S.D., n= 3. Treatment of cells under identical conditions without CHX is shown in (C).
Enhanced aggregate accumulation and apoptosis stimulated by the CQ+VOR combination requires superoxide generation
To confirm that inhibition of autophagy enhanced SAHA-mediated apoptosis, we knocked down the expression of Atg7, an essential gene required for autophagy by RNA interference (RNAi) (Fig. 4A). Following exposure to Atg7 siRNA, HCT8 cells were treated with VOR for 48 hrs. Knockdown of Atg7 significantly augmented VOR-induced apoptosis (Fig. 4A), suggesting that this pathway promotes drug resistance. Similar to CQ-treated cells, knockdown of Atg7 also resulted in an accumulation of ubiquitinated proteins, which was enhanced by the addition of VOR (Fig. 4B). We and other investigators have previously shown that HDAC6 is essential for BZ-mediated aggresome formation and that disrupting this process sensitizes cancer cells to apoptosis [21, 25, 26]. To evaluate the role of HDAC6 in colon cancer cells, we knocked down its expression using siRNA (Fig. 4C). Silencing of HDAC6 strongly enhanced BZ-mediated apoptosis (Fig. 4C) and disrupted aggresome formation (Fig. 4D). However, knockdown of HDAC6 only marginally increased apoptosis stimulated by CQ or 3-MA (Fig. 4C) and did not alter ubiquitinated protein localization following CQ exposure (Fig. 4D), suggesting that VOR enhances CQ and 3-MA-induced apoptosis via another mechanism.
Fig 4.
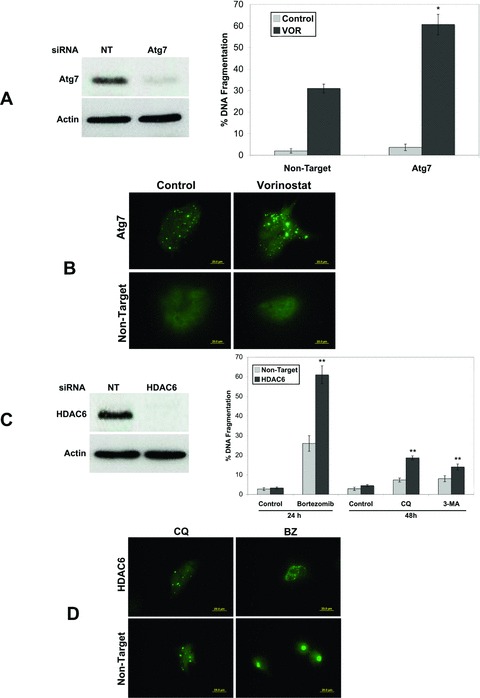
Role of Atg7 and HDAC6 in CQ+VOR-mediated apoptosis. (A) Atg7 knockdown enhances VOR-induced apoptosis. RNAi was performed as described in ‘Materials and methods’ and immunoblotting confirmed knockdown of Atg7. HCT8 cells transfected with non-target or Atg7 siRNA were treated with VOR for 48 hrs. Apoptosis was determined by PI staining and flow cytometry. Mean ± S.D., n= 3. *Indicates significant difference from non-target siRNA transfected cells treated with BZ. P < 0.05. (B) Knockdown of Atg7 causes ubiquitinated aggregate accumulation, which is enhanced by VOR. Following knockdown of Atg7 by RNAi, cells were treated with VOR for 48 hrs. Aggregate formation was determined by staining with an anti-ubiquitin antibody and images were captured by fluorescent microscopy. (C) Knockdown of HDAC6 enhances BZ-induced apoptosis, but only marginally increases CQ or 3-MA-mediated apoptosis. HCT8 cells were transfected with non-target or HDAC6 siRNA. Immunoblotting confirmed a decrease in HDAC6 levels. Cells were then treated with 100 nM BZ for 24 hrs or 50 μM CQ or 5 mM 3-MA for 48 hrs in the presence or absence of HDAC6 siRNA. Apoptosis was determined by PI staining and flow cytometry. Mean ± S.D., n= 3. **Indicates significant difference from non-target siRNA transfected cells treated with the same agent. P < 0.05. (D) Knockdown of HDAC6 disrupts BZ-mediated aggresome formation, but does not alter CQ-induced aggregate localization. Following knockdown of HDAC6 by RNAi, cells were treated with CQ or BZ for 48 and 24 hrs, respectively. Aggregate accumulation was detected by staining with an anti-ubiquitin antibody as described in the ‘Materials and methods’.
Several studies have reported that VOR induces oxidative stress and that this effect contributes to its anticancer activity [27, 28]. To investigate the possibility that VOR-mediated oxidative stress enhances CQ-mediated apoptosis, we measured superoxide levels following treatment with BZ+VOR and CQ+VOR. Superoxide was measured at time-points before significant levels of apoptosis occurred, 12 hrs for BZ+VOR and 24 hrs for CQ+VOR. Hydroethidine staining demonstrated that the CQ+VOR combination stimulated a large increase in superoxide (Fig. 5A and B) compared to BZ+VOR. To determine the significance of the CQ+VOR-mediated oxidative stress, cells were co-treated with antioxidant NAC. Treatment with NAC decreased the number of ubiquitinated protein aggregates caused by the CQ+VOR combination (Fig. 5C) and significantly reduced CQ+VOR-induced apoptosis (Fig. 5D).
Fig 5.
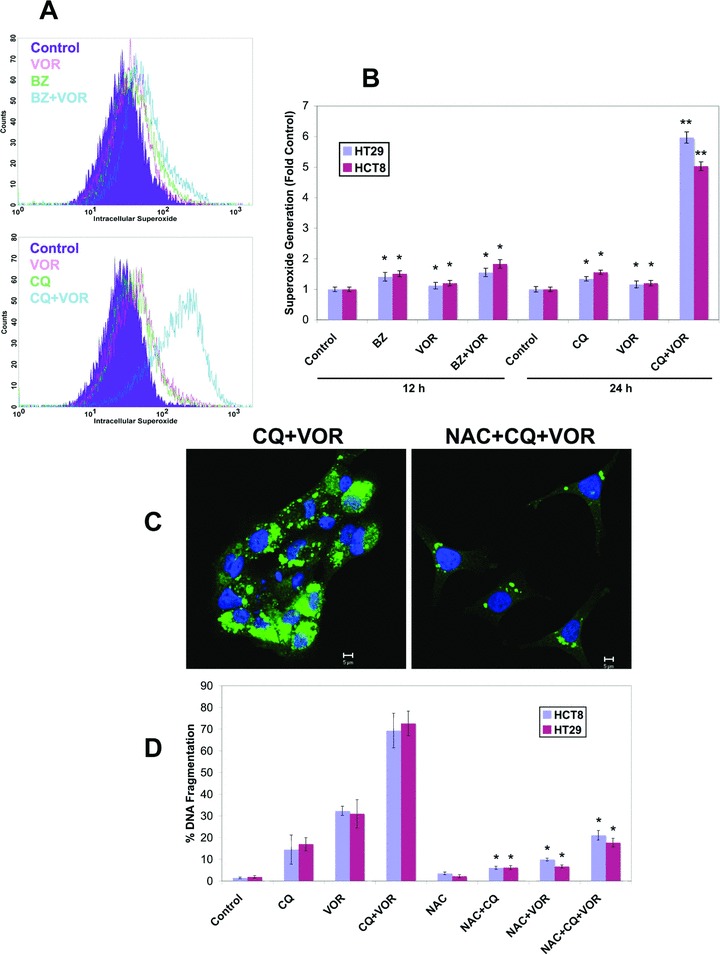
Treatment with the CQ/VOR combination stimulates a strong increase in superoxide production that contributes to ubiquitinated aggregate accumulation. (A) Flow cytometric analysis of superoxide content in HT29 cells treated with 100 nM BZ, 2.5 μM VOR, or both for 12 h, or 50 μM CQ, 2.5 μM VOR, or the combination for 48 hrs. Cells were labelled with hydroethidine (100 ng/ml) for 1 h and analyzed by flow cytometric analysis. (B) The CQ/VOR combination induces a strong increase in superoxide production. HCT8 and HT29 cells were treated with the 100 nM BZ, 2.5 μM VOR, 50 μM CQ or combinations for the indicated times. Cells were stained with hydroethidine and analyzed by flow cytometry. Mean ± S.D., n= 3. *Indicates significant difference from the control group. **Indicates a significant difference from single agent treatment groups. P < 0.05. (C) The antioxidant NAC reduces the levels of ubiquitinated aggregates. Cells were treated with 50 μM CQ and 2.5 μM VOR for 48 hrs with or without 10 mM NAC. Ubiquitinated aggregates were visualized with an anti-ubiquitin antibody (green). The nucleus was counterstained with ToPro-3 (blue). Images were generated by confocal microscopy. (D) Treatment with NAC strongly reduces apoptosis stimulated by the CQ/VOR combination. HT29 and HCT8 cells were treated with 10 mM NAC, 50 μM CQ, 2.5 μM VOR, or combinations for 48 hrs. Apoptosis was measured PI staining followed by flow cytometry. Mean ± S.D., n= 3. *Indicates a significant difference from cells treated without NAC. P < 0.05.
Effects of CQ and SAHA on HCT8 colon cancer tumours
In a final series of experiments we investigated the effects of CQ, VOR and the combination on tumour growth and cell death in the HCT8 colon cancer xenograft model. Established tumours were treated daily with 60 mg/kg CQ, 100 mg/kg VOR or both agents for 21 days. Combination therapy significantly reduced colon tumour weight as compared to therapy with either agent alone (Fig. 6A). Importantly, no signs of toxicity or weight loss were observed in mice in any treatment group (data not shown). Therapy with either CQ or VOR alone stimulated moderate increases in tumour cell apoptosis as measured by TUNEL staining, whereas combination therapy resulted in significantly higher levels of apoptosis (Fig. 6B and C). These data demonstrate that therapy of CQ in combination with VOR is well tolerated and induces tumour cell death and warrants further evaluation for the treatment of colon cancer.
Fig 6.
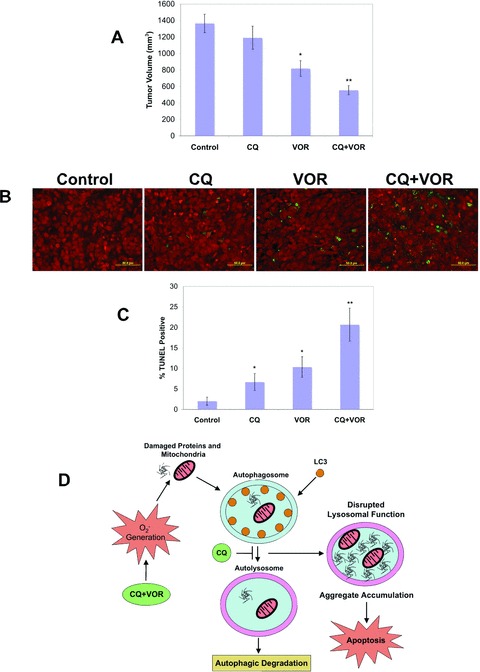
The CQ/VOR combination significantly reduces tumour burden and increases apoptosis in the HCT8 colon cancer xenograft model. (A) Effects of CQ and VOR on the growth of HCT8 xenografts. Following tumour formation, animals were pair-matched by tumour size and placed into groups of eight mice. Animals were treated by intraperitoneal injection of 60 mg/kg CQ, oral gavage of 100 mg/kg VOR, or both agents for 21 days. Tumour burden was assessed by measuring tumour volume on Day 21. Mean ± S.E.M. *Indicates a significant difference as compared to Control. **Indicates a significant difference compared to single agent groups. P < 0.05. (B, C) Combination therapy with CQ plus VOR produces a significant increase in tumour cell apoptosis in vivo. Apoptosis was measured by TUNEL staining of tissue sections (green). Tissues were counterstained with PI (red) to detect cell nuclei. TUNEL+ cells were quantified by manual counting of five independent fields. Mean ± S.D., n= 5. *Indicates significant difference as compared to levels observed in the Control. **Indicates a significant difference compared to either single agent group. P < 0.05. (D) Schematic representation of the mechanisms underlying apoptosis induced by the CQ/VOR combination. Treatment with the CQ/VOR combination induces an increase in superoxide generation that contributes to damaged proteins and organelles. Because CQ disrupts lysosomal function, it also blocks autophagic degradation. Inhibition of autophagy by CQ leads to an accumulation of ubiquitinated proteins, which is strongly enhanced by the addition of VOR due to an increase in oxidative stress. Aggregate accumulation induced by the CQ/VOR combination leads to apoptosis.
Discussion
The proteasomal and lysosomal systems are the two major protein degradation pathways in eukaryotic cells. Autophagy is the main mechanism by which cytoplasmic molecules are delivered to the lysosome [29] and is required for the turnover of cellular components activated under nutrient-deficient conditions [30]. Furthermore, autophagy has been implicated in cancer as a survival mechanism in response to chemotherapy and hypoxia [23, 31–34]. Inhibition of proteasomal degradation with BZ has demonstrated promising anticancer activity in numerous preclinical models and is approved for the treatment of multiple myeloma and mantle cell lymphoma [35]. Here we investigated the anticancer potential of inhibiting autophagic degradation for the treatment of colon cancer.
The lysosomotropic agent CQ disrupts lysosomal function and inhibits autophagy by interfering with the acid-dependent degradation of proteins within the autophagosome [7, 31]. Similar to BZ, treatment with CQ also leads to an accumulation of ubiquitin-conjugated proteins. However, CQ-mediated protein build-up occurs at a much slower rate than BZ, which is consistent with the idea that autophagic protein turnover degrades long-lived proteins, whereas the proteasome accounts for rapid short-lived protein degradation [36]. CQ treatment did not inhibit proteasomal activity (data not shown). In accordance with this result, CQ-mediated ubiquitin-conjugated protein accumulation differs from BZ in that these proteins remain within the lysosomal compartment and do not form aggresomes. Our data is in agreement with observations made in Atg7- or Atg5-deficient cells, which also formed ubiquitin-positive aggregates [36–38]. Proteasomes require unfolded proteins for degradation [39]. Because protein aggregates are difficult to unfold, autophagic degradation is the primary pathway for cells to remove these proteins. Collectively, our data suggest that autophagy is important for the turnover of some ubiquitinated proteins and is also likely to be the major route of elimination of ubiquitinated protein aggregates.
We and other investigators have previously shown that inhibition of HDAC6 pharmacologically with VOR or genetically with siRNA disrupts BZ-mediated aggresome formation and induces synergistic levels of apoptosis [3, 21, 25, 40]. Because CQ treatment also induced ubiquitinated protein accumulation, we compared the effect that VOR would have on BZ- and CQ-mediated ubiquitin-conjugated protein accumulation and apoptosis. Consistent with our data in other cancer types [3, 21], BZ stimulated aggresome formation in colon cancer cells, which was disrupted by the addition of VOR or targeted knockdown of HDAC6. Surprisingly, the addition of VOR led to a more pronounced aggregation of ubiquitinated proteins when given in combination with CQ. Our data suggests that protein aggregation plays an important role in CQ/VOR-mediated cell death, as high levels of apoptosis were detected following 48-hr, but not 24-hr treatment. Furthermore, inhibition of protein synthesis completely abrogated ubiquitinated aggregate formation and CQ/VOR-induced apoptosis.
To elucidate the mechanism(s) of CQ/VOR-mediated apoptosis, we knocked down the expression levels of Atg7, an essential gene required for autophagy [36], which dramatically enhanced VOR-induced apoptosis. Therefore, the ability of CQ to inhibit autophagy appears to sensitize colon cancer cells to VOR-mediated apoptosis. Because HDAC6 is essential for aggresome formation and plays a role in ubiquitinated protein accumulation [26], we investigated whether VOR’s inhibition of HDAC6 activity would augment apoptosis following treatment with autophagic inhibitors. Although HDAC6 knockdown strongly enhanced apoptosis stimulated by BZ, it only led to a minor increase in CQ or 3-MA-mediated apoptosis suggesting that VOR enhances cell death induced by autophagic inhibitors via a different mechanism. This result is consistent with the increased aggregation rather than aggresome disruption observed following treatment with the CQ/VOR combination.
Several investigators have demonstrated that VOR stimulates oxidative stress and that this significantly contributes to its mechanism of action [27, 28, 41, 42]. Our data show that CQ and VOR both induce modest levels of superoxide generation, but both agents together stimulate an extremely high level of this reactive oxygen species. Interestingly, the superoxide generation promoted by the CQ/VOR combination was detected as early as 24 hrs after treatment, which is before significant levels of ubiquitin-conjugated protein accumulation and apoptosis were observed. This suggests that superoxide production may be an essential initiating event during CQ/VOR-mediated apoptosis. We have previously shown in chronic myelogenous leukaemia cells that the CQ/VOR combination stimulated a large increase in cathepsin D levels that led to the degradation of thioredoxin (Trx) [23]. Because Trx is an important antioxidant, its degradation sensitizes cells to oxidative stress [43]. Consistent with oxidative stress playing an important role in CQ/VOR-induced apoptosis, treatment with the antioxidant NAC decreased the presence of ubiquitinated protein aggregates and significantly reduced apoptosis stimulated by the CQ/VOR combination. Based on these results, superoxide produced by VOR likely increases protein damage and enhances CQ-mediated aggregate formation. Studies in autophagic-deficient Atg7−/– cells determined that these cells accumulated defective mitochondria [36]. It is possible that these abnormal mitochondria are characterized by increased production of superoxide, which further contributes to oxidative stress. We are currently investigating this possibility. Taken together, our data show that superoxide generated by the CQ/VOR combination contributes to the formation of aggregates that cannot be removed due to a disruption in autophagic degradation (Fig. 6D).
To further address the therapeutic potential of the combination of CQ and VOR, we examined the activity of this combination in the HCT8 colon cancer xenograft model. The CQ/VOR combination significantly reduced tumour burden and enhanced tumour cell apoptosis compared to either single agent treatment. In addition, the combination was very well tolerated as no significant weight loss occurred in any animal throughout the duration of the study. A recent study demonstrated that inhibition of autophagy with CQ enhanced the anticancer activity of cyclophosphamide in a Myc-driven lymphoma model [31]. Because this alkylating agent is also known to induce oxidative stress [44], ubiquitinated aggregate accumulation may also contribute to the anticancer activity of this combination. Many anticancer drugs induce autophagy and disrupting this pathway may augment the efficacy of conventional chemotherapeutic agents and become an important part of cancer treatment. Although this is an emerging strategy, a small clinical study conducted in glioblastoma found that the median survival was increased twofold for patients receiving CQ with conventional therapy compared to controls, which lends further support to investigating autophagy inhibition as an approach to sensitize tumours to standard therapy [45].
Our findings demonstrate that blocking proteasomal or autophagic degradation with BZ and CQ, respectively, is detrimental to cancer cell survival. In addition, inhibition of HDAC activity with VOR greatly enhances apoptosis induced by either agent in colon cancer cells. Although VOR is able to enhance BZ-mediated apoptosis via multiple mechanisms, including enhanced oxidative stress and inhibition of angiogenesis, we have demonstrated that aggresome disruption through abrogation of HDAC6 activity is an important contributor to the efficacy of this combination. HDAC6 appears to play a lesser role during CQ-mediated apoptosis, which is likely due to ubiquitinated protein accumulation localized to the lysosomes instead of in a single perinuclear aggresome. However, VOR dramatically enhances CQ-mediated apoptosis by inducing high levels of superoxide that further increases ubiquitinated protein accumulation as a result of inhibition of autophagic degradation. Taken together, this study provides the framework for clinical trials of CQ and VOR in patients with colon cancer and potentially other malignancies. We are currently planning a clinical trial to investigate the promising activity of this drug combination.
Acknowledgments
This work was supported by the NIH Cancer Center Core Grant CA054174 and funds provided by the AT&T Endowed Chair at The Institute for Drug Development, Cancer Therapy and Research Center at The University of Texas Health Science Center at San Antonio, TX, USA.
References
- 1.Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004;23:3217–21. doi: 10.1038/sj.onc.1207548. [DOI] [PubMed] [Google Scholar]
- 2.Carew JS, Nawrocki ST, Cleveland JL. Modulating autophagy for therapeutic benefit. Autophagy. 2007;3:464–7. doi: 10.4161/auto.4311. [DOI] [PubMed] [Google Scholar]
- 3.Nawrocki ST, Carew JS, Maclean KH, et al. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008;112:2917–26. doi: 10.1182/blood-2007-12-130823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 8.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 10.Bursch W, Ellinger A, Kienzl H, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 11.Ertmer A, Huber V, Gilch S, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21:936–42. doi: 10.1038/sj.leu.2404606. [DOI] [PubMed] [Google Scholar]
- 12.Kanzawa T, Kondo Y, Ito H, et al. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–8. [PubMed] [Google Scholar]
- 13.Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 14.Shao Y, Gao Z, Marks PA, et al. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi H, Kondo Y, Fujiwara K, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 16.Khraishi MM, Singh G. The role of anti-malarials in rheumatoid arthritis – the American experience. Lupus. 1996;5:S41–4. [PubMed] [Google Scholar]
- 17.Myint HY, Tipmanee P, Nosten F, et al. A systematic overview of published antimalarial drug trials. Trans R Soc Trop Med Hyg. 2004;98:73–81. doi: 10.1016/s0035-9203(03)00014-2. [DOI] [PubMed] [Google Scholar]
- 18.Romanelli F, Smith KM, Hoven AD. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr Pharm Des. 2004;10:2643–8. doi: 10.2174/1381612043383791. [DOI] [PubMed] [Google Scholar]
- 19.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90:665–9. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition, and degradation capacity–formation of crinosomes. Exp Mol Pathol. 1987;47:346–62. doi: 10.1016/0014-4800(87)90018-9. [DOI] [PubMed] [Google Scholar]
- 21.Nawrocki ST, Carew JS, Pino MS, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–81. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 22.Nawrocki ST, Carew JS, Dunner K, Jr, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–9. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 23.Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nawrocki ST, Carew JS, Douglas L, et al. Histone deacetylase inhibitors enhance lexatumumab-induced apoptosis via a p21Cip1-dependent decrease in survivin levels. Cancer Res. 2007;67:6987–94. doi: 10.1158/0008-5472.CAN-07-0812. [DOI] [PubMed] [Google Scholar]
- 25.Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 27.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 28.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63:3637–45. [PubMed] [Google Scholar]
- 29.Fehrenbacher N, Jaattela M. Lysosomes as targets for cancer therapy. Cancer Res. 2005;65:2993–5. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 30.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 31.Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qadir MA, Kwok B, Dragowska WH, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112:389–403. doi: 10.1007/s10549-007-9873-4. [DOI] [PubMed] [Google Scholar]
- 34.Tiwari M, Bajpai VK, Sahasrabuddhe AA, et al. Inhibition of N-(4-hydroxyphenyl) retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis. 2008;29:600–9. doi: 10.1093/carcin/bgm264. [DOI] [PubMed] [Google Scholar]
- 35.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 36.Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 39.Baumeister W, Walz J, Zuhl F, et al. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 40.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 41.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 42.Fiskus W, Rao R, Fernandez P, et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood. 2008;112:2896–905. doi: 10.1182/blood-2007-10-116319. [DOI] [PubMed] [Google Scholar]
- 43.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–7. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Selvakumar E, Prahalathan C, Mythili Y, et al. Mitigation of oxidative stress in cyclophosphamide-challenged hepatic tissue by DL-alpha-lipoic acid. Mol Cell Biochem. 2005;272:179–85. doi: 10.1007/s11010-005-7322-4. [DOI] [PubMed] [Google Scholar]
- 45.Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144:337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]