

Abstract
The peritoneal cavity is recognized as an important site for autoreactive B cells prior to their transit to other immune tissues; however, little is known of the genes that may regulate this process. Mice lacking the receptor tyrosine kinase Mertk display a lupus-like autoimmune phenotype with splenomegaly and high autoantibodies titers. Here, we investigate whether Mertk regulates the composition of peritoneal cells that favor an autoimmune phenotype. We found an increase in the number of macrophages, DC, plasmacytoid DC, T cells and B cells in the peritoneal cavity of mertk−/− mice when compared to wild-type mice. This disparity in cell numbers was not due to changes in cell proliferation or cell death. In adoptive transfer experiments, we showed an increase in migration of labeled donor cells into the mertk−/− peritoneal cavity. In addition, bone marrow chimeric mice showed hematopoietic-derived factors were also critical for T cell migration. Consistent with this migration and the increase in the number of cells, we identified elevated expression of CXCL9, its receptor CXCR3, and IL-7 receptor on peritoneal cells from mertk−/− mice. To corroborate the migratory function of CXCR3 on cells, the depletion of CXCR3 donor cells significantly reduced the number of adoptively transferred cells that entered into the peritoneum of mertk−/− mice. This control of peritoneal cells numbers correlated with autoantibody production and was exclusively attributed to Mertk since mice lacking other family members, Axl or Tyro 3, did not display dysregulation in peritoneal cell numbers or the autoimmune phenotype.
Introduction
The peritoneal cavity is a site of leukocyte colonization in which the development and regulation of immune cells is poorly understood. The cells occupying the peritoneal cavity are similar to a lymph node in that some cells reside in the cavity while others are transient, passing through en route to other targeted tissues. Immune cells found in the peritoneal cavity include, macrophages, T cells, B cells, NK cells, mast cells and dendritic cells (DC)(1, 2). This heterogeneous population of cells is involved in both innate and adaptive immunity. While macrophages may be the predominant cell type of the peritoneal cavity, the B1-B cell is the largest lymphocyte population in this location. Peritoneal B1-B cells are the primary secretor of serum IgM, which targets pathogen-associated molecular patterns (PAMPs)(3, 4). B1-B cells can also produce low affinity autoantibody and their development and regulation has been the focus of autoimmune disease models(3, 5). Increases in B1-B cells are found in human autoimmune diseases as well as the NZB/NZW mouse model of lupus(3, 6).
More recently, an additional murine model of autoantibody production and lupus is found in the mertk−/− mice. Mice lacking Mertk as well as mice lacking all three receptor family members, Tyro3, Axl, and Mertk, (TAM−/−) display a lupus-like autoimmune phenotype characterized by splenomegaly and dysregulated lymphocyte activation as well as high autoantibody titers to dsDNA, ssDNA, nucleosomes, chromatin and the ribonucleoprotein, Smith antigen (Sm) (7–10). Also, Sm-specific B cells in 2–12H/mertk−/− transgenic mice display increased numbers of peritoneal B1-B cells as compared to 2–12H transgenic mice with wild-type Mertk(11). Furthermore, Sm-specific B cells injected into the peritoneal cavity of mertk−/− mice become activated, and selectively exited the peritoneal cavity. These Sm-specific B1- B cells appeared to have migrated to the lamina propria and mesenteric lymph node to become antibody-secreting cells(11). Lastly, ablation of peritoneal B-1 cells for extended periods abrogated autoantibodies in autoimmune-prone mice (12). These studies suggest the peritoneal cavity is an important transit site as autoreactive B cells migrate through prior to entering terminal immune sites.
Mertk is a receptor tyrosine kinase found on macrophages, DCs, and natural killer cells as well as other non-hematopoietic cells, however T and B lymphocytes lack expression under normal conditions (13–16). Mertk is a member of the Tyro3/Axl/Mertk (TAM) family of receptors. This family of receptors is characterized by an extracellular domain containing two Ig like and two fibronectin III like motifs and by an intracellular kinase domain with a characteristic KWIAIES motif(15, 17). Gas6 is a ligand for all three family members with a relative binding affinity of Axl>Tyro3≫Mertk(18). Gas6 is produced by macrophages and can also be found pre-bound to apoptotic thymocytes(19, 20). In addition to Gas6, Protein S, has been identified as a ligand for Tyro3(19, 21), and the oxidization and oligomerization of protein S on apoptotic cells permits binding with Mertk (22). These ligands bind phosphatidylserine which is exposed on the outer-membrane of apoptotic cells and provide a molecular bridge to the TAM family members. This subsequently triggers the phagocytosis of apoptotic cells by macrophages and dendritic cells, presumably through integrin-mediated, Rac1-activated signaling(10, 19, 23).
In addition to a defect in phagocytosis of apoptotic cells, macrophages and DCs lacking Mertk are hypersensitive to LPS stimulation, resulting in hyper-activation of NF-κB and subsequent over-production of TNF-α(9, 24, 25). However, Mertk and family members Axl and Tyro3 (TAM family) appear to play no role in bacterial phagocytosis or bacterial killing (26). Recently, Rothlin et. al. presented a mechanism by which the TAM family members cooperate with the type I interferon receptor to induce expression of the suppressor of cytokine signaling 1(SOCS1), which regulates TLR signaling and cytokine production(27). The TNF-α family member BAFF is also produced in greater amounts by DCs lacking Mertk or all three TAM receptors(8, 27). This is of particular interest to the autoimmune phenotype of the mertk−/− mice as excess BAFF can lead to an increased number of B cells and a propensity for autoimmune disease(28–30). However, DC from mertk−/− mice expressing high levels of BAFF did not increase survival of B cells, indicating other factors may be contributing(8).
There has been much speculation as to the reason for the autoimmune phenotype of mice lacking the TAM receptors. It is presumed that the lack of ability to phagocytize apoptotic cells results in higher availability of self-antigen and greater priming of autoreactive B cells. However, as in humans, it appears that the autoimmunity in these animals is more likely a multi-factorial development of molecular and cellular events that lead to a break in ignorance or tolerance.
In this report, we hypothesized that the cell populations in the peritoneal cavity maybe different in the autoimmune prone mertk−/− mice. We characterized peritoneal exudate cells (PEC) in wild-type and mertk−/− mice, and found an increase in all immune cell types in mertk−/− mice. We also characterize the activation status of the B cell population as well as the subpopulations of the T cells. We determined that the increase in cells was not due to aberrant division or defects in apoptosis, but rather an increased propensity for cells to migrate to the peritoneal cavity. We also determined that this migration was independent of cellular development in the mertk−/− mouse and show that neither hematopoietic nor non-hematopoietic factors exclusively contribute to this recruitment/migration. However, T cells are recruited by hematopoietic factors. We identify one chemokine and two receptors that are elevated in the mertk−/− peritoneal cells and they may contribute to increases in the migration of cells. Finally, we show that mice lacking Mertk family members, axl−/−, tyro3−/− and axl−/−/tyro3−/− showed no differences in resident peritoneal cell numbers compared to wild-type mice. Correspondingly, splenomegaly and autoantibody production were found only in mertk−/− mice and not other mutant mice.
Materials and Methods
Animals
All animals were housed in Specific Pathogen-Free facilities according to the guidelines of the UNC-Chapel Hill Institutional Animal Use and Care Committee. mertk−/− (also known as merkd or mertkkd mice) were generated as previously described(24). axl−/− and tyro3−/− were generously provided by Dr. Stephen P. Goff (Columbia U., New York) and Dr. Greg Lemke (Salk Institute for Biological Studies, San Diego) respectively. axl−/−/tyro3−/− mice were bred by crossing axl−/− and tyro3−/− mice. All animals are backcrossed at least 6 generations to C57BL/6J which serves as wild-type in all experiments. Three month old male mice were used unless otherwise indicated. Actin-green fluorescent protein (GFP) C57BL/6 transgenic mice were purchased from Jackson Labs.
Resident Peritoneal Exudate Cells (PEC)
Mice at indicated ages were euthanized with isofluorane. Mice were injected i.p. with 3mL of Versene, after 60 seconds of peritoneal massage cells were harvested. Cells were washed three times with 1X PBS prior to experimentation. Total number of cells was counted using a hemocytometer and Trypan Blue.
Flow Cytometry
Before labeling with antibody, cells were incubated with Fc Block (anti-CD16/CD32) from BD biosciences. Cells were then stained for surface expression using the following antibodies, anti-CD19-PE-Cy5, anti-CD5-PE, anti-B220-PE, anti-CD11b-PE-Cy5, anti-PDCA-1-FITC, anti-CD44-APC, anti-CD62L-PE, and anti-IL7R-PE antibodies were purchased from ebioscience. Anti-CD8-PE, anti-CD11b-FITC, and anti-CD4-FITC antibodies were bought from Caltag. Anti-CD11c-PE or PE-Cy7, anti-CD3-FITC, and anti-I-Ab-FITC antibodies were from BD/Pharmingen. Anti-F480-PE-Cy5 antibody was obtained from Serotec. Anti-CXCR3 antibody was obtained from Zymed. Secondary antibody to detect primary anti-CXCR3 antibody was anti-Rabbit-Alexa 405 and was obtained from Invitrogen. All washes and staining were done with 2% FCS in PBS and samples were analyzed using a Dako-Cyan flow cytometry and Summit 4.3 software. At least 15,000 cells were analyzed from each sample. Total number was calculated using percent cells positive for each stain.
Migration to Peritoneal Cavity
Resident PECs were obtained as described previously. Cells were washed with PBS and stained with Cell-Tracker green (Molecular Probes) according to manufacturers instructions. Three million cells were injected in 0.5mL 1X PBS via tail vein of naive recipient mice. 24 hours after injection, cells were harvested from the peritoneal cavity using 3 mLs of Versene to lavage out cells. PECs were analyzed by flow cytometry to identify percent of Cell Tracker green-positive cells in the collected samples. The number of cells that migrated was then calculated by multiplying the percent Cell Tracker green-positive cells by total number of cells harvested.
Where indicated, cells were stained for CXCR3 as above or left unstained. CXCR3-positive cells were removed by cell-sorting. In addition, as a control for damage during cell manipulation or cell-sorting unstained cells termed ‘all cells’ were subjected to the identical cell-sorting procedure. Total (all) cells or CXCR3-negative cells were then stained with Cell-Tracker green and adoptively transferred as described above.
Bone Marrow Transplant Chimeras
Recipient male mice at 4 weeks of age were lethally irradiated with 800 rads using either 137Cs gamma irradiator or X-ray irradiator. Twenty-four hours post irradiation, bone marrow cells were obtained from femurs and tibia of donor mice. Bone marrow cells were treated with red blood cell lysis buffer to remove red cells and 8 × 106 white cells were injected i.v. into irradiated recipient mice. Each irradiation experiment contained at least one actin-driven eGFP control donor mouse to assess hematopoietic reconstitution of chimeric mice. Percent reconstitution of chimeric mice at three months of age was assessed by quantifying the % GFP+ cells from the bone marrow and the peritoneum using flow cytometry. Reconstitution of chimeric mice with GFP+ cells was approximately 90% as expected.
BrdU Injection and Staining
Mice at the indicated ages were injected 24 and 48 hrs prior to harvest with 0.75mL of 10mg/mL BrdU (Sigma) in 0.9% NaCl to quantify proliferating cells. Cells were harvested and identified by staining with antibodies against cell surface markers. Cells were then permeabilized using Cytofix/Cytoperm buffer system (BD) and DNase treated to expose the BrdU. BrdU incorporation was visualized by using anti-BrdU-FITC antibody from BD. Cells were analyzed by flow cytometry using a Dako-Cyan flow cytometry and Summit 4.3 software.
Cell Death analysis
Resident PECs were harvested as described. Apoptosis was detected using TACS AnnexinV-FITC/propidium iodide (PI) Apoptosis Detection Kit (Trevigen) according to manufacturer’s instructions. For VAD-FMK staining, VAD-FMK-FITC (Promega) was diluted 1:1000 in PBS and 0.5mL was injected I.P. 1 hour prior to harvest of resident PEC. Both assays were analyzed by flow cytometry using a Dako-Cyan and Summit 4.3 software.
Real-Time PCR
Resident PECs from at least 4 mice of each genotype were pooled and RNA was purified according to manufacturer’s instructions using the RNeasy Mini Plus Kit (Qiagen). RNA was quantified and equal amounts were converted to cDNA using Superscript II reverse transcriptase (Invitrogen) using random hexamer primers. Equal amounts of cDNA were used for the Real-Time PCR reaction in 2x Sybr Green buffer (Applied Biosystems). Reactions were conducted in 96 well plates and were performed on the ABI 7500 Realtime PCR machine in the UNC Neuroscience Center Expression Profiling and SNP Genotyping Core Facilty. Primers were designed using FASTA sequences and primer express software3.0 (Applied Biosystems). GAPDH was used as the internal control. Primers set used are as follows: GAPDH, Forward TGTGTCCGTCGTGGATCTGA, Reverse CCTGCTTCACCACCTTCTTGA IL-5, Forward AGCACAGTGGTGAAAGAGACCTT, Reverse CATCGTCTCATTGCTTGTCAACA. IL-6, Forward CCACGGCCTTCCCTACTTC, Reverse TTGGGAGTGGTATCCTCTGTGA. IL-7, Forward GCCATGCCTTCTTGCTTCTC, Reverse AGCTATCTGTACCACTGAGTAGAGCATT. IL-9, Forward TTGCCTCTGTTTTGCTCTTCAG, Reverse CCCCATGTGGTGCTGCAT. IL-10, Forward GATGCCCCAGGCAGAGAA, Reverse CACCCAGGGAATTCAAATGC. BAFF, Forward CCCAAAACACTGCCCAACA, Reverse CTCATCTCCTTCTTCCAGCCTC. CXCL9, Forward AAGTCGTCGTCGTTCAAGGAA, Reverse AAGATTCAGGGTGCTTGTTGGT. CXCL13, Forward CCATCATGAGGTGGTGCAAAG, Reverse GGACTGTTTCCCCCAGGAA. CXCL14, Forward GGCCCAAGATCCGCTACA, Reverse CAGGCATTGTACCACTTGATGAAG. LFA, Forward CCTGAGGGTGGGCTGGAT, Reverse GCCAATTTCCTCCGGACAT. PECAM, Forward TGACCCAGCAACATTCACAGAT, Reverse CCGCTCTGCACTGGTATTCC. L-Selectin, Forward TGACGCCTGTCACAAACGA, Reverse GGCTGGCAAGAGGCTGTGT. CXCR5, Forward CCTTCGCTGGCGTAAAGTTC, Reverse CAGCCCAGCTTGGTCAGAA. CXCR3, Forward TCGGACTTTGCCTTTCTTCTG, Reverse CTCTCGTTTTCCCCATAATCGT. IL-7 Receptor, Forward GCCGCTGTACACAGTGCAA, Reverse AACTGTTTCTGGTGGGCTGACT.
Anti-nucleosome ELISA
ELISA plates (Dynatech) coated with 100μL of 40μg/mL histones (Immunovision) were incubated overnight at 4°C. Plates were then washed with boric-acid-buffered saline (BBS) and wells were coated with 100μL of 10μg/mL dsDNA (Sigma) in BBS with 0.5%BSA and 0.4% Tween for 3 hrs at room temperature. Wells were then washed with BBS. Samples and standards were loaded and incubated overnight at 4°C. Samples were detected with anti-mouse Ig-alkaline phosphatase and developed with p-nitrophenyl phosphate (Sigma). Wells were read at 405 nM.
Statistical analysis
Statistical analysis was performed using Graph Pad Prism software. Statistical tests used were one-way or two-way ANOVA with Bonferroni’s post-tests, or Students t-test. The appropriate test was applied to each data set, and the particular test used is indicated in the figure legends.
Results
In mertk−/− mice, there is an age-related increase in the production of auto-antibodies. In addition, activated B1-B cells are known to migrate more quickly out of the mertk−/− peritoneal cavity to become antibody secreting cells in the lamina propria and mesenteric lymph nodes(7–11). As cells from the peritoneal cavity are important for autoantibody production in other autoimmune mouse models(12), we first assessed whether the absence of Mertk affected the overall resident peritoneal cell population. We harvested cells from the peritoneal cavity, and found a significant increase in the total peritoneal cells(PEC) found in the mertk−/− mice as compared to wild-type mice (Figure 1A) at 1.5, 3, and 6 months of age. Flow cytometry identified the different cell types as F4/80+ macrophages, CD19+ B cells, and CD3+ T cells which were all found to have statistically significant increases when comparing the mertk−/− mice to wild-type mice across a time course of 1.5 to 6 months of age (Figure 1B, C, D).
Figure 1. Increase in peritoneal cellular populations in the mertk−/− mouse.

PEC were harvested from male mice at indicated ages and quantified by flow cytometry. A) Total number of PEC from wild-type (■) and mertk−/− (▲) mice were counted. B) The total number of F4/80+ macrophages, C) The total number of CD19+ B cells, and D) The total number of CD3+ T cells were also examined. *** p<0.0001 by 2-way ANOVA comparing wild-type to mertk−/− mice. n≥6 ** p<0.001 by 2-way ANOVA comparing wild-type to mertk−/− mice.
Further analyses of data collected from mice at 3 months of age are displayed in Table 1. Table 1 shows that in addition to differences in total number of Macrophages, B cells, and T cells shown in Figure 1, there are also statistically significant differences in percent of total cells that are Macrophages, B cells, and T cells between the genotypes. Analysis of the percentage of each peritoneal cell population showed macrophages from mertk−/− mice comprised less of the total population than wild-type mice; however, the percentage of B cells was slightly greater and the T cell percentage was nearly twice that of wild-type mice. Because the increase in immune cells occurs most abundantly at 3 months of age, our subsequent experiments focus on that time point.
Table 1.
Distribution of major peritoneal cell types by percentage and total number in wild-type and mertk−/− mice.
| F4/80+ Macrophage | CD19+ B cells | CD3+ T cells | ||||
|---|---|---|---|---|---|---|
| Genotype | Percent of total | Total cells × 105 | Percent of total | Total cells × 105 | Percent of total | Total cells × 105 |
| Wild-type | 33.4 ±5.5 | 8.2 ±1.6 | 50.5 ±3.9 | 13.3 ±3.9 | 16.2 ±2.6 | 4.3 ±1.6 |
| mertk−/− | 14.5 ±4.2§ | 18.3 ±10.8† | 55.3 ±5.7* | 59.3 ±16.1§ | 30.7 ±2.7§ | 31.4 ±6.3§ |
Peritoneal cells were harvested from WT mice and mertk−/− mice, immunostained, and flow cytometry performed to identify F4/80+ macrophages, CD19+ B cells, and CD3+ T cells. Percentages of total cells and the total number of cells are displayed.
p< 0.00005; n ≥ 12;
p< 0.01;
p< 0.05.
Our previous analysis of the spleen found an increase in the number of CD11c+ DC(8). We examined whether there is a similar increase in these cells in the peritoneal cavity of the mertk−/− mice. Indeed, we did find a statistically significant increase in the number of CD11c+ DC found in the peritoneal cavity of the mertk−/− mice (Figure 2A). Similarly, the number of plasmacytoid DCs, as identified by positive staining for CD11c, B220, and PDCA1, were also statistically increased in the peritoneum of mertk−/− mice as compared to wild-type mice (Figure 2B). The abundance of these cell types suggests the environment is conducive for the activation of the B1-B cells.
Figure 2. Increase in DC, pDC, and T cells by subset in mertk−/− peritoneal cavity.

Cells were stained and quantified by flow cytometry. A)PEC were stained for CD11c+ DC or B) CD11c+ B220+ PDCA1+ plasmacytoid DC. C) PEC were stained for CD4+ T cells and CD8+ T cells. D) PEC were stained for Effector T cells (CD44hi CD62Llo), Naive T cells (CD44lo CD62Lhi), and Central Memory T cells (CD44hi CD62Lhi) T cells (CD3+). E) PECs were stained for CD11b+ (macrophages) and MHC II. F) PECs were stained for F4/80+ macrophages and MHC II, CD80 or CD86. The sample number is n ≥4. Statistical significance was analyzed by Students t-test comparing samples from wild-type and mertk−/− mice. p-values are: *p<0.05; **p<0.005; and ***p<0.0001.
The increase in peritoneal lymphocyte populations shown in Figure 1 is particularly interesting because Mertk is not known to be expressed on unstimulated nonmalignant B and T cells (13–16, 25). In addition, the increase in these lymphoid cell types was not found in other lymphatic organs(8). Therefore, we characterized these lymphocytes further to determine whether one subpopulation could account for the increase in total number of cells. As shown in Figure 2C, both CD4+ and CD8+ T cells were present in higher numbers in the peritoneal cavity of mertk−/− mice compared wild-type mice. We then used CD44 and CD62L staining to determine the activation state of the T cells from the peritoneal cavity of both wild-type and mertk−/− mice. We found that there were statistically significant increases in effector (CD62L lo, CD44 hi), naive (CD62L hi, CD44 lo), and central memory T (CD62L hi, CD44 hi) cells. The largest increase was in the naive T cell population (Figure 2D). Thus, the absence of Mertk prevents proper regulation of the T cell population within the peritoneal cavity.
In addition, we also analyzed the macrophage populations. We found there were significantly more phagocytic macrophages as stained by CD11bhi, MHC II− expression found in the mertk−/− peritoneal cavity. However, similar numbers of non-phagocytic macrophages (CD11bhi MHC II+) that were presumably antigen-presenting cells were harvested from the mertk−/− and wild-type peritoneal cavities (Figure 2E). Over all there was not a large difference in activation status of these macrophages as similar levels of the co-stimulatory molecules CD80 and CD86 were found on both genotypes. However macrophage population from mice lacking Mertk showed a small but statistically significant increase in MHC II expression (Figure 2F).
We next analyzed the B cell populations which were also expanded in the mertk−/−mice (Figure 1C). Since the peritoneal cavity is known to contain a large number of B1 B cells, we used CD19, CD11b, and CD5 staining to analyze the B1a (Cd19+, CD11b+, CD5+) and B1b (CD19+, CD11b+, CD5−) subsets of the B1 (Cd19+, CD11b+) cells. As shown in Figure 3A, we found a statistically significant three-fold increase in the overall B1 population. Furthermore, in the absence of Mertk, there were over a four-fold increase in the number of B1b B cells and about a three-fold increase in the B1a B cell population. Thus, the increase in B1 B cells was due to an increase in both populations with a greater fold increase in B1b B cells. The number of CD19+ CD11b− B2 cells was negligible in comparison to the number of B1 cells; nonetheless, there was an increase of B2 cells in mertk−/− mice compared to wild-type mice (Data not shown). Furthermore, we wanted to determine whether the absence of Mertk affected the activation of the B cell population in the peritoneal cavity. Using the activation markers, MHC II, CD80, and CD86, we found that there were statistically significant decreases in the mean fluorescent intensity of MHC II and CD80 on CD19+ cells from mertk−/− mice (Figure 3B, C, D, E). Thus, while there was an increase in the number of B cells in the peritoneal cavity of mertk−/− mice, the B cells were less activated than those of wild-type mice.
Figure 3. Increase in B1 cell populations and lower B cell maturation status in mertk−/−peritoneal cavity.

A) PEC were stained for B1a (CD19+ CD11b+ CD5+) and B1b (CD19+ CD11b+ CD5−) cells. B) PEC were stained with CD19 to identify B cells and then double labeled with MHC II, CD80 and CD86. The Mean Fluorescence Intensity on CD19+ cells was graphed. The number of samples is n ≥4. Statistical significance by Students t-test was *p<0.05 and **p<0.005.
We hypothesized three reasons that could account for the increase in the number of cells in the peritoneal cavity of the mertk−/− mice. These included an increase in cell division, a decrease in cell death, and an increase in migration of cells into the peritoneal cavity of the mertk−/− mice. We first used BrdU incorporation to delineate cells undergoing proliferation within the peritoneal cavity of the mertk−/− mice. As shown in Figure 4A, an overall statistically significant decrease in the percent of dividing cells is evident when comparing cells collected from mertk−/− to wild-type mice at three different ages. However, the total number of cells dividing was not different when comparing the genotypes (Figure 4B). To assess whether excessive division was occurring elsewhere, we examined the bone marrow and found that there was no difference in cell division in the bone marrow (Figure 4C). While the spleen is a location for division of lymphocytes and leukocytes upon infection, we observed very few dividing cells in the spleen of both genotypes of mice under unstimulated conditions (data not shown). Thus, the large number of cells in the peritoneal cavity of mertk−/− mice was not due to local proliferation.
Figure 4. Increase in cells is not due to proliferation.

Proliferating PEC were identified by their incorporation of BrdU. PEC were collected after injection of BrdU I.P. and stained with anti-BrdU antibody. (A) Percent and (B) total number of BrdU+ cells across aged mice from 1.5 months to 6 months were examined. n≥6. C) Bone marrow cells from wild-type (WT) or mertk−/− (M−/−) mice were stained for BrdU incorporation and the data is expressed as the percent of BrdU+ cells, n ≥ 3 and representative of at least 2 separate experiments. Statistical significance is * p<0.05 by 2-way ANOVA. * p<0.05 by Bonferroni’s post-test.
We next examined the possibility that the large number of cells in the peritoneal cavity of mertk−/− mice was attributed to decreased in cell death We used several methods for measuring cell death at different stages, to provide a comprehensive profile. We first used VAD-FMK to analyze the number and percent of cells that were undergoing apoptosis. VAD-FMK-FITC binds to active caspase 3, a key component for the induction of apoptosis. We found the percent of cells that were VAD-FMK-FITC-positive were not statistically different (Figure 5A). However, while not reaching statistical significance, we found a trend toward an increase in the number of VAD-FMK+ cells in the peritoneal cavity of mertk−/− mice (Figure 5B). In addition to VAD-FMK-FITC staining, we also used Annexin V and PI staining to identify cells that were undergoing early apoptosis (Annexin V+) as well as those cells that were already in late apoptosis (PI+). We found no statistically significant differences in the percentage of cells that were undergoing apoptosis via Annexin V/PI staining (Figure 5C). However, there was a significant increase in the number of cells undergoing cell death at all stages of apoptosis in the peritoneal cavity of the mertk−/− mice (Figure 5D). We speculate this increase is likely due to an increase in total number of cells found in the peritoneal cavity of the mertk−/− mice. Taken together, these data suggest that a difference in cell death could not explain the increase in peritoneal cells found in the mertk−/− mice.
Figure 5. Increase in cells is not due to cell death.

3 month old mice were injected with VAD-FMK-FITC 1 hour prior to PEC harvest to detect caspase-positive cells. Cells were analyzed by flow cytometry. (A)The percent VAD-FMK+ cells, and (B) the total number of VAD-FMK+ cells were examined. PEC were harvested from 3 month old mice, stained with Annexin V-FITC and PI, and subsequently analyzed by flow cytometry. Data is expressed as (C) percent or (D) total number of Annexin-V+ or PI+ PEC. For each experiment, n ≥ 4. Statistical significance comparing samples from wild-type and mertk−/−mice is *** p<0.0001 by 2-way ANOVA. (**) p<0.001 by Bonnferoni’s post-test. (*) p<0.05 by Bonnferoni’s post-test.
Our third hypothesis for the large number of cells was an increase in migration of cells into the peritoneal cavity mertk−/− mice. To investigate this hypothesis, we labeled resident PEC from mertk−/− mice and adoptively transferred them into wild-type mice as well as in reciprocal combination to track migration into the peritoneal cavity. As controls, we injected wild-type PEC into wild-type mice and mertk−/− PEC into mertk−/−mice. We found that there was indeed an increase in the number of labeled cells recovered from the peritoneal cavity of mertk−/− mice (Figure 6). Interestingly the origin of the donor cells was not a factor since wild-type and mertk−/− donor cells migrated similarly to the peritoneal cavity of wild-type mice. Correspondingly, both donors migrated similarly to the mertk−/− peritoneal cavity. This data not only suggests that an increase in migration is the reason for the increase in cells found in the peritoneal cavity of the mertk−/− mice; but also that the environment of the mertk−/− mice influences the migration of cells.
Figure 6. Enhanced migration to mertk−/− peritoneal cavity.

Indicated donor PECs were harvested, stained with Cell Tracker green and injected via tail vein into recipient mice. PEC were harvested from 24 hrs after injection and analyzed by flow cytometry. The majority of donor cells recovered were lymphocytes based on forward scatter and side scatter analysis (data not shown). Data is expressed as number of Cell Tracker green positive cells recovered from recipient peritoneum. n ≥ 10. Statistical significance is * p<0.01 by Students t-test.
In order to differentiate between hematopoietic and non-hematopoietic sources for the increase in cells migrating to the peritoneal cavity of mertk−/− mice, we performed bone marrow transplants of mertk−/− bone marrow into wild-type mice and vice versa. If the hematopoietic cells from mertk−/− mice were responsible for the greater migration, we would expect that the wild-type mice reconstituted with mertk−/− bone marrow would have an increase in PEC. Conversely, we would expect that the mertk−/− mice reconstituted with bone marrow from wild-type mice would show a decrease in PEC. Control mertk−/−recipients reconstituted with bone marrow from mertk−/− donors showed the expected increase in numbers of cells in the peritoneal cavity compared to control wild-type mice reconstituted with wild-type bone marrow (Figure 7A). However wild-type and mertk−/−mice reconstituted with heterologous bone marrow showed a similar number of peritoneal cells. This indicates that factors from the immune cells and the host environment may be influencing migration into the peritoneal cavity.
Figure 7. Both hematopoietic and non-hematopoietic sources are required for cellular increase in mertk−/− peritoneal cavity.

Recipient 4 week old mice were lethally irradiated and given indicated donor bone marrow 1 day later. At 3 months of age PEC were collected and (A) total cell number was counted. Cells were then stained for (B) F4/80+ macrophage, (C) CD11c+ DC, (D) CD19+ B cells, and (E) CD3+ T cells. WT = wild-type M−/− = mertk−/− mice. Statistical significance using the Student’s t-test * p< 0.05 and ** p<0.0005.
We next analyzed the composition of the peritoneal cells and found no significant increases in F4/80+ macrophages, CD11c+ DCs, or CD19+ B cells (Figure 7B, C, D). However, a significant increase was found in CD3+ T cell number when wild-type recipients were given bone marrow from mertk−/− mice as compared to donor wild-type mice (Figure 7E). Correspondingly, there was a significant decrease in the number of T cells recovered from the peritoneal cavity of mertk−/− mice receiving wild-type donor cells as compared to the peritoneal cavity of mertk−/− mice receiving mertk−/− bone marrow. Concurrent with previously published data, the number of B cells recovered from the peritoneal cavity after bone marrow transplantation was dramatically reduced most likely due to the lack of B1 cells(3). B1 cells are not able to repopulate because their precursors are only available from fetal tissue and not adult bone marrow(3). Taken together, these data suggest that neither hematopoietic nor non-hematopoietic cells contribute solely to the overall increase in cells in found in the peritoneal cavity of the mertk−/− mice. However, the increase in T cells is most likely due to signals exclusively from the hematopoietic compartment.
The increase in migration of immune cells into the mertk−/− peritoneal cavity may be due to several candidate chemokines and cytokines. We hypothesized that the chemokine CXCL13 would be increased in mertk−/− cells since this chemokine is required for migration of B1 cells to the peritoneal cavity(31). However as shown in Figure 8A, real-time PCR of mRNA for CXCL13 was similar in both the mertk−/− and wild-type resident PEC. Likewise, IL-5, IL-6, IL-7, IL-9, IL-10, and BAFF were not different between the strains (Figure 8A or data not shown). However, a marginal increase in mRNA of the chemokine CXCL14 and a 3-fold increase in CXCL9 were observed in the mertk−/− population. In addition to the soluble factors, we also examined receptors present on migrating cells to determine if a certain receptor was up-regulated and contributing to the increase in number of cells found in the peritoneal cavity of mertk−/− mice (Figure 8B). We found the migration-associated receptors LFA, PECAM, and L-selectin were expressed similarly in cells from both genotypes (data not shown). Also, mRNA for CXCR5, the receptor for CXCL13, was similar in mertk−/− and wild-type cells (Figure 8B). Therefore this combination of ligand-receptor pairing is unlikely to explain the large numbers of cells in the peritoneal cavity of mertk−/− mice.
Figure 8. Expression of chemokines, cytokines, and migration receptors in PEC.

PEC were pooled from at least 4 three month old male mice. RNA was harvested and reverse transcribed to cDNA. A) Real-time PCR was performed for soluble molecules, IL-7, BAFF, CXCL9, CXCL13, and CXCL14. B) Real-time PCR was performed for receptors, CXCR5, CXCR3, and IL-7R. C) PEC from wild-type and mertk−/− mice were double-stained for CD3, CD19, or F480 and CXCR3. Representative histograms of CXCR3 expression gated on CD3+ T cells (D) or CD19+ B cells (E). F) PECs were stained and sorted on the basis of CXCR3 expression. Either CXCR3-negative sorted cells or all cells sorted from mertk−/− donors were stained with cell tracker green and adoptively transferred into mertk−/− recipients. G) PEC from wild-type and mertk−/− mice were double-stained for CD3, CD19, or F4/80 and IL-7R. H) Representative histograms of IL-7R expression CD3+ cells. Mean and standard deviation are graphed. n ≥ 3 per genotype per panel. Statistical significance are indicated, *p<0.05, **p<0.005 by the Student’s t test when comparing wild-type vs mertk−/− mice.
The receptor for CXCL9, CXCR3 is important to lymphocyte development and homing of T cells and B cells and its upregulation would be consistent with the increase of T and B cells within the peritoneal cavity of mertk−/− mice. In fact, the CXCR3 transcript was increased greater than 2-fold in cells from mertk−/− mice (Figure 8B). IL-7 receptor (IL-7R) is not known as a chemoattractant but it may promote the maintenance of lymphocytes (32). Similar to high CXCR3 expression, IL-7R mRNA was also found to be expressed over 4-fold greater in the cells harvested from mertk−/− mice (Figure 8B). This increase in mRNA for CXCR3 and IL7R was confirmed by flow cytometry (Figure 8C and D).
We next used flow cytometry to obtain a phenotypic profile of CXCR3-expressing peritoneal cells. Most of the expression of CXCR3 was on the cell surface of lymphocytes with T cells comprising the largest population in both wild-type and mertk−/−mice (Figure 8C). However, there was a significantly greater number of B cells (twofold) and T cells (nearly four-fold) expressing CXCR3 in the peritoneal cavity of mertk−/−mice. As a population, the percent of T cells and B cells from wild-type and mertk−/−mice did not show dramatically different expression levels of CXCR3 (Figure 8D and E). However, even though CXCR3-positive B cells are a small percentage of the total B cells (Figure 8E), when multiplied by the total number of B cells, the deductions show they comprise approximately 30% of the total CXCR3-positive cells in mertk−/− peritoneal cavity and about 45% in wild-type (Figure 8C). This is because B cells are the predominant cell found in the peritoneal cavity, comprising half of the cells in both wild-type and mertk−/− mice (Table 1 and 9B). Therefore, B cells are a substantial portion of CXCR3-expressing lymphocytes in the peritoneal cavity In contrast to B cells, a larger percentage of T cells are CXCR3-positive (Figure 8D); however, T cells only comprise 16% of the total peritoneal cell population in wild-type mice and 30% in mertk−/− mice (Table 1, and 9B). This results in T cells accounting for about 50% of the total CXCR3-positive cells in wild-type mice and 65% of the total CXCR3-positive cells in mertk−/−mice. Thus, there is a slightly larger number of CXCR3+ T cells than B cells in the peritoneal cavity of mertk−/− mice.
In addition, we demonstrate further that CXCR3 was functioning in migration by depleting CXCR3-expressing donor cells and determining whether numbers of cells migrating into the peritoneal cavity were reduced. Since donor cells, whether derived from wild-type or from mertk−/− mice, entered the peritoneal cavity of mertk−/− mice similarly as shown in Figure 6, we used mertk−/− mice as donors due to their greater numbers of resident cells. When donor peritoneal cells expressing CXCR3 were removed by cell-sorting and the remaining cells adoptively transferred into mertk−/− mice, fewer cells migrated to the peritoneal cavity than donor cells that contained CXCR3+ cells (Figure 8F). This data suggests that the expression of CXCR3 is at least partly responsible for the migration of cells into the peritoneal cavity (Figure 8F).
A similar expression pattern to CXCR3 was observed for the number of B and T cells expressing IL-7R: however, only T cells were significantly different (Figure 8G). Similar to CXCR3, as a population, T cells from wild-type and mertk−/− mice did not show different expression levels of IL-7R(Figure 8H). In contrast, there were very few macrophages expressing either CXCR3 or IL-7R receptor (Figure 8C and G). Thus, we have identified potential ligands and receptors that appear to regulate cell populations by the presence or absence of Mertk.
After finding the absence of Mertk led to an increase in PECs, we wanted to determine if knock-out mice lacking the other TAM family members, Axl or Tyro3, had a similar increase in PEC number. Interestingly, we found that there were no statistically significant differences in the total number of cells collected from the peritoneal cavity of the other knock-out mice when compared to wild-type mice (Figure 9A). Along these lines, there were no differences compared to wild-type mice in the number of CD19+ B cells, CD3+ T cells, or F4/80+ macrophages found in the axl−/−, tyro3−/− and axl−/−/tyro3−/−double knock-out mice (Figure 9B). Previously, we and others have shown mertk−/− mice produce autoantibodies to several self-antigens including dsDNA and nucleosomes (7–10). Since the peritoneal cavity is an important reservoir for the B1 B cell population which is known to play a role in autoantibody formation, we assessed anti-nucleosome antibody production of these mice. Although autoantibodies had been attributed to the absence of all three family members(9), mertk−/− mice were the only mutant strain to show elevated levels of anti-nucleosome antibody (Figure 9C). In addition, increased splenic weights were only associated with mertk−/− mice whereas axl−/− and tyro3−/− mice were similar to wild-type mice (Figure 9D). This suggests that Mertk is the critical TAM family receptor that dictates the composition of cells in the peritoneal cavity and is important in regulating the production of autoantibody.
Figure 9. Increase in peritoneal cells and autoantibody production is due to Mertk and not Axl or Tyro3.
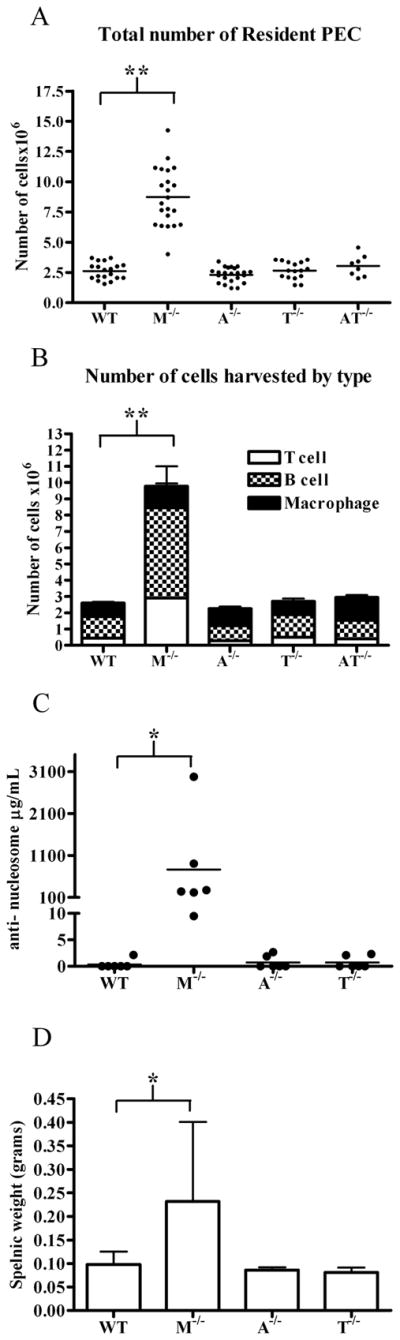
PEC were harvested from 3 month old male mice from wild-type (WT), mertk−/− (M−/−), axl−/− (A−/−), tyro3−/− (T−/−), axl−/−/tyro3−/− (AT−/−). A) Total cells were counted and stained for B) F4/80+ macrophage, CD19+ B cells and CD3+ T cells. C) Anti-nucleosome antibody titers were determined by ELISA from serum of mice 6 months of age or older D) Splenic weight was measured from mice 6 months of age or older. n ≥ 6 No statistically significant differences were found by ANOVA with post-tests between WT, A−/−, T−/− and AT−/− peritoneal cells. Statistical significance is indicated in comparisons with samples from wild type and mertk−/− mice, *p<.01 **p<.001 by ANOVA post-test.
Discussion
Mice lacking Mertk manifest a lupus like syndrome including splenomegaly, nephropathy, and autoantibody production to many different auto-antigens(7–11). Autoimmune diseases in mouse models are often correlated with enlarged lymphocyte compartments. We previously demonstrated that splenic weight was increased in mertk−/−mice, however only the DC populations were significantly elevated(8). In this study, we characterized an increase in total cell number found in the peritoneal cavity of mertk−/−mice, and found this increase to be in multiple cell types including cells involved in both innate and adaptive immunity.
Mertk is expressed on macrophages and dendritic cells. Although the number of DC and pDC were elevated in mertk−/− mice, they represent a small proportion of the resident PECs. Resident peritoneal macrophages were elevated in mertk−/− mice; however, when the macrophage-eliciting reagent, thioglycollate, was injected into the peritoneal cavity, similar numbers of elicited cells are recruited after 3 days (data not shown). This suggests that the upper limit of elicited cells was not restricted in wild-type mice and it appears only during normal homeostasis that the absence of functioning Mertk is evident. Furthermore, our analyses demonstrate that only Mertk regulated the resident peritoneal cell numbers and the other family members Axl or Tyro3 were not important. This increase in peritoneal immune cells correlated well with the increased production of autoantibodies since only mertk−/− mice showed excessive levels. Our data suggests that in the absence of Mertk, migration of cells into the peritoneal cavity is dysregulated and this may be due to an elevation of specific chemokines and receptors associated with leukocyte homing and survival. These findings provide insights for the propensity of mertk−/− mice to develop a lupus like syndrome highlighted by the inability to clear apoptotic cells normally and by the increased production of autoantibodies.
Interestingly, we found that mertk−/− mice had an age-dependent increase in PECs. This difference between wild-type and mertk−/− mice was most evident at 3 months of age (Figure 1). In the absence of Mertk, we found an increase in the lymphocyte compartment of both B cells and T cells from the peritoneal cavity (Figure 1). In the mertk−/− lymphocyte population, CD19+ B cells were over 5-fold greater in number than wild-type, and CD3+ T cells were over 6-fold greater (Figure 1). Further analyses indicated that the B1 B cells, the B1a and B1b cells were elevated in mertk−/− mice compared to wild type mice (Figure 3). An increase in B1 cell number was also found in the peritoneal cavity of the Sm-specific 2–12H-mertk−/− mice (11). In the T cell population, the CD4+ and CD8+ T cell compartments were elevated in the mertk−/− mice although they were at equal ratios to that of wild-type mice (Figure 2C). The largest T cell increase was found in the naive T cells, shown in Figure 2D. Lastly, there were elevated numbers of effector and memory T cells as indicated by the distribution of CD44 and CD62L (Figure 2D). Thus, it appears in the absence of Mertk, T lymphocytes accumulate in greater numbers in the peritoneal cavity and this accumulation appears to be dependent on hematopoietic-derived factors (Figure 7). It is plausible that the increase in T cells may increase the frequency that a T cell may aid antigen activation of B cells.
While the peritoneum acts as a lymphoid organ, it lacks the architecture similar to that of a lymph node or spleen, thus presenting unconventional possibilities for antigen presentation and subsequent lymphocyte activation. This may be particularly true for an animal with an increased lymphocyte population and greater numbers of antigen-presenting cells with in the peritoneal cavity. Although B1-B cells normally produce T independent, low-affinity auto antibodies(3); the mertk−/− mice present a unique scenario with increased levels of B1-B cells as well as T cells in the peritoneum (Figure 1, 2, 3). Thus, this creates an environment with greater likelihood that these B cells would receive T cell help, undergo class switching, and begin producing higher affinity autoantibodies.
B1-B cells are critical for the autoimmune hemolytic anemia found in the NZB and NZB/W mice, as hypotonic ablation of peritoneal B1-B cells decreases autoantibody titers and prevents disease development in aged animals (12). Interestingly, these and other autoimmune mice display increased B1 cell levels in the peritoneal cavity prior to disease development (6, 33, 34). Previously published data from our lab and others indicates that autoimmune disease in the mertk−/− mice is not manifested until greater than 3 months of age(7–9). Similar to the NZB mice, we have found a dramatic elevation of peritoneal B cells by 3 months of age in mertk−/− mice (Figure 1C) prior to an age when autoantibodies are prevalent (Figure 9C)(7–10). The influence of Mertk on the lymphocyte populations may be indirect as T cells normally do not express Mertk (16) and our analysis of the LPS-stimulated and unstimulated B cell population found no detectable expression of Mertk by flow cytometry (data not shown). However, Mertk has been detected recently on B cells in mice undergoing chronic graft-versus-host disease (cGVHD) and it is presumed that Mertk may help activate the alloreactive B cells in wild-type mice which produced antibodies to alloantigens and nuclear autoantigens. Interestingly, mice lacking Mertk could not generate alloantibodies or produce anti-dsDNA autoantibody in this cGVHD model which is contrary to their development of spontaneous autoimmunity in older animals (7, 10, 35). This suggests that B cell mechanisms regulating allograft responses may be different than those involved in spontaneous autoimmune responses; however, similar to their findings, we also found no difference in mixed lymphocyte reaction or on cell-mediated lympholysis assays (data not shown). In addition, B cells from mertk−/− mice undergoing cGVHD are slightly less active by MHC II expression similar to our finding in Figure 3; however, the implications of this small reduction in MHC II are not clear. Thus, the peritoneal cavity is an important site for autoreactive cells and it would be interesting to determine if the B1-B cells are critical for autoantibody production in the mertk−/− mice similar to the NZB strain.
At 3 months of age mertk−/− mice have nearly four times the number of B1 cells compared to their wild-type counterparts (Figure 3A). Based on previous work, mertk−/−mice possess fewer peritoneal autoreactive anti-Sm B1 B cells compared to the wild type (Non-Tg) animals. This is likely due to the fact that autoreactive B1 B cells become antigen-activated in the mertk−/− peritoneum and then migrate out to the lamina propria and mesenteric lymph nodes to become antibody-secreting cells (11). Thus, the peritoneal cavity appears to be a recruitment area for the migration of T and B cells, DC, and macrophages that optimizes the priming of autoreactive B1-B cells. In the absence of Mertk, it is plausible that the presence of apoptotic cells may not be adequately cleared by the Mertk-deficient macrophages(10), resulting in exposure of B cells and T cells to self-antigens. Over time this constant activation of autoreactive B cells accumulates and likely contributes to the increasing proportion of autoantibody-secreting cells and the escalation of autoantibody titers associated with the Mertk defect (7, 8).
The large number of cells in the peritoneal cavity of mertk−/− mice is likely due to an increase in migration (Figure 6) rather than an increase in local proliferation (Figure 4) or an inhibition of cell death (Figure 5). In support of this, we examined a list of candidate cytokines and chemokines as well as receptors known to be involved in homing and migration of cells. While most of the molecules investigated did not show differences, we identified the chemokine CXCL14 as having a marginal increase in the mertk−/− mice (Figure 8A). CXCL14 is a chemokine responsible for migration of macrophages to adipose tissue and also affects insulin uptake(36). This is particularly interesting because the mertk−/− mice have a larger peritoneal fat pad as compared to wild-type mice (data not shown).
Most relevant to cell migration, we identified a more dramatic over-expression of the chemokine CXCL9 in the PECs of the mertk−/− mice which coincided with an increased expression of the cognate receptor CXCR3 (Figure 8A, B). CXCL9 is a chemokine that can act as a chemoattractant for CXCR3-expressing T cells, B cells and plasmacytoid DCs (pDC)(37–39). Type I IFNs are known to induce CXCL9 expression(37) and one common finding in patients diagnosed with systemic lupus erythematosus is increased in production of type I IFN which maybe a result of DNA/immune complexes activating pDC (40, 41). Thus, it is plausible that the greater numbers of pDC (Figure 2B), T cells and B cells (Figure 1, 2, 3) found in the peritoneal cavity of the mertk−/− mice are a consequence of greater amounts of CXCL9: however, a definitive role for CXCL9/CXCR3 on cell migration into the peritoneal cavity of mertk−/−mice will require further experimentation.
CXCR3 has also been implicated in the kidney pathology found in the MRL/lpr lupus mouse model, as MRL/lpr mice lacking CXCR3 show less T cell infiltration and nephritis(42). It was not investigated whether the MRL/lpr mice lacking CXCR3 also had fewer peritoneal cells. Here in our study, we also find that CXCR3 is important for the migration and accumulation of cells in our lupus model of the mertk−/− mice (Figure 8F). In the adoptive transfer of CXCR3-depleted peritoneal cells, the fewer numbers of migrating cells is attributed to the lack of CXCR3+ cells. Since the control group (all cells) were subjected to the same procedural conditions as the CXCR3− cells, the significant reduction is likely due to the lack of CXCR3-expressing cells. Therefore, CXCR3 is at least partly responsible for the migration of cells into the peritoneal cavity, although other factors are likely operating.
The other receptor that was notably increased in the mertk−/− PEC was the IL-7 receptor. Mice lacking the IL-7 receptor have low peritoneal B cell levels(32); however mice lacking the ligand, IL-7, have normal numbers of peritoneal B cell suggesting the importance of another ligand for the IL-7 receptor in the peritoneal B cell population(43). In addition, the chemokine critical for B cell homing to the peritoneal cavity, CXCL13, has been shown to act synergistically with the IL-7 receptor in development of lymph nodes, particularly the mesenteric lymph node(44). This is of interest because while we did not find differences in expression levels of CXCL13 (Figure 8A), we did find an increase in expression of IL-7 receptor (Figure 8B and 8G), presenting the potential for the synergy of CXCL13 and IL-7 receptor in establishing the peritoneal cavity lymph system as well. Thus, although we have not detected Mertk on B cells (data not shown), Mertk may be indirectly influencing the B cell populations through the IL-7 receptor and perhaps account for the increase in total B cell and B1 cell numbers in the peritoneal cavity.
Equally intriguing is the expression of IL-7 receptor on T cells and we found a four-fold greater number in the absence of Mertk (Figure 8G). It is plausible that IL-7R may allow greater numbers of T cells similar to B cells by increasing the survival of T cells within the peritoneal cavity or dampen their egress. This appears to be particularly true in the naive T cell population since they show the greatest representation (Figure 2D). Furthermore, IL-7 receptor has been associated with an increase in survival of CD8+ effector T cells that convert to memory T cells (45) and indeed, we find elevated numbers of effector T cells and central memory T cells in the peritoneal cavity of mertk−/−mice compared to wild-type mice (Figure 2C and 2D). Taken together, this data indicates that Mertk has a profound affect on the regulation of T cell populations within the peritoneal cavity.
Although Mertk is not normally expressed on lymphocytes, we observed increased numbers of T and B cells. We deduce that typically, Mertk-expressing cells may be regulating the expression of chemoattractants and cytokines to prevent excessive accumulation of peritoneal cells that may promote autoimmune disease. Work presented here identifies an increase in the cellular populations found in the peritoneal cavity of mertk−/− mice. We show that this increase is not due to one particular cell type but rather all cell types investigated. We also determined this increase in B cells, T cells and antigen-presenting cells is due to enhanced migration/recruitment into the peritoneal cavity, not a change in local proliferation or death of cells. Our data suggest that Mertk may be increasing the accumulation of immune cells by upregulating CXCL9 and its receptor CXCR3 as well as IL-7R. Consequently, by establishing an abnormally large number of these immune cells in the peritoneal cavity, the likelihood of interaction and support of autoreactive B cells is favored. We find only Mertk to be critical for the regulation of peritoneal cell recruitment as mice lacking Axl and Tyro3 do not show a similar increase (Figure 9). This is consistent with our current findings that splenomegalia and the onset of high autoantibody titers are found only in mertk−/− mice and not axl−/− and tyro3−/− mice. Our report provides a platform to study additional mechanisms that are controlled by Mertk and it appears that Mertk is a primary regulator driving the autoimmune phenotype.
Acknowledgments
This work was funded in part by National Institute for Allergy and Infectious Disease Grant AI50736
The authors would like to thank Stephen Goff for the axl−/− mice and Greg Lemke for the tyro3−/− mice. We would also like to thank Roland Tisch and Stephen Clarke for helpful discussions. We would like to acknowledge Jeff Frelinger for CD62L and CD44 antibodies and Jenny Ting for pDCA-1 antibody. We would like to thank Paul Gholke and Paul Hiatt for technical assistance. We would also like to acknowledge the UNC Flow Cytometry Core Facility as well as the UNC Neuroscience Functional Genomics Core Facility.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subject to the editorial proofreading by the JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Broche F, Tellado JM. Defense mechanisms of the peritoneal cavity. Curr Opin Crit Care. 2001;7:105–116. doi: 10.1097/00075198-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Finlay-Jones JJ, Davies KV, Sturm LP, Kenny PA, Hart PH. Inflammatory processes in a murine model of intra-abdominal abscess formation. J Leukoc Biol. 1999;66:583–587. doi: 10.1002/jlb.66.4.583. [DOI] [PubMed] [Google Scholar]
- 3.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu Rev Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- 4.Tumang JR, Hastings WD, Bai C, Rothstein TL. Peritoneal and splenic B-1 cells are separable by phenotypic, functional, and transcriptomic characteristics. Eur J Immunol. 2004;34:2158–2167. doi: 10.1002/eji.200424819. [DOI] [PubMed] [Google Scholar]
- 5.Viau M, Zouali M. B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. 2005;114:17–26. doi: 10.1016/j.clim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gohlke PR, Williams JC, Vilen BJ, Dillon SR, Tisch R, Matsushima GK. The receptor tyrosine kinase MerTK regulates dendritic cell production of BAFF. Autoimmunity. 2009;42:183–197. doi: 10.1080/08916930802668586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 10.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 11.Qian Y, Wang H, Clarke SH. Impaired clearance of apoptotic cells induces the activation of autoreactive anti-Sm marginal zone and B-1 B cells. J Immunol. 2004;172:625–635. doi: 10.4049/jimmunol.172.1.625. [DOI] [PubMed] [Google Scholar]
- 12.Murakami M, Yoshioka H, Shirai T, Tsubata T, Honjo T. Prevention of autoimmune symptoms in autoimmune-prone mice by elimination of B-1 cells. Int Immunol. 1995;7:877–882. doi: 10.1093/intimm/7.5.877. [DOI] [PubMed] [Google Scholar]
- 13.Behrens EM, Gadue P, Gong SY, Garrett S, Stein PL, Cohen PL. The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol. 2003;33:2160–2167. doi: 10.1002/eji.200324076. [DOI] [PubMed] [Google Scholar]
- 14.Caraux A, Lu Q, Fernandez N, Riou S, Di Santo JP, Raulet DH, Lemke G, Roth C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat Immunol. 2006;7:747–754. doi: 10.1038/ni1353. [DOI] [PubMed] [Google Scholar]
- 15.Graham DK, Bowman GW, Dawson TL, Stanford WL, Earp HS, Snodgrass HR. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene. 1995;10:2349–2359. [PubMed] [Google Scholar]
- 16.Graham DK, Salzberg DB, Kurtzberg J, Sather S, Matsushima GK, Keating AK, Liang X, Lovell MA, Williams SA, Dawson TL, Schell MJ, Anwar AA, Snodgrass HR, Earp HS. Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2006;12:2662–2669. doi: 10.1158/1078-0432.CCR-05-2208. [DOI] [PubMed] [Google Scholar]
- 17.Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994;5:647–657. [PubMed] [Google Scholar]
- 18.Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- 19.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–5642. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 20.Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, Huang Y, Mathews CE, Earp HS, Matsushima G, Wang B, Tisch R. MerTK is required for apoptotic cell-induced T cell tolerance. J Exp Med. 2008;205:219–232. doi: 10.1084/jem.20062293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 22.Uehara H, Shacter E. Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinase-mediated phagocytosis of apoptotic cells. J Immunol. 2008;180:2522–2530. doi: 10.4049/jimmunol.180.4.2522. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 24.Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999;162:3498–3503. [PubMed] [Google Scholar]
- 25.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, Earp HS, Matsushima G, Baldwin AS, Jr, Tisch RM. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood. 2007;109:653–660. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams JC, Craven RR, Earp HS, Kawula TH, Matsushima GK. TAM receptors are dispensable in the phagocytosis and killing of bacteria. Cell Immunol. 2009;259:128–134. doi: 10.1016/j.cellimm.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 28.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 29.Khare SD, Sarosi I, Xia XZ, McCabe S, Miner K, Solovyev I, Hawkins N, Kelley M, Chang D, Van G, Ross L, Delaney J, Wang L, Lacey D, Boyle WJ, Hsu H. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3370–3375. doi: 10.1073/pnas.050580697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansel KM, Harris RB, Cyster JG. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity. 2002;16:67–76. doi: 10.1016/s1074-7613(01)00257-6. [DOI] [PubMed] [Google Scholar]
- 32.Hesslein DG, Yang SY, Schatz DG. Origins of peripheral B cells in IL-7 receptor-deficient mice. Mol Immunol. 2006;43:326–334. doi: 10.1016/j.molimm.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Ito T, Ishikawa S, Sato T, Akadegawa K, Yurino H, Kitabatake M, Hontsu S, Ezaki T, Kimura H, Matsushima K. Defective B1 cell homing to the peritoneal cavity and preferential recruitment of B1 cells in the target organs in a murine model for systemic lupus erythematosus. J Immunol. 2004;172:3628–3634. doi: 10.4049/jimmunol.172.6.3628. [DOI] [PubMed] [Google Scholar]
- 34.Xu Z, Butfiloski EJ, Sobel ES, Morel L. Mechanisms of peritoneal B-1a cells accumulation induced by murine lupus susceptibility locus Sle2. J Immunol. 2004;173:6050–6058. doi: 10.4049/jimmunol.173.10.6050. [DOI] [PubMed] [Google Scholar]
- 35.Shao WH, Eisenberg RA, Cohen PL. The Mer receptor tyrosine kinase is required for the loss of B cell tolerance in the chronic graft-versus-host disease model of systemic lupus erythematosus. J Immunol. 2008;180:7728–7735. doi: 10.4049/jimmunol.180.11.7728. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Iida K, Okimura Y, Kaji H, Chihara K. CXCL14 enhances insulin-dependent glucose uptake in adipocytes and is related to high-fat diet-induced obesity. Biochem Biophys Res Commun. 2007;364:1037–1042. doi: 10.1016/j.bbrc.2007.10.120. [DOI] [PubMed] [Google Scholar]
- 37.Wenzel J, Worenkamper E, Freutel S, Henze S, Haller O, Bieber T, Tuting T. Enhanced type I interferon signalling promotes Th1-biased inflammation in cutaneous lupus erythematosus. J Pathol. 2005;205:435–442. doi: 10.1002/path.1721. [DOI] [PubMed] [Google Scholar]
- 38.Krukemeyer MG, Moeller J, Morawietz L, Rudolph B, Neumann U, Theruvath T, Neuhaus P, Krenn V. Description of B lymphocytes and plasma cells, complement, and chemokines/receptors in acute liver allograft rejection. Transplantation. 2004;78:65–70. doi: 10.1097/01.tp.0000132324.14207.8b. [DOI] [PubMed] [Google Scholar]
- 39.Yoneyama H, Matsuno K, Matsushimaa K. Migration of dendritic cells. Int J Hematol. 2005;81:204–207. doi: 10.1532/IJH97.04164. [DOI] [PubMed] [Google Scholar]
- 40.Vallin H, Blomberg S, Alm GV, Cederblad B, Ronnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999;115:196–202. doi: 10.1046/j.1365-2249.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallin H, Perers A, Alm GV, Ronnblom L. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol. 1999;163:6306–6313. [PubMed] [Google Scholar]
- 42.Steinmetz OM, Turner JE, Paust HJ, Lindner M, Peters A, Heiss K, Velden J, Hopfer H, Fehr S, Krieger T, Meyer-Schwesinger C, Meyer TN, Helmchen U, Mittrucker HW, Stahl RA, Panzer U. CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis. J Immunol. 2009;183:4693–4704. doi: 10.4049/jimmunol.0802626. [DOI] [PubMed] [Google Scholar]
- 43.Carvalho TL, Mota-Santos T, Cumano A, Demengeot J, Vieira P. Arrested B lymphopoiesis and persistence of activated B cells in adult interleukin 7(−/)- mice. J Exp Med. 2001;194:1141–1150. doi: 10.1084/jem.194.8.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. J Exp Med. 2003;197:1191–1198. doi: 10.1084/jem.20021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]