

Abstract
Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) “autonomy” (T286-autophosphorylation-induced Ca2+-independent activity) is required for long-term potentiation (LTP) and for learning and memory, as demonstrated by CaMKII T286A mutant mice. The >20-year-old hypothesis that CaMKII stimulation is required for LTP induction, while CaMKII autonomy is required for LTP maintenance was recently supported using the cell-penetrating fusion-peptide inhibitor antCN27. However, we demonstrate here that ant/penetratin fusion to CN27 compromised CaMKII-selectivity, by enhancing a previously unnoticed direct binding of CaM to ant/penetratin. In contrast to antCN27, the improved cell-penetrating inhibitor tatCN21 (5 μm) showed neither CaM binding nor inhibition of basal synaptic transmission. In vitro, tatCN21 inhibited stimulated and autonomous CaMKII activity with equal potency. In rat hippocampal slices, tatCN21 inhibited LTP induction, but not LTP maintenance. Correspondingly, tatCN21 also inhibited learning, but not memory storage or retrieval in a mouse in vivo model. Thus, CaMKII autonomy provides a short-term molecular memory that is important in the signal computation leading to memory formation, but is not required as long-term memory store.
Introduction
Mechanisms by which unstable molecules could store stable memories have been proposed in the 1980s (Crick, 1984; Lisman, 1985), and the model still reflects current opinion (Ogasawara and Kawato, 2009). Shortly after, a mechanism within neurons that could fit this memory bill was described: The CaM-dependent protein kinase II (CaMKII) (Schulman and Greengard, 1978; Yamauchi and Fujisawa, 1980) can autophosphorylate at T286 during a Ca2+/CaM stimulus, which in turn generates “autonomous” CaMKII activity that is no longer dependent on the initial Ca2+-signal (Lou et al., 1986; Miller and Kennedy, 1986; Schworer et al., 1986). Long-term potentiation of synaptic strength (LTP) was already established as likely cellular substrate for learning and memory (Bliss and Lomo, 1973; for review, see Malenka and Nicoll, 1999; Lisman et al., 2002; Lee and Silva, 2009). Together, these findings laid the groundwork for the “CaMKII autonomy hypothesis of memory storage,” postulating that CaMKII stimulation is important for LTP induction and learning, while autonomous CaMKII activity is critical for LTP maintenance and memory storage. Indeed, inhibition and knock-out demonstrated a vital role of CaMKII in LTP and learning (Malinow et al., 1989; Silva et al., 1992a,b), by increasing number (Hayashi et al., 2000) and single channel conductance (Benke et al., 1998; Derkach et al., 1999) of synaptic AMPA-type glutamate receptors. Importantly, CaMKII T286A mutant mice were also impaired in LTP and learning and memory (Giese et al., 1998), demonstrating critical importance of CaMKII autonomy. However, these studies in mutant mice did not allow distinction between functions in LTP induction versus maintenance or in memory acquisition versus storage/retrieval. Additionally, recent studies showed that autonomous CaMKII is not fully active and can be significantly further stimulated by Ca2+/CaM signals (Coultrap et al., 2010). The initial inhibitor studies indicated requirement of both CaMKII and PKC activity for LTP induction but not for LTP maintenance (Malinow et al., 1989). The importance of both kinases in LTP induction stands unchallenged, however, subsequent reports implicated autonomous forms of CaMKII (Feng, 1995; Wang and Kelly, 1996; Sanhueza et al., 2007) and PKC (Sacktor et al., 1993; Ling et al., 2002; Pastalkova et al., 2006) in LTP maintenance. Technical issues have been cited as possible reasons for the discrepancy (Otmakhov et al., 1997; Chen et al., 2001; Sanhueza et al., 2007), in case of PKC regarding isoform-selectivity of the inhibitors used (Ling et al., 2002; Pastalkova et al., 2006). In contrast to the traditional CaMKII inhibitors KN93 and KN62, the novel cell-penetrating fusion-peptide tatCN21 is highly CaMKII-selective (Vest et al., 2007). Most importantly, while KN93 can block generation of autonomous CaMKII activity by inhibiting autophosphorylation (for instance when KN93 is present during LTP induction), KN93 does not block autonomous activity once it is generated (for instance after LTP induction) (Vest et al., 2010). By contrast, tatCN21 blocks both stimulated and autonomous CaMKII activity (Vest et al., 2007). Thus, tatCN21 allowed directly testing the CaMKII autonomy hypothesis of memory storage. Our results show that neither stimulated nor autonomous CaMKII activity is required for LTP maintenance or for memory storage/retrieval in vivo. Together with the studies in T286A mutant mice (Giese et al., 1998), our results additionally indicate CaMKII autonomy is instead required for LTP induction and learning. This implicates CaMKII autonomy in the computation rather than storage of information, consistent with its biochemical properties in frequency detection of Ca2+-oscillations (De Koninck and Schulman, 1998; Bayer et al., 2002). As an important byproduct, our results provide a cautionary example that peptide fusion can cause undesired gain-of-function (in a previous inhibitor version, antCN27).
Materials and Methods
Materials.
CaMKII and CaM were purified as described previously (Bayer et al., 2001); CaMKIV and CaMKK were purchased from SignalChem. Peptides were obtained from GenScript or CHI Scientific; the scrambled CN21 sequence fused to tat in the tatCtrl control peptide was VKEPRIDGKPVRLRGQKSDRI.
Kinase activity assays.
Phosphate incorporation into peptide substrates was assessed as described previously (Bayer et al., 2001; Vest et al., 2007). Stimulated kinase assays were done at 30°C for 1 min and contained 2.5 nm kinase subunits, 50 mm PIPES, pH 7.0–7.2, 0.1% BSA, 10 mm MgCl, 100 μm [γ-32P]ATP (∼1mCi/μmol), 45 μm substrate peptide (see Fig. 3; or 75 μm), 1 mm CaCl2, and 1 μm CaM or as indicated. For assessment of autonomous activity, CaMKII was prephosphorylated at T286 on ice, and activity was measured in presence of 0.5 mm EGTA (instead of CaCl2 and CaM). CaMKIV was prephosphorylated by CaMKK for full stimulation. The kinase panel was tested using a commercial service (Millipore), and the conditions and substrates used were described in detail previously (Vest et al., 2007).
Figure 3.

tatCN21 inhibits stimulated and autonomous CaMKII activity with equally high potency and efficacy. A, tatCN21 potently inhibited Ca2+/CaM-stimulated CaMKII (2.5 nm) activity toward the peptide substrate syntide 2 (75 μm). The calculated IC50 was ∼40 nm. B, tatCN21 inhibited stimulated and autonomous CaMKII activity with equal potency (as indicated by identical inhibition at 40 nm, the IC50 calculated from A) and efficacy (as indicated by complete block of inhibition at 5 μm, 120-fold of IC50), while stimulated activity of the related CaMKIV was not affected at all. Error bars indicate SEM in all panels.
Biochemical assays of protein–protein interactions.
Transfer of peptides or proteins onto PVDF membrane by slot blot or after SDS-PAGE, followed either by overlay with biotinylated CaM or immunodetection was done as described previously (Bayer et al., 2006; Vest et al., 2007). Fluorescence change of 15 nm TA-CaM (kind gift by Dr. Katalin Török) after addition of increasing amounts of CaMKII-derived CaM binding domain peptide (CBD; 0–3.2 μm) was measured in a spectrofluorometer (Fluorolog3; Horiba Jobin Yvon) at 365 nm excitation and 415 nm emission wavelength as described previously (Vest et al., 2007), in presence of 2 μm ant, antCN27, tatCN21, or no additional peptide.
Electrophysiology.
Hippocampal slices (400 μm) were prepared from male Sprague Dawley rats (6–7 weeks old) in ice-cold cutting solution containing the following (in mm): 220 sucrose, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 12 MgSO4, 0.2 CaCl2, and 10 glucose equilibrated with 95% O2-5% CO2. Slices were allowed to recover in artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2.5 CaCl2 and 10 glucose equilibrated with 95% O2-5% CO2 at 31°C for 90 min. For recording, slices were placed into a recirculating chamber (15 ml total volume) for conservation of drug. Stimulating and recording electrodes were placed in the CA1 synaptic field, and field EPSPs (fEPSPs) were recorded at 20 s intervals. Stimulus intensity was adjusted to elicit 40–50% maximal fEPSPs. High-frequency stimulation (HFS) consisted of 2 trains of 1 s 100 Hz stimulation, at the test stimulation intensity.
Behavioral studies.
During a training session, 12-week-old male 129/B6 F1 hybrid mice (Taconic Farms Inc) were placed in the conditioning chamber 2 min before the onset of a footshock (0.5 mA, 2 s), and returned to their home cage 1 min after. During a test session 24 h later, mice were placed in the same conditioning chamber, and conditioning was assessed by measuring freezing behavior for 3 min using an automated system (Med Associates) (software is based on the algorithm described by Anagnostaras et al., 2000). Baseline freezing behavior before conditioning was assessed during the learning session. Drug administration was done at different time points, as indicated.
Results
Ant/penetratin but not tat fusion to CN inhibitors compromised selectivity
In contrast to the traditional CaMKII inhibitor KN93, new peptide inhibitors derived from the endogenous inhibitory protein CaM-KIIN (Chang et al., 1998; Vest et al., 2007) (CN peptides; Fig. 1A) can be used to address the CaMKII autonomy hypothesis of memory storage, as they inhibit not only Ca2+/CaM-stimulated but also autonomous CaMKII activity. Previously, we made a CN inhibitor cell-penetrating by fusion to the ant/penetratin sequence (antCN27) (Fink et al., 2003; Illario et al., 2003), and use of this inhibitor later appeared to indicate involvement of CaMKII autonomy in LTP maintenance (in a study that elegantly circumvented the serious problem of run-down of basal transmission caused by antCN27) (Sanhueza et al., 2007). However, surprisingly, ant fusion with CN peptides generated an additional CaM-directed mode of inhibition, which further enhanced CaMKII inhibition (Fig. 1B,C) but also compromised kinase selectivity, as shown by additional strong inhibition of CaMKIV (Fig. 1D) (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Ant/penetratin but not tat fusion compromised selectivity of CN inhibitor peptides by an additional CaM-directed mode of inhibition. A, The ant and tat sequences fused to the N terminus of CNs; (K) was replaced by the first K of CN27 in the fusion. B, Extent of CaMKII inhibition by 1 μm antCN27 and the reverse sequence control antRev depended on the CaM concentration, indicating an additional CaM-directed mode of inhibition generated by ant fusion. AC2 phosphorylation by CaMKII was measured. Error bars show SEM. C, tatCN21 (1 μm) strongly inhibited CaMKII-mediated AC3 phosphorylation at all CaM concentrations, while the reverse and scrambled sequence controls tatRev, and tatCtrl had no effect compared with assays without tat peptide. D, antCN27 blocked CaMKII activity, but also reduced CaMKIV activity by ∼70% when tested against a panel of different kinases.
CN fusion enhanced a previously unnoticed CaM-binding to ant/penetratin
The additional mode of inhibition by antCN27 was mediated by its direct binding to CaM, as biotinylated CaM efficiently bound to immobilized ant peptide, in addition to immobilized CaMKII or CBD, but not to CN27 or the tat peptide (Fig. 2A). Indeed, antCN27 also interfered with Ca2+-dependent binding of biotinylated CaM to calcineurin A (CaN-A), CaMKIIα, and other proteins from rat brain extract immobilized on PVDF membranes (Fig. 2B; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The ant peptide alone also interfered with such CaM binding, but somewhat less efficiently than antCN27 (Fig. 2B). Neither antCN27 nor ant affected protein binding of CaM that was independent of Ca2+ (Fig. 2B). Binding of CaM to ant and antCN27 was further investigated using a TA-labeled CaM that shows reduced fluorescence after binding to CaMKII or CBD (Török et al., 2001; Vest et al., 2007), which has a kd for CaM of ∼0.05 nm (Török et al., 2001) (see also Fig. 2C; supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Surprisingly, binding to ant did not reduce TA-CaM fluorescence (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), preventing direct affinity measurement, but enabling competition experiments (Fig. 2C). Competition with CBD indicated ∼1 nm and ∼5 nm kd of antCN27 and ant for binding to CaM, respectively (Fig. 2C). Thus, the previously unrecognized direct binding of CaM to ant/penetratin was significantly further enhanced by ant fusion to CN27, which did not bind CaM on its own. Loss-of-function is a commonly considered possible side-effect of peptide or protein fusion. These results provide a cautionary example that fusion can also cause gain-of-function, in this case potentially affecting all CaM-mediated signaling.
Figure 2.
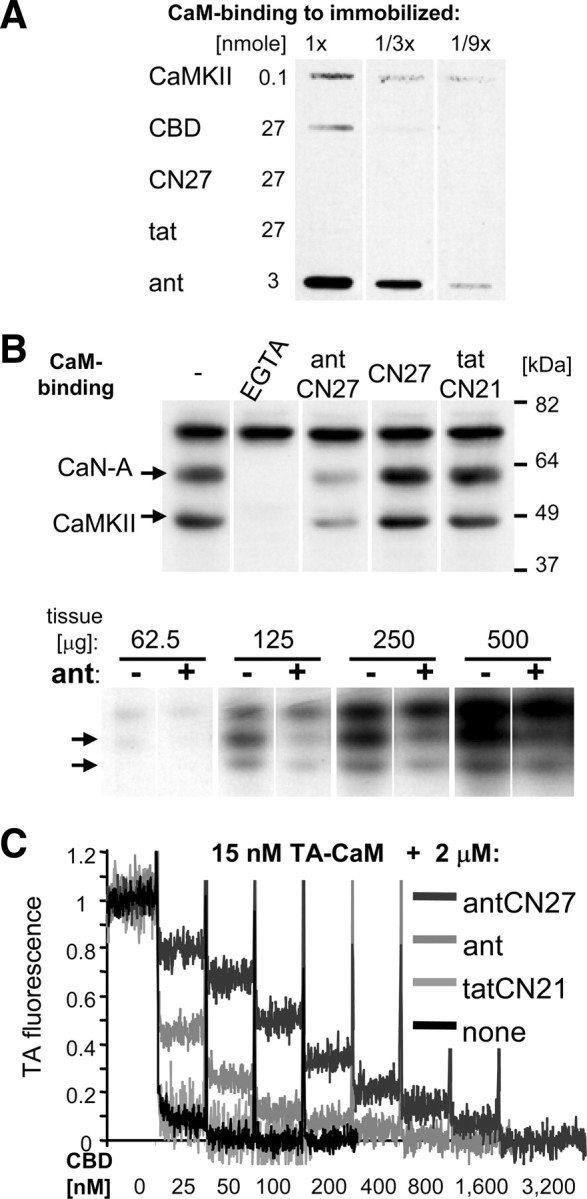
Fusion with a CN peptide enhanced the previously unrecognized direct CaM binding to ant/penetratin. A, Binding of biotinylated CaM (25 nm in TBS, pH 7.5, 1 mm CaCl2) to proteins (CaMKII) and peptides (CBD, CN27, tat, and ant) immobilized by slot blot, in three different dilutions of the amount indicated. B, Interference of peptides (5 μm) with CaM binding (conditions as in A) to brain proteins in a blot overlay assay. The major Ca2+-dependent CaM-binding proteins were CaN-A and CaMKIIα. AntCN27 (top) and ant (bottom) affect Ca2+-dependent CaM binding, but not binding to a protein also detected after Ca2+ was chelated by EGTA. C, antCN27 and ant, but not tatCN21, compete with CBD for binding of TA-labeled CaM. Fluorescence of TA-CaM is reduced after addition of CBD (but not ant; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In presence of antCN27 or ant (but not tatCN21), significantly more CBD has to be added for the same reduction in fluorescence. Fluorescence (λex = 335 nm; λem = 415 nm; 1 s sample time) was monitored for 150 s after each addition of CBD.
tatCN21 is a viable alternative to antCN27
Tat fusion of the newly identified minimal inhibitory region of CaM-KIIN, CN21 (Vest et al., 2007), provided a viable alternative to antCN27, as the tatCN21 peptide did not interfere with general CaM signaling (see Figs. 1C, 2A–C; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). tatCN21 potently inhibited CaMKII activity in vitro, with an IC50 of ∼40 nm (Fig. 3A). Most importantly, tatCN21 inhibited both stimulated and autonomous CaMKII activity with the same potency and efficacy (Fig. 3B). Additionally, tatCN21 retained CaMKII-selectivity when tested against the same kinase panel as shown in Figure 1D (Vest et al., 2007). In contrast to antCN27, even 5 μm tatCN21 (120-fold IC50 of CaMKII inhibition) did not cause any inhibition of the closely related kinase CaMKIV (Fig. 3B).
tatCN21 inhibits induction but not maintenance of LTP
In contrast to antCN27 (Sanhueza et al., 2007), tatCN21 did not cause run down of basal transmission in field recordings of EPSPs (fEPSPs) in rat hippocampal CA1 neurons in acute slices (Fig. 4A). tatCN21 (5 μm) (∼50-fold IC50 of CaMKII inhibition) in the perfusion solution did not affect presynaptic measures, such as paired pulse facilitation or fiber volleys (Fig. 4B,C; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). However, tatCN21 blocked generation of LTP when present during its induction by HFS (Fig. 4A; supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material), demonstrating effectiveness of the inhibitor. By contrast, when applied 15 min after HFS, tatCN21 had no effect on maintenance of LTP (Fig. 4D; supplemental Fig. 5C,D, available at www.jneurosci.org as supplemental material). These results indicate that neither stimulated nor autonomous activity is required for LTP maintenance. Combined with previous studies in CaMKII T286A mutant mice (Giese et al., 1998), the results also indicate that LTP induction requires not only stimulated but also autonomous CaMKII activity.
Figure 4.
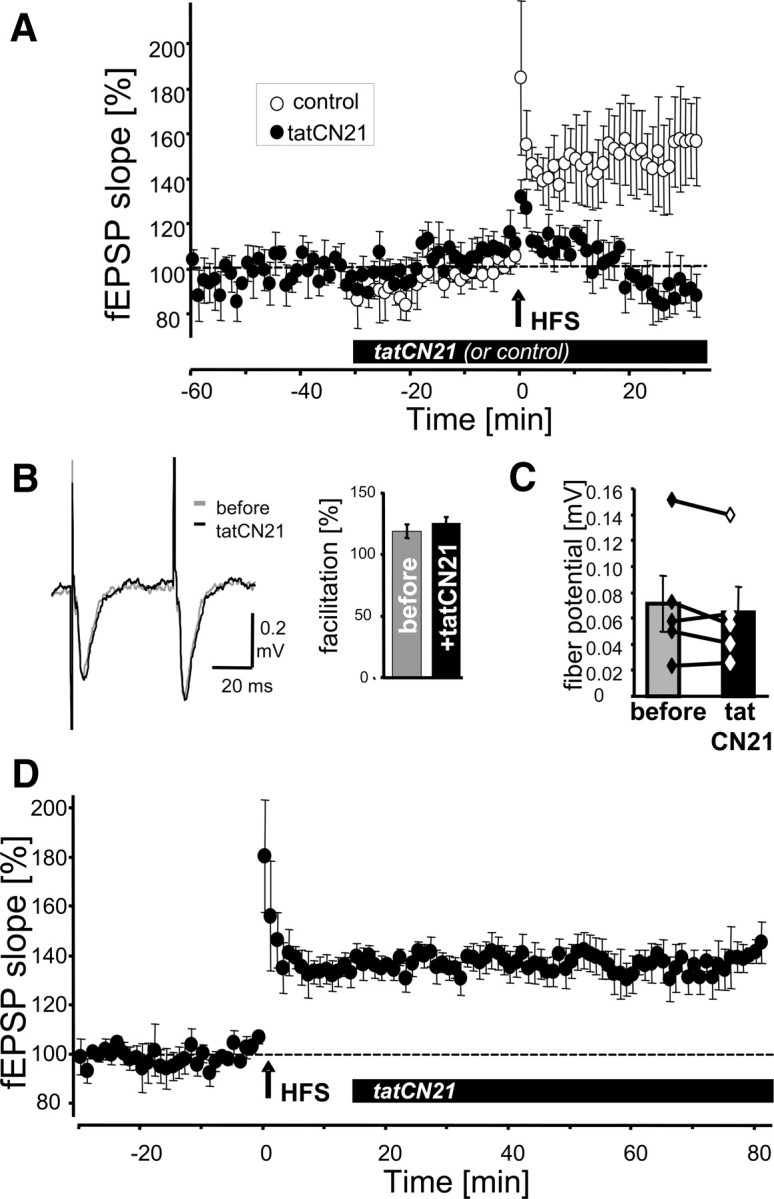
tatCN21 blocks induction but not maintenance of LTP. A, Perfusion of acute hippocampal slices (from 6- to 7-week-old rats) with tatCN21 (5 μm) did not affect basal transmission in CA1 but blocked LTP induction by high-HFS (2 × 1 s, 100 Hz) (n = 5), compared with untreated control (n = 5). fEPSP slope over time is shown as percentage of baseline. B, Paired pulse facilitation was the same before and after 20 min perfusion with tatCN21. Shown are sample traces from one experiment (left) and quantification of five experiments (right). C, Presynaptic fiber potential was unaffected by 20 min perfusion with tatCN21 (p > 0.2, paired t test). D, tatCN21 (5 μm) perfusion 15 min after LTP induction by HFS did not interfere with LTP maintenance. fEPSP slope is shown as percentage of baseline.
tatCN21 inhibits acquisition but not storage/retrieval of contextual fear memory
As tat fusion peptides penetrate the blood brain barrier (Schwarze et al., 1999; Aarts et al., 2002), tatCN21 also provided a means to examine the effects of inhibiting autonomous CaMKII activity on learning versus memory in vivo. The learning paradigm chosen was a form of hippocampus- and NMDA-receptor-dependent contextual fear conditioning in mice (Young et al., 1994). This paradigm is very robust and it includes important behavioral controls that can distinguish between effects directly on learning versus effects on other behaviors required for learning, thereby minimizing the potential for confounding drug effects not directly on learning and memory itself. Moreover, CaMKII has been shown to be involved in this behavioral task (Silva et al., 1992a; Rodrigues et al., 2004; Irvine et al., 2005). Briefly, during a training-session, mice received a foot-shock within a conditioning chamber, and, during a test session 24 h later, freezing behavior was measured in response to placement into the chamber again, but without foot-shock (Fig. 5A). tatCN21 injection did not affect baseline freezing or freezing immediately after the shock during the training session itself (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). However, as expected for CaMKII inhibition, systemic application of tatCN21 but not the scrambled sequence control tatCtrl (10 mg/kg, i.p.), 1 h before the training session significantly reduced learning, as measured by reduced freezing responses in the test session (Fig. 5B). In contrast, tatCN21 injection 1 h before the test session had no effect on freezing behavior (Fig. 5C). This finding indicates that pretraining injection of tatCN21 did not affect fear mechanisms, but instead directly interfered with memory formation. More importantly, these data show that pre test injection of tatCN21 did not erase established memory and did not interfere with its retrieval.
Figure 5.

tatCN21 interfered with learning but not memory in a hippocampus-dependent contextual fear conditioning paradigm. A, Intraperitoneal injection of tatCN21 (10 mg/kg) was done either 1 h before the training session (pre) or the test session 24 h after training (post). B, tatCN21 (n = 10) injection before the training session significantly reduced freezing behavior in the test session, compared with saline (n = 14) or the tatCtrl scrambled sequence control (n = 7) (**p < 0.05 in Bonferroni's multiple-comparisons test, after ANOVA with p < 0.023) (left). C, Injection of tatCN21 (n = 12) before the test session did not affect freezing behavior compared with injection of saline (n = 10) and tatCtrl control (n = 8) (p > 0.6, ANOVA) (right). Error bars indicate SEM in all panels.
Discussion
The necessity of CaMKII in LTP and in learning/memory is well established (Malinow et al., 1989; Silva et al., 1992a,b), and T286A mutant mice demonstrated involvement of autonomous CaMKII activity (Giese et al., 1998; for review, see Malenka and Nicoll, 1999; Lisman et al., 2002; Lee and Silva, 2009). Our results implicate the function of CaMKII autonomy as computational device (as discussed below) necessary for LTP induction and learning, but not (as was previously proposed) as a long-term storage device involved in LTP maintenance or memory retention. As an important byproduct, our results provide a cautionary example that fusion of peptides can generate unexpected and undesired “gain-of-function”, in this case compromising the selectivity of a previously used CaMKII inhibitor. Undesired gain-of-function by fusion is a well accepted principle in cancer biology (with the prominent fusion example of bcr-abl; for review, see Hantschel and Superti-Furga, 2004; Weisberg et al., 2007). However, in designing fusion proteins or peptides as research tools, only “loss-of-function” is generally considered. Obviously, it is much harder to actually test for a specific gain-of-function (without knowledge which function this may be), but our cautionary example should at least encourage to carefully evaluate results obtained with fusion peptides, especially with ant/penetratin, which was found to bind to Ca2+/CaM even on its own.
The “systems functions” of CaMKII autonomy in LTP induction and learning (but not LTP maintenance or memory storage) are remarkably well paralleled by the well described biochemical properties of CaMKII, which support molecular short-term memory and computation (but not long-term molecular memory). Generation of autonomy by T286 autophosphorylation requires binding of at least two Ca2+/CaM to neighboring subunits of a holoenzyme: binding of one CaM to the subunit acting as kinase, to activate it, binding of the other CaM to the subunit acting as substrate, to make T286 accessible for phosphorylation (Hanson et al., 1994; Rich and Schulman, 1998), as T286 is buried in the structure of the inactive kinase (Rosenberg et al., 2005). This mechanism does not allow long-term molecular memory (beyond the activation of phosphatases), as rephosphorylation of T286 can only occur during a Ca2+-stimulus, due to the substrate-directed role of CaM. Thus, CaMKII does not fulfill the requirements for memory storage by a self-perpetuating kinase system originally postulated by John Lisman (1985) before the discovery of CaMKII autonomy (Lou et al., 1986; Miller and Kennedy, 1986; Schworer et al., 1986). However, the dual function of CaM in T286 phosphorylation bestows short-term molecular memory and the ability to decode oscillation frequencies of Ca2+ signals upon CaMKII, with autonomy generated much more readily by high-frequency stimulations (De Koninck and Schulman, 1998; Bayer et al., 2002). For a synapse, high stimulation frequencies induce LTP, while low-frequency stimulation can actually induce long term depression (LTD) (for review, see Malenka and Bear, 2004; Kessels and Malinow, 2009). Remarkably, according to this study, the requirement for autonomous CaMKII activity in LTP (Giese et al., 1998) is indeed in the induction phase rather than in maintenance, with frequency dependence of LTP induction again paralleled by the biochemical frequency dependence of generation of CaMKII autonomy.
But why is T286 autophosphorylation critical in LTP induction and memory acquisition, if CaMKII is also activated by Ca2+/CaM-binding alone? Several abundant synaptic proteins compete for CaM binding, including CaMKII and the phosphatase calcineurin (PP2B), and T286-phosphorylation transforms CaMKII from one of the lowest to one of the highest affinity CaM-targets within the cell (Meyer et al., 1992). Individual CaM molecules do not dissociate from T286-phosphorylated CaMKII during a Ca2+-stimulus, while they rapidly dissociate and reassociate from un-phosphorylated CaMKII (Meyer et al., 1992). Thus, synaptic CaMKII may reach its full activity only after T286 autophosphorylation. Additionally, even a relatively short-term extension of only partial CaMKII activity beyond the initial Ca2+-stimulus may significantly overcome the counteraction by calcineurin, which is also Ca2+-activated and highly abundant at the synapse (for review see Xia and Storm, 2005). Additionally, while T286 phosphorylation is not required for binding to the NMDA-type glutamate receptor subunit NR2B and synaptic translocation seen after stimulation (Shen and Meyer, 1999; Bayer et al., 2001), it does increase the amount of CaMKII that persists at synapses after the stimulus (Bayer et al., 2006). Indeed, such translocation occurs during LTP (Otmakhov et al., 2004) and binding to NR2B regulates plasticity (Barria and Malinow, 2005). The differences in both activity and localization between CaMKII wild type and T286A are not dramatic, but together, they appear to have a profound effect on LTP induction and ability to form memories.
Previous studies on LTP maintenance also saw no effect of other inhibitors, which were derived from the CaMKII autoinhibitory region (Malinow et al., 1989; Otmakhov et al., 1997; Chen et al., 2001). However, one of these reports (Chen et al., 2001) found a very low potency of this inhibitor (IC50 of ∼30 μm) (Chen et al., 2001). Later studies by the same group using antCN27 were thought to indicate a role of autonomy in LTP maintenance, and elegantly circumvented the problem that antCN27 directly caused significant rundown of transmission (Sanhueza et al., 2007). However, both rundown and the LTP maintenance effect are caused by off-target effects created by the ant fusion in the antCN27 inhibitor, as other CaMKII inhibitors, including tatCN21, do not result in any rundown (while effectively inhibiting LTP induction; in this study and in refs. Malinow et al., 1989; Otmakhov et al., 1997; Chen et al., 2001). Remarkably, the only exceptions were CaMKII inhibitors that additionally bind Ca2+/CaM (Waxham et al., 1993), a side-effect demonstrated here for antCN27. Importantly, tatCN21 proved suitable even for systemic in vivo application, enabling assessment of autonomy involvement in learning versus memory. We used contextual fear conditioning in the studies described here because with this behavioral paradigm it is possible to distinguish between the effects on the acquisition (i.e., learning) or stability of information (i.e., memory). Additionally, this learning paradigm has been shown to require hippocampal NMDA receptor activity (Young et al., 1994) and CaMKII autophosphorylation at T286A (Irvine et al., 2005). Indeed, tatCN21 significantly reduced learning when injected before training, but did not affect memory storage or retrieval when injected later, before the test session. Thus, CaMKII autonomy is not required for storing contextual conditioning information, neither in hippocampus nor amygdala (the two brain regions known to be involved). In this learning task, T286A mutant mice were able to overcome impaired conditioning by more intensive training (Irvine et al., 2005), also consistent with involvement of CaMKII autonomy in learning but not in storage of memory once it is formed. A potential caveat was that more extensive training with repeated stimuli may evoke a different form of memory (possibly correlated to human nondeclarative memory), while memory after rapid learning (possibly correlated to declarative memory) could still have been dependent on CaMKII autonomy. However, combination of this genetic approach and our pharmacological results, together with strong arguments based on the biochemical mechanism of autonomy, provide very convincing evidence that phospho-T286-induced CaMKII autonomy is indeed important for memory acquisition but not storage. In further support of the widely accepted notion of LTP as an important cellular template for learning and memory (for review, see Lee and Silva, 2009) is the finding of a similar requirement of autonomy in LTP induction but not maintenance.
Could CaMKII store memories by mechanisms other than its autonomy? Two alternative mechanisms have been proposed previously: (1) T286 autophosphorylation effects other than autonomy, such as CaM sequestration (Meyer et al., 1992; Deisseroth et al., 1995) or directly changing charge distribution synapses (Tompa and Friedrich, 1998). Application of tatCN21 after LTP induction would neither cause T286 dephosphorylation nor reduce CaM affinity (Vest et al., 2007). However, remarkably, based on its inhibitory mechanism, T286 autophosphorylation is the only CaMKII reaction that tatCN21 would not efficiently inhibit during LTP induction (Vest et al., 2007) (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, as tatCN21 efficiently inhibits LTP induction and learning, T286 autophosphorylation alone (without generation of CaMKII activity) is not sufficient to establish LTP. Furthermore, the regulation of T286 autophosphorylation does not support long-term storage, as discussed above. (2) Another possible mechanism for memory storage could be a stimulus-induced persistent change in the synaptic localization of CaMKII by regulated binding to the NMDA-receptor subunit NR2B (Shen and Meyer, 1999; Strack et al., 2000; Bayer et al., 2001, 2006). Indeed, CaMKII translocates during LTP (Otmakhov et al., 2004) and its binding to NR2B is important for synaptic plasticity (Barria and Malinow, 2005). However, while 5 μm tatCN21 efficiently inhibits the binding of CaMKII to NR2B (Vest et al., 2007), it does not reverse the interaction once fully established (Vest et al., 2010). Thus, our experiments do not rule out the possibility that persistent binding of CaMKII to NR2B may participate in the maintenance of LTP and/or storage of neuronal memory.
In summary, our findings implicate CaMKII autonomy a computational rather than a storage mechanism. While CaMKII autonomy does not provide long-term memory storage itself, it constitutes a form of molecular short-term memory that allows processing of temporal stimulation patterns and is indeed required for functional generation of long-term memory.
Footnotes
The research was supported by National Institutes of Health Grants P30NS048154 (University of Colorado Denver Center grant), R01NS040701 (to M.D.A.), R37AG013622 (to A.J.S.), and R01NS052644 (to K.U.B.). TA-CaM was a kind gift from Katalin Török (St. George's University of London).
The University of Colorado is currently seeking patent protection for tatCN21.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Josselyn SA, Frankland PW, Silva AJ. Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice. Learn Mem. 2000;7:58–72. doi: 10.1101/lm.7.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Schulman H. Alternative splicing modulates the frequency-dependent response of CaMKII to Ca(2+) oscillations. EMBO J. 2002;21:3590–3597. doi: 10.1093/emboj/cdf360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke TA, Lüthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- Coultrap SJ, Buard I, Kulbe JR, Dell'Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/CaM. J Biol Chem. 2010;285:17930–17937. doi: 10.1074/jbc.M109.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Schulman H, Tsien RW. Synaptic plasticity: a molecular mechanism for metaplasticity. Curr Biol. 1995;5:1334–1338. doi: 10.1016/s0960-9822(95)00262-4. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng TP. The involvement of PKC and multifunctional CaM kinase II of the postsynaptic neuron in induction and maintenance of long-term potentiation. Prog Brain Res. 1995;105:55–63. doi: 10.1016/s0079-6123(08)63283-5. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Bayer KU, Di Matola T, Fenzi G, Rossi G, Vitale M. Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem. 2003;278:45101–45108. doi: 10.1074/jbc.M305355200. [DOI] [PubMed] [Google Scholar]
- Irvine EE, Vernon J, Giese KP. AlphaCaMKII autophosphorylation contributes to rapid learning but is not necessary for memory. Nat Neurosci. 2005;8:411–412. doi: 10.1038/nn1431. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci U S A. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lou LL, Lloyd SJ, Schulman H. Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc Natl Acad Sci U S A. 1986;83:9497–9501. doi: 10.1073/pnas.83.24.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Ogasawara H, Kawato M. Bistable switches for synaptic plasticity. Sci Signal. 2009;2:pe7. doi: 10.1126/scisignal.256pe7. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Rich RC, Schulman H. Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1998;273:28424–28429. doi: 10.1074/jbc.273.43.28424. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci U S A. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–5199. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman H, Greengard P. Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature. 1978;271:478–479. doi: 10.1038/271478a0. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Schworer CM, Colbran RJ, Soderling TR. Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J Biol Chem. 1986;261:8581–8584. [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Tompa P, Friedrich P. Synaptic metaplasticity and the local charge effect in postsynaptic densities. Trends Neurosci. 1998;21:97–102. doi: 10.1016/s0166-2236(97)01176-4. [DOI] [PubMed] [Google Scholar]
- Török K, Tzortzopoulos A, Grabarek Z, Best SL, Thorogate R. Dual effect of ATP in the activation mechanism of brain Ca(2+)/calmodulin-dependent protein kinase II by Ca(2+)/calmodulin. Biochemistry. 2001;40:14878–14890. doi: 10.1021/bi010920+. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O'Leary H, Port JD, Bayer KU. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O'Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel CaMKII inhibitor. J Biol Chem. 2010 doi: 10.1074/jbc.M109.008617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. The balance between postsynaptic Ca(2+)-dependent protein kinase and phosphatase activities controlling synaptic strength. Learn Mem. 1996;3:170–181. doi: 10.1101/lm.3.2-3.170. [DOI] [PubMed] [Google Scholar]
- Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Fujisawa H. Evidence for three distinct forms of calmodulin-dependent protein kinases from rat brain. FEBS Lett. 1980;116:141–144. doi: 10.1016/0014-5793(80)80628-4. [DOI] [PubMed] [Google Scholar]
- Young SL, Bohenek DL, Fanselow MS. NMDA processes mediate anterograde amnesia of contextual fear conditioning induced by hippocampal damage: immunization against amnesia by context preexposure. Behav Neurosci. 1994;108:19–29. doi: 10.1037//0735-7044.108.1.19. [DOI] [PubMed] [Google Scholar]