

Abstract
Stimulation of hepatic stellate cells (HSCs) by platelet-derived growth factor (PDGF) and transforming growth factor-β1 (TGF-β1) is an essential pathway of proliferation and fibrogenesis, respectively, in liver fibrosis. We provide evidence that PTK787/ZK222584 (PTK/ZK), a potent tyrosine kinase inhibitor that blocks vascular endothelial growth factor receptor (VEGFR), significantly inhibits PDGF receptor expression, as well as PDGF-simulated HSC proliferation, migration and phosphorylation of ERK1/2, Akt and p70S6 kinase. Interestingly, PTK/ZK also antagonizes the TGF-β1-induced expression of VEGF and VEGFR1. Furthermore, PTK/ZK downregulates TGF-β receptor expression, which is associated with reduced Akt, ERK and p38MAPK phosphorylation. Furthermore, PDGF-induced TGF-β1 expression is inhibited by PTK/ZK. These findings provide evidence that PTK/ZK targets multiple essential pathways of stellate cell activation that provoke proliferation and fibrogenesis. Our study underscores the potential use of PTK/ZK as an antifibrotic drug in chronic liver disease.
Keywords: Akt, HSC, PDGF, PTK787/ZK22258, TGF-β1
Liver fibrosis is a pathological response of the liver to a variety of chronic stimuli. Hepatic stellate cells (HSCs) play an essential role in the development of liver fibrosis. After chronic liver injury, HSCs undergo a process of activation, developing a myofibroblast-like phenotype that proliferates and becomes fibrogenic1,2 and produces increased extracellular matrix proteins.3
Several cytokine mediates are central to the fibrotic process, including platelet-derived growth factor (PDGF) and transforming growth factor-β1 (TGF-β1). PDGF is the most potent proliferative cytokine toward HSCs, whereas TGF-β1 primarily functions in the stimulation of extracellular matrix production.3 In the liver, expression of PDGF and its receptors is increased both in experimental fibrosis in rats and in human fibrotic liver.4–6 Both PDGF-B and PDGFR-β are rapidly induced in vivo and in culture.4,7 Moreover, the genetic overexpression of PDGF leads to liver fibrosis in mice.8 Remarkably, very few studies have assessed the impact of PDGF antagonism on hepatic fibrosis.9 Recent reports using imatinib mesylate (Gleevec) show promise,10–12 but it is uncertain whether PDGFR is its main target of action in these studies.
TGF-β1 is the most potent stimulus to hepatic fibrogenesis.13 Increased levels of TGF-β have been described in chronic liver diseases, and activated HSCs represent a major cellular source of TGF-β in injured liver. In HSCs, TGF-β promotes HSC transformation into myofibroblasts, simulates the synthesis of extracellular matrix proteins and inhibits their degradation. Antagonism of TGF-β1 signaling pathways markedly decreases fibrosis in experimental models.14,15
Because of their combined roles in fibrosis, inhibiting PDGF and TGF-β1 signal transduction is an attractive target for antifibrotic therapy. An emerging strategy is to generate small-molecule inhibitors of receptor tyrosine kinase domains. PTK787/ZK22258 (PTK/ZK) is a potent tyrosine kinase inhibitor of both vascular endothelial growth factor receptor 1 (VEGFR1, also named Flt-1) and VEGFR2 (Flk-1), and also inhibits the tyrosine kinase activity of PDGFR-β, Flt-4, c-kit and c-fms, although with less potency.16 PTK/ZK inhibits endothelial cell migration and proliferation without cytotoxic or antiproliferative effects on cells that do not express VEGF receptors.16 Oral administration of PTK/ZK at a dose of 25–100 mg/kg/day was previously shown to inhibit tumor growth in human cancer xenografts, including hepatocellular carcinoma.16–18 Very recently, we reported that PTK/ZK inhibited liver fibrosis in mice and downregulated stellate cell activation.19 In this study, we uncover the molecular mechanisms of PTK/ZK in attenuating HSC activation.
MATERIALS AND METHODS
Drug
PTK/ZK (succinate salt) was provided by Novartis Pharma AG. (Basel, Switzerland). A stock solution of 50 mM PTK/ZK was prepared in DMSO, and the concentration of DMSO for all assays did not exceed 0.1%.18,19 PTK/ZK (dihydrochloride salt) was synthesized as previously described.20 Dihydrochloride salt was dissolved in distilled water.
HSC Isolation and Culture
HSCs were purified from normal rats purchased from the Laboratory Animal Unit. Nonparenchymal cell suspension was obtained by a single-step density gradient centrifugation with Nycodenz, characterized and cultured as described in detail previously.21 Experimental manipulations were performed with cells at passage 4–7. Research protocol was approved by the Institutional Ethics Committee.
Effect of PTK/ZK on PDGFR-β Expression on HSCs by Flow Cytometry Analysis
HSCs were pretreated overnight with PTK/ZK at various concentrations before labeling for PDGFR-β antibody. HSCs were incubated with PDGFR-β antibody (R&D Systems, Minneapolis, MN, USA) for 45 min at 4°C, washed with ice-cold PBS and then incubated with anti-mouse PE (BD PharMingen, San Diego, CA, USA) for 30 min. Cells were washed and then subjected to flow cytometry analysis by FACS calibur (Becton Dickinson, San Jose, CA, USA). Mouse IgG1 isotype (R&D Systems) was included as a negative control.
Effects of PTK/ZK on PDGF-Induced HSC Proliferation
Proliferation of HSCs was measured by bromodeoxyuridine (BrdU) incorporation using a BrdU labeling and detection kit (Roche Diagnostics Corporation, Indianapolis, IN, USA). Cells were plated at a density of 2 × 103 cells/well into 96-well plates and were cultured overnight, followed by washing of cells with PBS twice and replacing the growth medium with a medium containing 0.1% FBS. PTK/ZK in serial dilutions was added 3 h before PDGF (10 ng/ml) (R&D Systems) and incubated with cells for 48 h. BrdU labeling solution was added to cells, followed by incubation for another 16 h before fixation, and addition of nucleases, anti-BrdU-POD and peroxidase substrate. The absorbance at 405 nm (with a reference wavelength at 490 nm) was measured using an ELISA plate reader (Molecular Devices Corp., Sunnyvale, CA, USA).
HSC Migration Assay
The migratory capacity of HSCs was investigated using a BIOCOAT MATRIGEL Invasion Chamber (Becton Dickinson). Confluent HSCs at the top chamber were incubated in serum-free medium for 24 h. The lower chamber was filled with PDGF (10 ng/ml) in the presence or absence of PTK/ZK at incremental concentrations. After incubation for 24 h, cells from the upper surface of membranes were completely removed with gentle swabbing. The remaining migrated cells on the lower surface of membranes were fixed and stained with hematoxylin and eosin. Cellular migration was determined by counting the number of stained cells on membranes in five randomly selected fields at high power. HSCs that migrated through the membrane were enumerated by flow cytometry as described before.22
HSC Wound-Healing Assay
For measurement of cell migration during wound healing, rat HSCs were seeded in 6-well plates and grown to confluence in a growth medium containing 10% FBS. Confluent HSCs were deprived of serum for 24 h, and then disrupted to generate a linear wound, followed by incubation in a medium containing PDGF (10 ng/ml) (R&D Systems) in the presence or absence of increasing doses of PTK/ZK for 20 h. HSCs were subsequently fixed and observed under phase contrast microscopy. For evaluation of wound closure under different experimental conditions, five randomly selected points along each wound were marked, and the vertical distance of migrating cells from the initial wound was measured. Experiments were carried out in duplicate, and five fields of each well were recorded.
Western Blot
For analysis of the expression of PDGFR-β, TGF-β type I receptor (TGFβ RI) and TGF-β type II receptor (TGFβ RII), HSCs were treated with PTK/ZK at incremental concentrations in the medium with 1% FBS for 24 h and washed twice with PBS before lysis. To evaluate the effect of TGF-β1 on the phosphorylation of Smad2, Akt, ERK and p38MAPK in HSCs, HSCs were serum deprived overnight, and then stimulated with TGF-β1 at various concentrations for 20 min. To examine the effect of PTK/ZK on PDGF-induced expression of ERK, Akt and p70S6 kinase, or TGF-β1-induced expression of Akt, ERK and p38MAPK, HSCs were serum deprived overnight, and were then treated with PTK/ZK for 3 h, followed by stimulation with PDGF 10 ng/ml or TGF-β1 1 ng/ml for 10 or 20 min, respectively, before lysis. To detect the Flt-1 expression induced by TGF-β, HSCs grown to subconfluence were rendered quiescent in serum-free medium for 24 h, followed by treatment with TGF-β1 at various concentrations for 20 h. In the experiments using PTK/ZK, cells were preincubated for 4 h in the absence or presence of 5 μM, or 10 μM PTK/ZK, before treatment with TGF-β1. After blocking, membranes were immunoblotted with the following antibodies: PDGFRβ, TGF-β1, TGFβRI, TGFβRII, Smad2 (Santa Cruz Biotechnology), ERK, phospho-ERK, Akt, phospho-Akt, p70S6 kinase, phospho-p70S6 kinase, p38 MAPK, phospho-p38 MAPK, Raf, phospho-Raf (Cell Signaling) and phospho-Smad2 (Chemicon) for 18 h at 4°C, followed by detection using an HRP-conjugated secondary antibody (Santa Cruz Biotechnology) (1 h at room temperature). Immunoreactive protein bands were visualized by the ECL system (Amersham Biosciences).
Analysis of VEGF and Flt-1 mRNA Expression in HSCs
HSCs were cultured for 24 h in a serum-free medium and were then stimulated with TGF-β1 for 12 h at indicated concentrations. For the effect of PTK/ZK, cells were incubated with PTK/ZK at 5 μM, or 10 μM PTK/ZK for 4 h, before treatment with TGF-β1. Total RNA was isolated from HSCs using an RNAeasy Mini kit (QIAGEN Inc. Hilden, Germany), according to the manufacturer’s instructions. For cDNA synthesis, Taqman reverse transcription reagents were used as described in the manufacturer’s protocol (PE Applied Biosystems, Foster City, California, USA). The mRNA expression of VEGF and Flt-1 was evaluated using quantitive real-time PCR using TaqMan Gene Expression Assays and Taqman Universal PCR Master Mix kit (PE Applied Biosystems) on a PE Applied Biosystems 7700 Sequence Detector. The amplification conditions on the PE Applied Biosystems 7700 sequence detector were the following: 2 min at 50°C, 10 min at 95°C, 40 cycles of 95°C for 15 s and 60°C for 1 min for all reactions. Relative gene expression was calculated using 18S as internal control.
Statistical Analysis
Continuous data were expressed as mean±standard error (s.e.) of mean. One-way ANOVA was used where appropriate. A P-value <0.05 was considered as statistically significant.
RESULTS
PTK/ZK Downregulates PDGFR-β Expression in HSCs
Flow cytometry analysis on gated active cells (Figure 1a) confirmed the expression of PDGFR-β by HSCs (Figure 1b), as determined by both the percentage of PDGFR-β-positive cells and PDGFR-β expression levels (mean channel fluorescence) (Figure 1c); PDGFR-β expression was confirmed using western blot (Figure 1d). PTK/ZK treatment significantly suppressed the PDGFR-β expression as measured by both flow cytometry (Figure 1b and c) and western blot (Figure 1d).
Figure 1.

PTK/ZK inhibited the expression of PDGFRβ on HSCs. (a) HSCs were treated with PTK/ZK at 5, 10 or 20 μM overnight and were labeled with anti-PDGFRβ, followed by anti-mouse PE and then analyzed by flow cytometry. Analyzing on gated active HSCs (a), PDGF-induced upregulation of PDGFRβ was inhibited by PTK/ZK. Representative data are shown in b. Expression of PDGFRβ was also presented as percentages of cells positive for the receptor and as mean channel fluorescence (expression levels) on gated active HSCs. PTK/ZK significantly inhibited the surface expression of PDGFRβ (c). Data are presented as mean±s.e. from three experiments, *P<0.05, **P<0.01. (d) Expression of PDGFRβ in total cell extracts was determined by western blot analysis. PTK/ZK significantly suppressed the PDGFRβ expression. The results of a representative study are shown. Two additional experiments yielded similar results.
PTK/ZK Inhibits PDGF-Induced HSC Proliferation and Migration
As shown in Figure 2a, PDGF induced a marked increase in HSC proliferation, which was significantly inhibited by PTK/ZK in a dose-dependent manner. Incubation of HSCs with PDGF also dramatically increased HSC migration in BIOCOAT MATRIGEL chamber systems, whereas PTK/ZK significantly blocked the migration of HSC induced by PDGF (Figure 2b, P<0.01). Figure 2c shows wounded HSC monolayers incubated in a medium containing PDGF without or with PTK/ZK. PDGF induced cell migration leading to wound closure 20 h after wounding. In the presence of PTK/ZK, PDGF-induced HSC migration in the wounded area was inhibited.
Figure 2.

Effect of PTK/ZK on HSC proliferation and migration induced by PDGF. (a) HSCs were cultured overnight in medium with 0.1% FBS before the addition of PTK/ZK at various concentrations, followed by PDGF (10 ng/ml) stimulation. Cell proliferation was measured by BrdU incorporation. Each experiment was carried out in quadruplicate and the data represent the mean±s.e. of six experiments. (b) Effect of PTK/ZK on HSC migration induced by PDGF. HSCs were incubated in serum-free medium for 24 h in the upper compartment of the BIOCOAT Chamber and were examined for migration in the lower compartment in the presence of various concentrations of PTK/ZK with or without PDGF. Cells that migrated to the lower compartment were quantified by cell counting. Data are expressed as a percentage of control from three experiments. **P<0.01. (c) Wound-healing assay. HSCs were grown to confluence and were disrupted to generate a linear wound, and then incubated in medium containing PDGF in the presence or absence of PTK/ZK for 20 h. Experiments were carried out in duplicate.
PTK/ZK Treatment Inhibits PDGF-Induced Activation of Raf, ERK1/2, Akt and p70S6 Kinase
To assess the effect of PTK/ZK on Raf, ERK1/2, Akt and p70S6 kinase phosphorylation in HSCs, cultured activated HSCs were serum starved for 24 h and were then treated with 5 or 10 μM PTK/ZK for 3 h. Cells were subsequently stimulated with 10 ng/ml PDGF for 10 min. Western blot analysis showed that 10 ng/ml PDGF led to phosphorylation of Raf, ERK1/2, Akt and p70S6 kinase. PTK/ZK significantly blocked Raf, ERK1/2, Akt and p70S6 kinase phosphorylation (Figure 3).
Figure 3.

Effect of PTK/ZK on Raf, ERK, Akt and p70S6 kinase activation induced by PDGF in HSCs. HSCs were treated for 3 h with PTK/ZK where indicated, and then stimulated with 10 ng/ml PDGF for 10 min. Cell lysates were analyzed for total Raf and phospho-Raf, total ERK and phospho-ERK, total Akt and phospho-Akt, total p70S6 kinase and phospho-p70S6 kinase levels by western blot. A representative study from three experiments is shown.
PTK/ZK Inhibits VEGF mRNA Induced by TGF-β1
Based on its importance as a fibrogenic cytokine, we examined the effect of TGF-β1 on the expression of VEGF mRNA in HSCs. HSCs treated with TGF-β1 expressed higher VEGF mRNA (Figure 4a). PTK/ZK antagonized the augmented VEGF mRNA expression induced by TGF-β1 (Figure 4b).
Figure 4.

Analysis of VEGF gene expression in TGF-β1-treated HSCs. (a) HSCs were cultured for 24 h in a serum-free medium and were then stimulated with TGF-β1 for 12 h at indicated concentrations. Total RNA was isolated from HSCs using an RNAeasy Mini kit, and VEGF gene expression was measured by real-time PCR. TGF-β1 significantly induced VEGF mRNA expression. VEGF values were normalized to 18S mRNA values. Data are expressed as mean±s.e., *P<0.05. (b) Effect of PTK/ZK on VEGF mRNA induced by TGF-β1. PTK/ZK at 5 or 10 μM concentration was added 4 h before TGF-β1 (1 ng/ml). The VEGF gene expression level was significantly suppressed by PTK/ZK. Data are presented as mean±s.e., *P<0.05.
PTK/ZK Reduces Flt-1 mRNA and Protein Expression Induced by TGF-β1
Analyzed by real-time PCR, Flt-1 mRNA expression increased significantly after TGF-β1 treatment compared with that in untreated cells (Figure 5a). This induction was significantly suppressed by PTK/ZK (Figure 5b). The results were further confirmed at the protein level; TGF-β1-induced upregulation of Flt-1 expression was suppressed by PTK/ZK (Figure 5c and d).
Figure 5.

(a) TGF-β1 stimulated Flt-1 mRNA expression in HSCs. HSCs were deprived of serum for 24 h before incubation with TGF-β1 at various concentrations for 12 h. Total RNA was extracted and Flt-1 expression was determined by real-time PCR. TGF-β1 significantly induced Flt-1 mRNA expression. Flt-1 values were normalized to 18S mRNA values (a). Data are presented as mean±s.e., *P<0.05. (b) Flt-1 mRNA expression induced by TGF-β1 was inhibited by PTK/ZK. HSCs were treated with PTK/ZK at 5 or 10 μM for 4 h before stimulation with TGF-β1. Flt-1 mRNA expression by HSCs was measured by real-time PCR. PTK/ZK suppressed Flt-1 mRNA expression induced by TGF-β1 in HSCs. Results shown are normalized to 18S mRNA. Data are mean±s.e., *P<0.05. (c) TGF-β1 upregulated Flt-1 protein expression in HSCs. Primary cultured HSCs were starved for 24 h in serum-free medium, and were then incubated with TGF-β1 at incremental concentrations for 20 h. HSCs were extracted with a cell lysis buffer and analyzed by western blot. The Flt-1 protein level was significantly upregulated by TGF-β1. The results of a representative study are shown. Two additional experiments yielded similar results. (d) PTK/ZK reduced Flt-1 protein expression induced by TGF-β1. PTK/ZK was added in growth-arrested HSCs 4 h before TGF-β1, followed by TGF-β1 induction for 20 h. Cell lysis from HSCs was subjected to western blot analysis, and the results shown are a representative study from three experiments.
PTK/ZK Downregulates both TGFβRI and TGFβRII Expression in HSCs
Using western blot analysis, we detected the expression of both TGFβRI and TGFβRII in HSCs (Figure 6). PTK/ZK treatment resulted in the downregulation of TGFβRI and TGFβRII (Figure 6).
Figure 6.
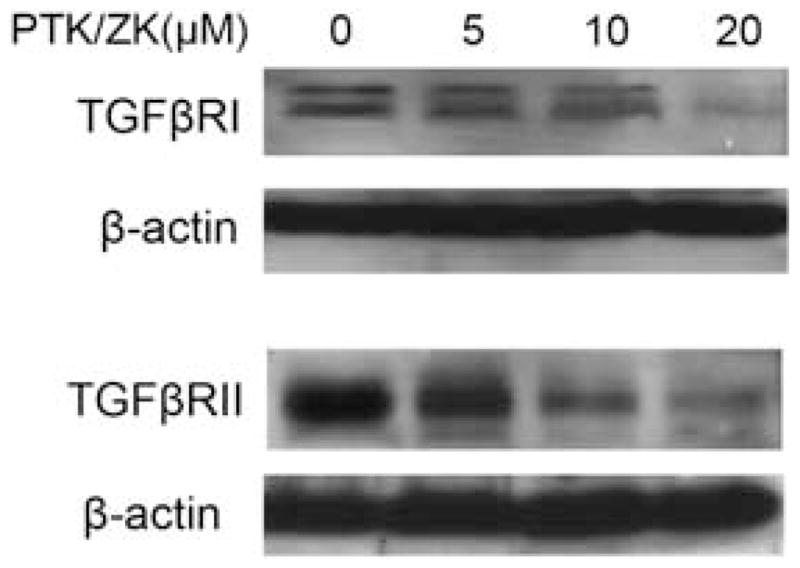
Effect of PTK/ZK on the expression of TGFβRI and TGFβRII in HSCs. HSCs were treated with PTK/ZK at indicated concentrations and cultured in the medium containing 1% FBS for 24 h. Cell lysates were subjected to western blot for analysis of TGFβRI and TGFβRII proteins. A representative study from three experiments is shown.
PTK/ZK Suppresses the TGF-β1-Stimulated Phosphorylation of Akt, ERK and p38MAPK, but not Smad2 in HSCs
We observed that TGF-β1 signaled through Akt, ERK and p38MAPK pathways (Figure 7a), consistent with earlier evidence indicating that TGF-β1 activates the PI3K-Akt, ERK and p38MAPK pathways in stellate cells.23–25 In HSCs exposed to TGF-β1, PTK/ZK inhibited Akt, ERK and p38MAPK phosphorylation (Figure 7b). Although Smads are the preferred substrates and signal transducers of TGF-β receptors, PTK/ZK did not show a significant inhibition of Smad2 phosphorylation induced by TGF-β (Figure 8a and b).
Figure 7.

PTK/ZK inhibited Akt, ERK and p38MAPK phosphorylation in TGFβ1-induced HSCs. (a) HSCs were serum starved for 24 h and were then exposed to TGF-β1 at different concentrations for 20 min. (b) HSCs were pretreated with PTK/ZK at 5 or 10 μM for 3 h, and then exposed to 1 ng/ml of TGF-β1 for 20 min. Cell lysates were subjected to SDS-PAGE, followed by western blot analysis using antibodies against phospho-Akt, Akt, phospho-ERK, ERK, phospho-p38MAPK and p38MAPK. The SDS-PAGE profile represents one experiment out of three.
Figure 8.

PTK/ZK inhibited the activation of HSC induced by TGF-β1 independent of Smad2. (a) HSCs were cultured in a serum-free medium for 24 h and were then stimulated with TGF-β1 at incremental concentrations for 20 min. The expression of phospho-Smad2 and Smad2 in total cell extracts was determined by western blot analysis. Results are from a representative study. Two additional experiments yielded similar results. (b) HSCs were incubated with PTK/ZK at 5 or 10 μM for 3 h, and were then stimulated with 1 ng/ml of TGF-β1 for 20 min. Cell lysates were subjected to SDS-PAGE, followed by western blot analysis using antibodies against phospho-Smad2 and Smad2. The SDS-PAGE profile represents one experiment out of six. The relative expression of phospho-Smad2 was calculated by the ratio of band intensity relative to β-actin. Data are expressed as mean±s.e., *P<0.05.
PTK/ZK Inhibits TGF-β1 Expression in Response to PDGF Stimulation
An increased TGF-β1 expression in HSCs was observed after PDGF-BB (10 ng/ml) induction for 16 h. The elevated TGF-β1 expression could be inhibited by PTK/ZK added 3 h before TGF-β1 (Figure 9).
Figure 9.

The effect of PTK/ZK on TGF-β1 expression induced by PDGF. HSCs were growth arrested before addition of PTK/ZK for 3 h, and were subsequently stimulated with 10 ng/ml of PDGF overnight. Total proteins were extracted and TGF-β1 expression was analyzed by western blot. PDGF stimulation resulted in an increased expression of TGF-β1, which could be suppressed by PTK/ZK. Representative data from three experiments are shown.
DISCUSSION
Interest in treating hepatic fibrosis has continued to accelerate. PTK/ZK, a potent receptor tyrosine kinase inhibitor, was initially developed as a potent antiangiogenic agent binding directly to the ATP-binding sites of VEGF receptors, but it also inhibits PDGFR-β with less potency.16 Our study demonstrated that PTK/ZK not only inhibits liver cancer18 but also liver fibrosis both in vivo and in vitro.19 In a separate report, PTK/ZK inhibits HSC activation by attenuating HSC proliferation, migration and collagen synthesis through the VEGF pathway.19 In this study, we further explore molecular targets of PTK/ZK in HSCs. This study addresses the novel mechanisms and molecular signaling pathways of PTK/ZK as an antifibrotic agent, which were not included in our earlier publication.19
This study has revealed that PTK/ZK inhibits PDGFR-β expression in activated HSCs, and proliferation and motility of activated HSCs induced by PDGF, as well as activation of Raf, ERK, Akt and p70S6 kinase stimulated by PDGF. In HSCs, it was very likely that the activation of Ras-Raf-ERK, induced by PDGF binding to PDGFR-β, was the signal involved in the mitogenic response to PDGF.26 ERK activation induced by PDGF was also associated with HSC proliferation and migration,27 whereas Akt activation not only stimulates HSC proliferation and migration but also increases collagen production by HSC.28 In addition, Akt signaling also mediated HSC survival and resistance to apoptosis.29 The p70S6 kinase is a downstream target of Akt, and is activated by mitogens and growth factors in a PI3K-dependent manner. In HSCs, p70S6 kinase is critical to cell proliferation, collagen expression and cell cycle control after PDGF stimulation.30 Our findings uncover a molecular link between PTK/ZK as a PDGF receptor tyrosine kinase inhibitor and ERK-, Akt- and p70S6 kinase-mediated HSC proliferation, migration, collagen expression, apoptosis and cell cycle distribution.
Our data have extended previous studies examining the role of TGF-β1 in HSC activation in three important areas. First, we report that TGF-β1 stimulates VEGF gene expression in HSCs in a dose-dependent manner, whereas previous studies indicated that TGF-β treatment induces VEGF mRNA in fibroblasts and epithelial cells, but not in endothelial cells.31 Second, we demonstrate that TGF-β1 also induces both VEGFR1 (Flt-1) gene and protein expression in HSCs. TGF-β1 induction of Flt-1 was reported previously in bovine retinal endothelial cells.32 Third, PTK/ZK inhibited both VEGF and VEGFR1 expression induced by TGF-β1. Interestingly, PTK/ZK also suppressed the expression of both TGFβRI and TGFβRII. The signaling of the TGF-β family is mediated through TGFβRI and TGFβRII to phosphorylate receptor-activated Smad, which is the best characterized downstream target of the TGF-β pathway.33 In addition to the Smad-mediated canonical TGF-β signaling pathway, evidence over the past few years suggested that TGF-β may signal through non-Smad pathways to mediate cellular effects. For example, TGF-β can activate ERK,34 PI3K/Akt and p38MAPK.23–25,35,36 It is reported that p38MAPK could be activated by TGFβR to mediate Smad-independent TGF-β responses.35 Furthermore, PI3K/Akt activity could be potently induced by the activation of TGFβRI.36 Indeed, we observed that TGF-β1 stimulated the phosphorylation of Akt, ERK and p38MAPK, which could be inhibited by PTK/ZK. It is reported that the phosphorylation of p38MAPK is augmented in activated HSC, which is associated with increased collagen production.25 In addition, p38MAPK is also involved in TGF-β-stimulated synthesis of VEGF in aortic smooth muscle cells.37 Furthermore, both Akt and p38MAPK are involved in TGF-β1-downregulated matrix metalloproteinase-13 (MMP-13) expression, as well as in upregulated type I collagen expression.23 MMP-13 plays an important role in the resolution of liver fibrogenesis augmented by macrophages through increased matrix degradation.6 Therefore, PTK/ZK inhibits HSC activation by complex mechanisms involving Akt, ERK, p70S6kinase, as well as p38MAPK. Unlike PDGF, we could not detect Raf activation upon TGF-β1 stimulation. Therefore, PTK/ZK might exert an inhibitory effect on ERK induced by TGF-β1, rather than through the Ras–Raf cascade. Although Smad signaling is important in fibrogenesis,24,38 PTK/ZK failed to significantly inhibit Smad2 signaling induced by TGF-β. As the antibody we used interacts not only with Smad2 but also with Smad3 upon addition of TGF-β, it is unlikely that PTK/ZK inhibited both Smad2 and Smad3 activation by TGF-β. Consistent with our findings, Wang et al39 also showed that renal fibrosis was ameliorated through a non-Smad TGF-β pathway by imatinib mesylate. Several reasons may account for the failure of PTK/ZK to inhibit Smad2/3 phosphorylation. First, Smad2/3 is a receptor substrate not only for TGF-β/TGFβR but also for Activin/Activin receptor (ActR).40,41 Activin and its receptors are expressed in both hepatic and pancreatic stellate cells, and activin serves as an autocrine activator for stellate cell activation.42,43 Although PTK/ZK inhibited TGFβR, Smad2/3 could still be activated through Activin/ActR. Second, Smad phosphorylation is directly mediated through TGF-βRI; however, other kinase pathways also regulate Smad signaling. For example, phosphorylation of Smad2 can also result from a stimulation of EGF or hepatocyte growth factor (HGF), which acts through its own cognate receptor tyrosine kinase receptors.44,45 Indeed, HGF is expressed in cultured HSCs,46 and therefore Smad2 could still be activated through HGF, even if PTK/ZK blocked TGFβ/TGFβR signaling. Third, phosphorylation of Smad2/3 could also be activated directly by IGF-binding protein-3 (IGFBP-3),47 although IGFBP-3 reportedly acts through TGFβRI in smooth muscle cells.47 IGFBP-3 is expressed by HSCs, and an increased expression of IGFBP-3 is observed during transformation to myofibroblast-like cells in culture.48 PTK/ZK at 5 or 10 μM only partially inhibited the expression of TGFβRI; therefore, phospho-Smad2 could still be induced by an incomplete inhibition of the pathway by PTK/ZK and by the possible presence of alternative Smad2 phosphorylation events that are not sensitive to PTK/ZK.
In support of our findings, Bisping et al49 also reported that BIBF 1000, a novel receptor tyrosine kinase inhibitor targeting VEGFR1 through VEGFR3, FGFR1, FGFR3, as well as PDGFRα, also interferes with TGF-β-mediated effects in bone marrow stromal cells. However, the mechanism by which the receptor tyrosine kinase inhibitor interfered with signaling pathways triggered by TGF-β was not elucidated. In an earlier study, PDGF reportedly upregulates TGF-β1 expression in human mesangial cells and modulates mesangial cell proliferation and mesangial matrix production.50 As the mesangial cell is a cell type in kidney related to HSC, we investigated whether PDGF stimulates TGF-β1 expression in activated HSCs. Indeed, we found that PDGF stimulated TGF-β1 expression, and the induced TGF-β1 expression was suppressed by PTK/ZK. The mechanism by which PTK/ZK inhibited TGF-β1-mediated effects could also be through a blockade of the PDGF/PDGFR system by PTK/ZK. Therefore, PTK/ZK suppresses HSC proliferation and survival not only through a direct inhibition of VEGFR,19 PDGFR and TGFβR-mediated Akt activation but also through an indirect inhibition of the TGF-β1-mediated Akt pathway. In HSCs, inhibition of proliferation and collagen production by PTK/ZK was related to the inhibition of VEGF, PDGF and TGF-β1 signaling and their downstream target, Akt.
In conclusion, this study provides a critical molecular analysis of a comprehensive analysis that uncovers the attenuating development of liver fibrosis through the inhibition of PDGF and TGF-β1, two most potent stimuli of liver fibrosis, by a receptor tyrosine kinase inhibitor PTK/ZK, which is completely distinct from our published work.19 Furthermore, it is unlikely that PTK/ZK has a toxic effect on hepatocytes as immortalized human hepatocytes were not affected by PTK/ZK in our previous report.18 Importantly, PTK/ZK is capable of inhibiting liver fibrosis and blocking VEGF,19 as well as PDGF and TGF-β1-regulated HSC activation. Thus, PTK/ZK may involve the blockage of multiple essential signal pathways in connection with HSC activation that provoke proliferation and fibrogenesis. Our study underscores the therapeutic potential of PTK/ZK as an antifibrotic drug for patients with chronic liver disease. We believe that this study provides even more compelling molecular mechanisms that are distinct from our earlier work.19 It establishes a novel approach and reveals novel insights into understanding and treating hepatic fibrosis.
Acknowledgments
The authors thank Professor Mien-Chie Hung (Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.), Dr Jeremy Hughes (MRC Center for Inflammation Research, University of Edinburgh, UK) and Dr Nai-Sum Wong (Department of Biochemistry, The University of Hong Kong) for their valuable advice and comments, as well as Xueming Qian for technical assistance. We acknowledge financial support from the Hong Kong Research Grants Council (Projects: PolyU 5407/06M; PolyU 5638/07M); Shenzhen Bureau of Science, Technology and Information (Shenzhen Key Laboratory Advancement Scheme); Small Project Funding Programme of the University of Hong Kong; and NIH Grant DK56621.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest. The authors do not have any financial interest related to the development of PTK/ZK.
References
- 1.Baroni GS, D’Ambrosio L, Curto P, et al. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–1199. doi: 10.1002/hep.510230538. [DOI] [PubMed] [Google Scholar]
- 2.Geerts A, Lazou JM, De Bleser P, et al. Tissue distribution, quantitation and proliferation kinetics of fat-storing cells in carbon tetrachloride-injured rat liver. Hepatology. 1991;13:1193–1202. [PubMed] [Google Scholar]
- 3.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 4.Pinzani M, Milani S, Grappone C, et al. Expression of platelet-derived growth factor in a model of acute liver injury. Hepatology. 1994;19:701–707. doi: 10.1002/hep.1840190323. [DOI] [PubMed] [Google Scholar]
- 5.Pinzani M, Milani S, Herbst H, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong L, Yamasaki G, Johnson RJ, et al. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–1569. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Czochra P, Klopcic B, Meyer E, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalo T, Beljaars L, van de Bovenkamp M, et al. Local inhibition of liver fibrosis by specific delivery of a platelet-derived growth factor kinase inhibitor to hepatic stellate cells. J Pharmacol Exp Ther. 2007;321:856–865. doi: 10.1124/jpet.106.114496. [DOI] [PubMed] [Google Scholar]
- 10.Yoshiji H, Kuriyama S, Noguchi R, et al. Amelioration of liver fibrogenesis by dual inhibition of PDGF and TGF-beta with a combination of imatinib mesylate and ACE inhibitor in rats. Int J Mol Med. 2006;17:899–904. [PubMed] [Google Scholar]
- 11.Neef M, Ledermann M, Saegesser H, et al. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J Hepatol. 2006;44:167–175. doi: 10.1016/j.jhep.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 12.Kinnman N, Francoz C, Barbu V, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest. 2003;83:163–173. doi: 10.1097/01.lab.0000054178.01162.e4. [DOI] [PubMed] [Google Scholar]
- 13.Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Hu H, Yin JQ. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26:8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 15.George J, Roulot D, Koteliansky VE, et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood JM, Bold G, Buchdunger E, et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178–2189. [PubMed] [Google Scholar]
- 17.Drevs J, Hofmann I, Hugenschmidt H, et al. Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000;60:4819–4824. [PubMed] [Google Scholar]
- 18.Liu Y, Poon RT, Li Q, et al. Both antiangiogenesis- and angiogenesis-independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2005;65:3691–3699. doi: 10.1158/0008-5472.CAN-04-3462. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Lui ELH, Friedman S, et al. PTK787/ZK22258 attenuates stellate cell activation and hepatic fibrosis in vivo by inhibiting VEGF signaling. Lab Invest. 2009;89:209–221. doi: 10.1038/labinvest.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bold G, Altmann KH, Frei J, et al. New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis. J Med Chem. 2000;43:2310–2323. doi: 10.1021/jm9909443. [DOI] [PubMed] [Google Scholar]
- 21.Schafer S, Zerbe O, Gressner AM. The synthesis of proteoglycans in fat-storing cells of rat liver. Hepatology. 1987;7:680–687. doi: 10.1002/hep.1840070411. [DOI] [PubMed] [Google Scholar]
- 22.Wang XM, Yu DM, McCaughan GW, et al. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology. 2005;42:935–945. doi: 10.1002/hep.20853. [DOI] [PubMed] [Google Scholar]
- 23.Lechuga CG, Hernández-Nazara ZH, Domínguez Rosales JA, et al. TGF-beta1 modulates matrix metalloproteinase-13 expression in hepatic stellate cells by complex mechanisms involving p38MAPK, PI3-kinase, AKT, and p70S6k. Am J Physiol Gastrointest Liver Physiol. 2004;287:G974–G987. doi: 10.1152/ajpgi.00264.2003. [DOI] [PubMed] [Google Scholar]
- 24.Ohnishi H, Miyata T, Yasuda H, et al. Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in transforming growth factor-beta1 regulation of pancreatic stellate cellular functions. J Biol Chem. 2004;279:8873–8878. doi: 10.1074/jbc.M309698200. [DOI] [PubMed] [Google Scholar]
- 25.Varela-Rey M, Montiel-Duarte C, Osés-Prieto JA, et al. p38 MAPK mediates the regulation of alpha1(I) procollagen mRNA levels by TNF-alpha and TGF-beta in a cell line of rat hepatic stellate cells. FEBS Lett. 2002;528:133–138. doi: 10.1016/s0014-5793(02)03276-3. [DOI] [PubMed] [Google Scholar]
- 26.Pinzani M, Marra F, Caligiuri A, et al. Inhibition by pentoxifylline of extracellular signal-regulated kinase activation by platelet-derived growth factor in hepatic stellate cells. Br J Pharmacol. 1996;119:1117–1124. doi: 10.1111/j.1476-5381.1996.tb16012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marra F, Arrighi MC, Fazi M, et al. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor’s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology. 1999;30:951–958. doi: 10.1002/hep.510300406. [DOI] [PubMed] [Google Scholar]
- 28.Reif S, Lang A, Lindquist JN, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278:8083–8090. doi: 10.1074/jbc.M212927200. [DOI] [PubMed] [Google Scholar]
- 29.Lafdil F, Chobert MN, Couchie D, et al. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology. 2006;44:228–239. doi: 10.1002/hep.21237. [DOI] [PubMed] [Google Scholar]
- 30.Gabele E, Reif S, Tsukada S, et al. The role of p70S6K in hepatic stellate cell collagen gene expression and cell proliferation. J Biol Chem. 2005;280:13374–13382. doi: 10.1074/jbc.M409444200. [DOI] [PubMed] [Google Scholar]
- 31.Pertovaara L, Kaipainen A, Mustonen T, et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem. 1994;269:6271–6274. [PubMed] [Google Scholar]
- 32.Shih SC, Ju M, Liu N, et al. Transforming growth factor beta1 induction of vascular endothelial growth factor receptor 1: mechanism of pericyte-induced vascular survival in vivo. Proc Natl Acad Sci USA. 2003;100:15859–15864. doi: 10.1073/pnas.2136855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 34.Le Pabic H, L’Helgoualc’h A, Coutant A, et al. Involvement of the serine/threonine p70S6 kinase in TGF-beta1-induced ADAM12 expression in cultured human hepatic stellate cells. J Hepatol. 2005;43:1038–1044. doi: 10.1016/j.jhep.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 35.Yu L, Hébert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yi JY, Shin I, Arteaga CL. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J Biol Chem. 2005;280:10870–10876. doi: 10.1074/jbc.M413223200. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto T, Kozawa O, Tanabe K, et al. Involvement of p38 MAP kinase in TGF-beta stimulated VEGF synthesis in aortic smooth muscle cells. J Cell Biochem. 2001;82:591–598. doi: 10.1002/jcb.1179. [DOI] [PubMed] [Google Scholar]
- 38.Li J, Campanale NV, Liang RJ, et al. Inhibition of p38 mitogen-activated protein kinase and transforming growth factor-beta1/Smad signaling pathways modulates the development of fibrosis in adriamycin-induced nephropathy. Am J Pathol. 2006;169:1527–1540. doi: 10.2353/ajpath.2006.060169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S, Wilkes MC, Leof EB, et al. Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 40.Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 41.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 42.Wada W, Kuwano H, Hasegawa Y, et al. The dependence of transforming growth factor-beta-induced collagen production on autocrine factor activin A in hepatic stellate cells. Endocrinology. 2004;145:2753–2759. doi: 10.1210/en.2003-1663. [DOI] [PubMed] [Google Scholar]
- 43.Ohnishi N, Miyata T, Ohnishi H, et al. Activin A is an autocrine activator of rat pancreatic stellate cells: potential therapeutic role of follistatin for pancreatic fibrosis. Gut. 2003;52:1487–1493. doi: 10.1136/gut.52.10.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 45.de Caestecker MP, Parks WT, Frank CJ, et al. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998;12:1587–1592. doi: 10.1101/gad.12.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skrtic S, Wallenius V, Ekberg S, et al. Hepatocyte-stimulated expression of hepatocyte growth factor (HGF) in cultured rat hepatic stellate cells. J Hepatol. 1999;30:115–124. doi: 10.1016/s0168-8278(99)80015-4. [DOI] [PubMed] [Google Scholar]
- 47.Kuemmerle JF, Murthy KS, Bowers JG. IGFBP-3 activates TGF-beta receptors and directly inhibits growth in human intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G795–G802. doi: 10.1152/ajpgi.00009.2004. [DOI] [PubMed] [Google Scholar]
- 48.Scharf JG, Knittel T, Dombrowski F, et al. Characterization of the IGF axis components in isolated rat hepatic stellate cells. Hepatology. 1998;27:1275–1284. doi: 10.1002/hep.510270513. [DOI] [PubMed] [Google Scholar]
- 49.Bisping G, Kropff M, Wenning D, et al. Targeting receptor kinases by a novel indolinone derivative in multiple myeloma: abrogation of stroma-derived interleukin-6 secretion and induction of apoptosis in cytogenetically defined subgroups. Blood. 2006;107:2079–2089. doi: 10.1182/blood-2004-11-4250. [DOI] [PubMed] [Google Scholar]
- 50.Di Paolo S, Gesualdo L, Ranieri E, et al. High glucose concentration induces the overexpression of transforming growth factor-beta through the activation of a platelet-derived growth factor loop in human mesangial cells. Am J Pathol. 1996;149:2095–2106. [PMC free article] [PubMed] [Google Scholar]