

Abstract
Neointima formation is a common feature of atherosclerosis and restenosis after balloon angioplasty. To find a new target to suppress neointima formation, we investigated the possible role of midkine (MK), a heparin-binding growth factor with neurotrophic and chemotactic activities, in neointima formation. MK expression increased during neointima formation caused by intraluminal balloon injury of the rat carotid artery. Neointima formation in a restenosis model was strongly suppressed in MK-deficient mice. Continuous administration of MK protein to MK-deficient mice restored neointima formation. Leukocyte recruitment to the vascular walls after injury was markedly decreased in MK-deficient mice. Soluble MK as well as that bound to the substratum induced migration of macrophages in vitro. These results indicate that MK plays a critical role in neointima formation at least in part owing to its ability to mediate leukocyte recruitment.
Introduction
The complex processes leading to atherosclerosis and postangioplasty restenosis are being increasing clarified (1, 2). A common feature of these vascular pathological processes is intimal lesion formation. Responding to signals delivered from the injured endothelial cells and accumulating inflammatory cells, smooth muscle cells migrate from the media to the intima and proliferate to form intimal lesions.
Suppression of intimal lesion formation is important from the viewpoint of prevention of restenosis and atherosclerosis. For clinical application, inhibition of growth factor action (3–7) is promising, and PDGF has become one focus of such an approach (3, 4).
The choice of a growth factor as the target is important, so that a good therapeutic effect can be obtained. We considered that the growth factor of choice should preferably have the following properties: (a) it is essential for neointima formation, namely, restenosis is suppressed in mice deficient in this factor; (b) its distribution is restricted; and (c) its level of expression is increased during neointima formation.
Here, we report that a heparin-binding growth factor, midkine (MK) (8, 9), fulfills these requirements. MK has 50% sequence identity with pleiotrophin/heparin-binding growth-associated molecule (PTN/HB-GAM) and is distinct from fibroblast growth factors (10–12). Expression of MK in adult tissue is severely restricted, and in the mouse, only the kidney and the uterus express this molecule at high levels (13). Both MK and PTN/HB-GAM have neurotrophic activities and are considered to be involved in neurogenesis and tumor progression (14–17).
Methods
Rat models.
Male Sprague-Dawley rats (15–20 weeks old, 350–400 g) that had been fed normal rodent chow were anesthetized with ketamine (45 mg/kg). Intraluminal balloon injury in the carotid artery was performed as described by Clowes et al. (18). Three (n = 3), 7 (n = 4), and 14 days (n = 3) after balloon injury, the carotid arteries were taken for histological analysis. For RT-PCR and Western blotting analysis, samples 3 hours (n = 3), 3 (n = 3), 7 (n = 3) and 14 days (n = 3) after balloon injury and control (n = 3) carotid arteries were excised and carefully cleaned of the surrounding tissue. The tissue was homogenized immediately for isolation of total RNA or protein as described previously (19).
Mouse models.
MK-deficient mice were generated as described elsewhere in detail (15). The mice were fed normal rodent chow. To monitor intimal lesion formation, adult male 129-SV mice and male MK-deficient mice with the 129-SV genetic background were used in the carotid ligation model (10–15 weeks old, 25–30 g). The age of the mutant mice and the control mice was set to be identical. After anesthesia with Nembutal (Abbott Laboratories, North Chicago, Illinois, USA), the carotid artery was ligated near the carotid bifurcation as described by Kumar and Lindner (20).
In the pump study, MK protein in saline (0.8 mg/mL) (n = 10), human albumin (Wako Pure Chemical Industries, Osaka, Japan) in saline (0.8 mg/mL) (n = 10), or saline (n = 10) was infused using an osmotic pump (Alza Corporation, Palo Alto, California, USA) into MK-deficient mice. The pumps infused a total of 90 μL continuously over a period of 7 days. The pumps were implanted under the abdominal skin and exchanged 7 days after the initial implantation. The serum cholesterol level and lipoprotein profile were determined by SRL Inc. (Tokyo, Japan) with kits for clinical use.
Morphometry of mouse arteries.
The extent of intimal lesion proliferation was quantified by examining 10 hematoxylin and eosin–stained cross-sections of each left carotid artery within 5 mm of the ligation site, as described by Kumar and Lindner (20). The circumferences of the external elastic lamina, internal elastic lamina and lumen, and the areas of the intima and media were measured using C. Imaging Series Simple (Compix Inc., Tualatin, Oregon, USA). Statistical analysis was carried out by the Mann-Whitney test.
MK protein and antibodies.
To generate human MK protein, an expression vector for yeast (Pichia pastoris GS115; Research Corporation Technologies, Tucson, Arizona, USA) was constructed by inserting a cDNA fragment covering the open reading frame of human MK (21) into pHIL-D4 (Invitrogen, Carlsbad, California, USA). After transfection of the expression vector into yeast, selection with histidine and G418 was carried out. The human MK protein was purified from the yeast by anion exchange chromatography and affinity chromatography on a heparin column. The purified human protein exhibited neurotrophic activity comparable to that of mouse MK produced in L cells (22). Antibodies against bacteria-produced mouse MK (23) were raised by injection of the purified protein into rabbits and purified by a combination of affinity chromatography on protein-A and MK columns (23). The antibodies were specific to MK and did not react to PTN/HB-GAM.
PCR.
One microgram of total RNA was reverse-transcribed by Superscript II (GIBCO BRL, Rockville, Maryland, USA). The samples were denatured at 94°C for 1 minute; the PCR was performed for 28 cycles of 30 seconds at 94°C, 30 seconds at 55°C, and 30 seconds at 72°C. The oligonucleotides used for amplification were as follows: Rat MK cDNA sequence (EMBL/Gene Bank Accession number AB025023) was used to design the oligonucleotides for MK: forward, ATGCAGCACCGAAGTTTCTTC; reverse, TCAGTCCTTT CCTTTTCCTTT. For rat GAPDH: forward, GACCACAGTCCATGCCATCAC; reverse, GTAGCCGTATTCATTGTCATACC. Competitive PCR of MK and GAPDH was performed using a Competitive DNA Construction Kit (Takara, Osaka, Japan) according to the manufacturer’s protocol.
Western blotting analysis.
Proteins extracted from the carotid arteries that corresponded to an original weight of 10 mg were separated by 15% SDS-PAGE. MK protein was detected by Western blotting with anti-mouse MK antibody using an ECL kit (Amersham, Buckinghamshire, United Kingdom) (19).
Histological specimens.
Carotid arteries were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 5-μm sections. To make 5-μm cryosections of carotid arteries, mice were perfused with 3.0% paraformaldehyde in PBS, and tissues were rinsed overnight in 20% sucrose before embedding in OCT compound (Tissue-Tek, Torrance, California, USA).
Immunohistochemistry.
The procedure for immunostaining of MK in paraffin sections was as described previously (24). The specificity of the immunostaining for MK was confirmed by absorption of the anti-MK antibodies with recombinant MK, followed by heparin-Sepharose affinity chromatography as described previously (25).
A rat mAb against the mouse leukocyte common antigen CD45 (PharMingen, San Diego, California, USA) was used to determine the presence of inflammatory cells in paraffin sections. In addition to incubation with unconjugated goat anti-rat IgG (The Jackson Laboratory, Bar Harbor, Maine, USA) as the second antibody, biotinyl-tyramide and streptavidin-horseradish peroxidase (NEN Life Science Products, Boston, Massachusetts, USA) incubation was performed to enhance the signal. Cryosections were stained with a monoclonal rat anti-mouse monocyte-macrophage marker MOMA-2 (Biosource International, Camarillo, California, USA), a monoclonal rat anti-mouse neutrophil marker 7/4 (Serotec Ltd., Oxford, United Kingdom), or the anti-CD45 followed by detection with fluorescent isothiocyanate-rabbit anti-rat IgG (Zymed Laboratories, Inc., South San Francisco, California, USA).
Culture of macrophages and migration assay.
Macrophages were collected from the mouse peritoneum as described by Xie et al. (26). The migration of macrophages was assayed by a modification of the Boyden chamber method using Transwell chambers (Costar, Cambridge, Massachusetts, USA) with pores of 5.0-μm diameter as described by Brown et al. (27). Aliquots of 1.0 × 106 cells/mL in RPMI1640 medium containing 5% FBS (100 μL) were placed in the upper chamber. Then, 600 μL of RPMI1640 medium containing 5% FBS and MK was placed in the lower chamber. Alternatively, the lower surface of the filter was coated with MK in PBS.
Culture of aortic smooth muscle cells and migration assay.
Rat aortic smooth muscle cells were prepared as described by Basson et al. (28). The tissue explants were cultured in DMEM supplemented with 10% FBS. Smooth muscle cells at the fifth to eighth passage were used for the migration assay performed as described for macrophage migration. Conditioned medium of smooth muscle cells was collected after culturing 2 days and used instead of the assay medium. MTT (3-[4,5-dimethylthiazoil-2yl]-2,5-diphenyltetrazolium bromide) assay was performed according to Fukuzawa et al. (29).
Results
MK expression during neointima formation.
A rat model using intraluminal balloon injury of the carotid artery (18) was used to determine the mode of MK expression during neointima formation, as rat samples are relatively large and more suitable for quantitative analysis than those of the mouse. We first cloned the rat MK cDNA by RT-PCR. The deduced amino acid sequence (EMBL/GenBank accession number AB025023) showed 95% identity with mouse MK and 87% with human MK. MK RNA level was monitored by RT-PCR (Figure 1a) and more quantitatively by competitive PCR (Figure 1b). MK RNA was expressed at a low level in uninjured arteries (Figure 1, a and b). This weak expression continued until 3 days after balloon injury (abbreviated as day 3), although a slight decrease in the expression was observed after 3 hours (Figure 1b). On day 14, when neointima formation was completed, MK RNA level was slightly decreased compared with that on day 7 (Figure 1b). MK expression reached a maximum on day 7, with induction of more than 10-fold compared with the control level (Figure 1b). Western blotting analysis confirmed that MK protein expression was similar to that of MK RNA expression (Figure 1c). Immunohistochemical staining revealed that MK protein was present only in endothelial cells before injury (Figure 2b), but was strongly expressed in the neointimal region on day 7 (Figure 2d) and day 14 (Figure 2g). Therefore, the mode of MK expression implies a possible role in neointima formation.
Figure 1.

Increased MK expression in the carotid artery after balloon injury. (a) RT-PCR products for MK and GAPDH were separated on 1% agarose gels. (b) Competitive RT-PCR for MK and GAPDH. (c) Proteins extracted from the carotid arteries that corresponded to an original weight of 10 mg were separated by 15% SDS-PAGE. MK protein was detected with anti-mouse MK antibody on Western blotting.
Figure 2.

MK protein localization in the rat model. (a and b) Untreated carotid artery; (c–e) 7 days after balloon injury; (f and g) 14 days after balloon injury. (a, c, f) Hematoxylin and eosin staining; (b, d, g) MK-immunostaining; (e) control immunostaining with preabsorbed antibodies. MK protein was detected in the endothelial cells (arrow in b) in untreated samples (b). Seven and 14 days after balloon injury, MK protein expression was mainly localized in intimal lesions (d, g). Arrowheads in c–g indicate internal elastic lamina above which intimal lesions are located. The negative staining with preabsorbed antibodies indicated that the immunostaining with MK antibodies was specific (e). a and b; c, d, and e; and f and g have identical magnification, respectively. Each scale bar is 50 μm.
The role of MK in neointima formation determined in MK-deficient mice.
Recently, a mouse model of neointima formation induced after cessation of blood flow by ligation of the carotid artery was established (20). The spatio-temporal profile of neointima formation in this model is similar to that in the more widely used rat balloon injury model (18). In both cases, neointima formation is completed by day 14.
We used the mouse model to determine whether neointima formation was affected in mice deficient in the MK gene (Mdk). The MK gene–deficient (Mdk–/–) mice were fertile and showed no apparent abnormalities except that postnatal development of the hippocampus was delayed and the young mice exhibited abnormal behavior (15).
We compared neointima formation in Mdk–/– mice and in control wild-type mice (Mdk+/+). Both Mdk+/+ and Mdk–/– mice were 129/SV genetic background and were the same age. In Mdk+/+ mice, neointima formation was evident as assessed on day 14 (Figure 3b), whereas it was almost completely suppressed in Mdk–/– mice (Figure 3c). This was also true on day 28 (data not shown). For quantitative analysis, we examined specimens from 10 Mdk+/+ and 10 Mdk–/– mice on day 14. The mean ratio of the areas of the neointima and media was significantly decreased in Mdk–/– mice (Figure 4a). In addition, the circumference of the lumen was significantly increased in Mdk–/– mice (Figure 4b). The circumference of the external elastic lamina was comparable (Figure 4b), indicating that artery samples of comparable sizes were used for analysis. On the other hand, thickening of the media took place in Mdk–/– mice (Figure 3c) as in Mdk+/+ (Figure 3b). The serum cholesterol level and lipoprotein profiles were not different between Mdk+/+ and Mdk–/– mice (data not shown).
Figure 3.

Intimal lesion formation was suppressed in MK-deficient mice, and exogenous MK caused its resumption. The results of hematoxylin and eosin staining of samples 14 days after ligation (b–e) and a control (a) are shown. (a) Control (without ligation); (b) wild-type mice (Mdk+/+); (c) MK-deficient mice (Mdk–/–); (d) Mdk–/– mice treated with saline; (e) Mdk–/– mice treated with MK protein. Arrowheads indicate the internal elastic lamina. Asterisks indicate intimal lesions. Bar, 100 μm.
Figure 4.

Morphometric analysis of neointima formation. (a and b) Comparison between Mdk+/+ and Mdk–/– mice; (c) comparison between Mdk–/– mice treated with either saline, albumin, or MK protein. (a) The mean ratios between the areas of the intimal lesion and the media (IL/M) and standard deviations for Mdk+/+ and Mdk–/–. The difference between Mdk+/+ and Mdk–/– mice was statistically significant (P < 0.001; Mann-Whitney test). (b) Circumferences of the external elastic lamina (EEL) and lumen (Lumen). The circumference of the lumen was significantly different between Mdk+/+ and Mdk–/– mice (P < 0.001; Mann-Whitney test). In all plots, the open columns indicate Mdk+/+ mice, and the filled columns indicate Mdk–/–. NS, not significant. (c) The mean ratios (IL/M) and standard deviations for Mdk–/– mice treated with saline, human albumin, or MK. The open column indicates mice treated with MK; the gray column indicates mice treated with human albumin; and the filled column indicates mice treated with saline. The difference between the MK-treated group and those treated with human albumin or saline was statistically significant (P < 0.001; Mann-Whitney test).
To confirm further the direct role of MK in neointima formation, MK protein was continuously infused at 0.021 mg/kg per hour into Mdk–/– mice after carotid artery ligation using an osmotic pump. Neointima formation was then evaluated on day 14. We found that neointima formation was restored by MK administration (Figure 3e), whereas infusion with saline (Figure 3d) or human albumin in saline (data not shown) did not produce any effect. The mean ratio of the areas of the intima and the media was significantly different between the MK-treated group and those treated with human albumin or saline (Figure 4c).
Inflammatory leukocyte recruitment and smooth muscle cell migration.
Because inflammatory leukocytes, especially macrophages, play important roles in neointima formation (7, 30), we examined leukocyte infiltration by staining for CD45, a common inflammatory leukocyte antigen. As shown in Figure 5a, several CD45-positive cells were seen adhering to endothelial cells on day 3 in Mdk+/+ mice. Many CD45-positive cells were detected in the intimal lesion on day 7 (Figure 5c). However, such cells were scarcely detected in Mdk–/– mice on day 3 or 7 (Figure 5, b and d).
Figure 5.
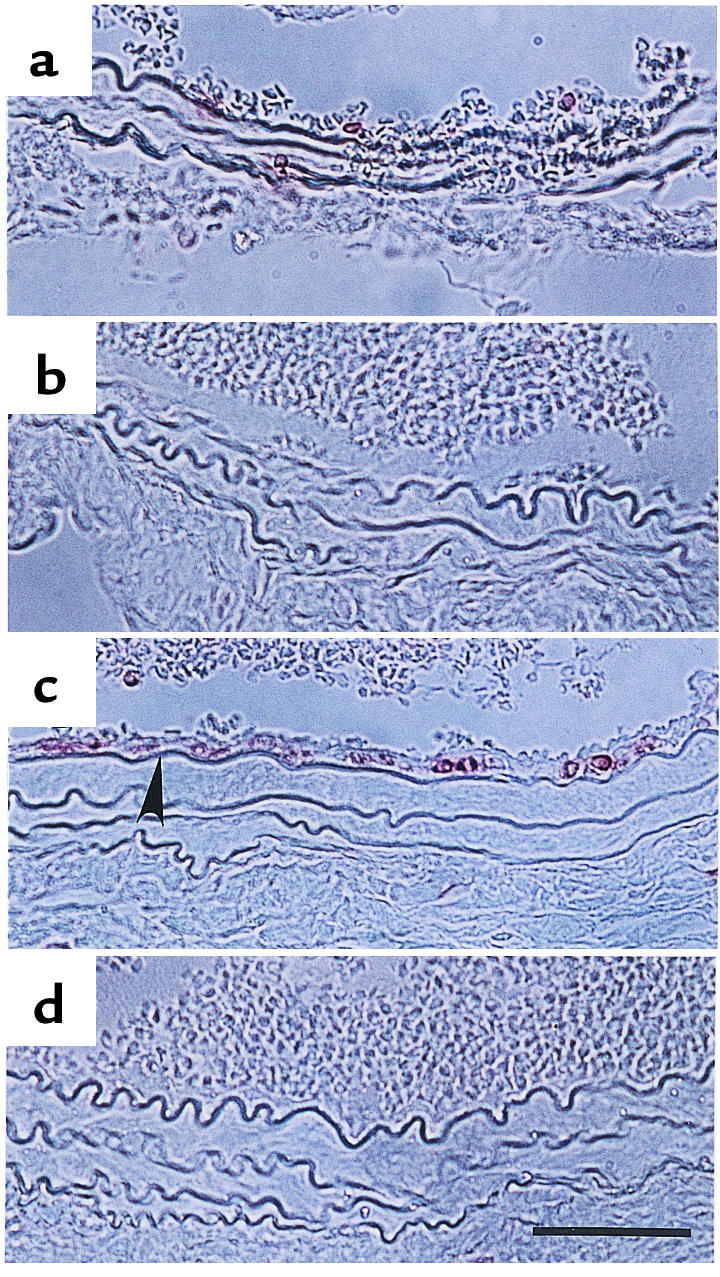
CD45 staining of paraffin sections from the mouse carotid artery after ligation. (a and b) 3 days after ligation; (c and d) 7 days after ligation. (a and c) Mdk+/+ mice; (b and d) Mdk–/– mice. Arrowheads indicate CD45-positive cells. Bar, 50 μm.
Given that CD45-positive cells consist of neutrophils and macrophages/monocytes, we studied the nature of CD45-positive cells by using cryosections of the specimen from Mdk+/+ and Mdk–/– mice on day 7. Cells positive in MOMA-2, a monocyte-macrophage marker (Figure 6a), and cells positive in 7/4, a neutrophil marker (Figure 6c), accumulated in the intimal region in wild-type mice. The number of MOMA-2–positive cells were more than 7/4-positive cells. Scarcely any MOMA-2–positive cells or 7/4-positive cells were present in the specimens from Mdk–/– mice (Figure 6, b and d).
Figure 6.
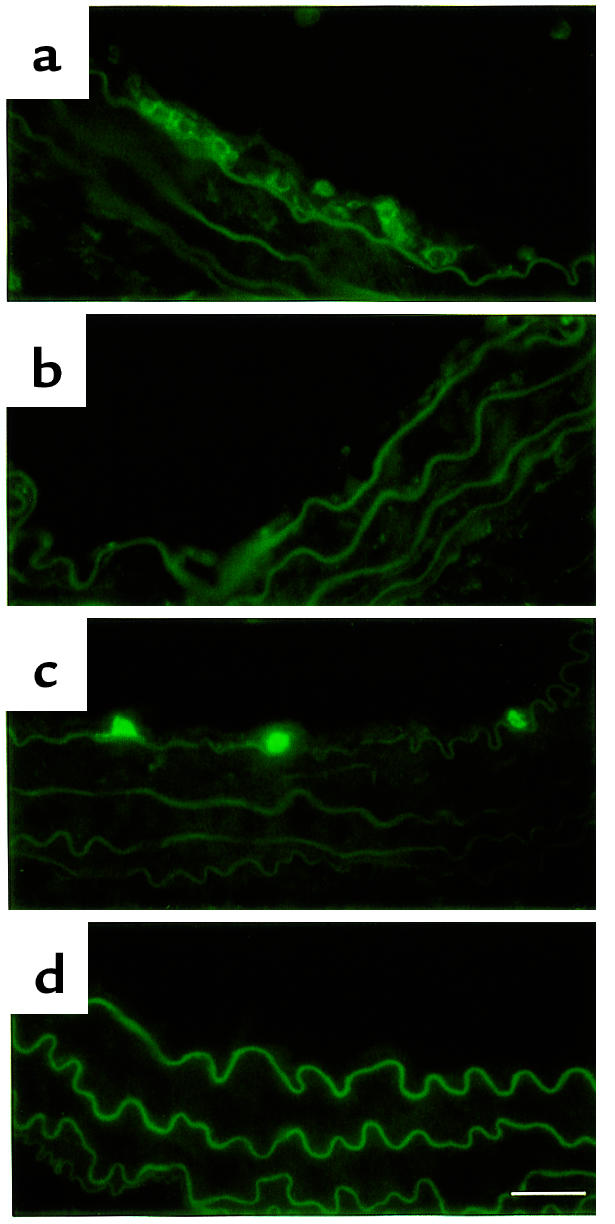
Expression of a macrophage monocyte marker and a neutrophil marker in inflammatory cells that accumulated in the intimal lesion 7 days after ligation. (a and b) Expression of MOMA-2, a monocyte-macrophage marker; (c and d) expression of 7/4, a neutrophil marker. (a and c) Mdk+/+ mice; (b and d) Mdk–/– mice. Bar, 25 μm.
To confirm MK’s ability to stimulate chemotaxis, we carried out Boyden chamber assay. At 100 ng/mL, MK increased macrophage chemotaxis by 2.5-fold (Figure 7a). When the lower surface of the filter of the assay chamber was coated with high concentration of MK, migration was enhanced by 4-fold (Figure 7b).
Figure 7.

Enhanced migration of macrophages (a and b) and smooth muscle cells (c) by MK. Values shown are means ± SD of migrated cell number observed in 10 fields at ×400. (a) MK was added at the indicated concentrations of 0–1,000 ng/mL to the assay well containing macrophages. (b) The filters in the assay well containing macrophages were coated on 1 side with MK at concentrations of 1–200 μg/mL. (c) MK at 100 ng/mL was added to the assay well containing smooth muscle cells. hMK, human MK produced in yeast. mMK, mouse MK produced by the baculovirus expression system (39). CM, conditioned medium of smooth muscle cells.
The effect of MK on smooth muscle cells was also determined by modified Boyden chamber assay. Soluble MK at the concentration of 100 ng/mL increased the migration by 1.8-fold, and this effect was significant (P < 0.01) (Figure 7c). In conditioned medium from smooth muscles cells, smooth muscle cell migration increased and addition of MK further increased the migration by 2.0-fold (Figure 7c). MK did not promote smooth muscle cell proliferation as assessed by MTT assay (data not shown).
Discussion
Using MK-deficient mice, we found that MK plays a crucial role in neointima formation. This conclusion was supported by the finding that administration of MK to MK-deficient mice caused resumption of neointima formation. MK was also found at the sites of neointima formation before and during the event. Quantitative analysis of MK mRNA in the rat system showed that MK mRNA level increases during the process of neointima formation.
Inflammatory cells that migrate to the vessel walls are thought to secrete factors that promote the migration and/or proliferation of smooth muscle cells (1, 2). The importance of inflammatory cell recruitment in neointima formation was verified using P-selectin–deficient mice, in which neointima formation was suppressed with concomitant suppression in recruitment of CD45-positive cells (30). Furthermore, when mice deficient for the receptor of macrophage chemotactic protein-1 were crossed with apo E–deficient mice, development of atherosclerosis occurring in the latter was significantly suppressed (7). We found that the recruitment of CD45-positive inflammatory cells was almost completely eliminated in Mdk–/– mice. It is unlikely that this was due to altered characteristics of inflammatory cells in Mdk–/– mice because MK enhanced migration of macrophages derived from Mdk–/– mice in a modified Boyden chamber assay (data not shown). MK is known to promote the chemotaxis of neutrophils (31), and in the present study we showed that MK promoted the migration of macrophages. PTN/HB-GAM is chemotactic to neurons (32) and osteoblasts (33). Therefore, it is highly likely that the recruitment of inflammatory cells is a critical role of MK in neointima formation.
The central issue in neointima formation is the response of smooth muscle cells. In the rat balloon injury model, the response of smooth muscle cells to neointima-inducing insult is thought to consist of 3 steps: namely, replication within the media, migration from the media to the intima, and proliferation within the neointima (2). Neointima formation in the mouse ligation model proceeds through the same steps (20, 30). Basic fibroblast growth factor released from dying smooth muscle cells appears to participate in the first step (34). MK-deficient mice exhibited thickening of the media after ligation, which was comparable to that in wild-type controls. Thus, the initial response to the neointima-inducing insult is not affected by MK deficiency, but the second and/or final steps were affected. We did not detect growth-promoting activity of MK toward cultured smooth muscle cells, although other researchers previously reported moderate activity (35). However, MK exhibited significant migration-promoting activity toward cultured smooth muscle cells. Therefore, MK may promote neointima formation through 2 routes: recruitment of inflammatory cells that secrete factors acting on smooth muscle cells, and direct action on smooth muscle cells. These 2 routes might collaborate to exert the full function of MK in vivo.
Several reagents, including heparin, a somatostatin analogue, and inhibitors of angiotensinogen converting enzyme, have been clinically used in attempts to suppress restenosis, but these attempts failed because of side effects or species-related differences (36–38). Identification of MK as a molecule involved in neointima formation will enable development of new approaches for preventing restenosis and possibly atherosclerosis through the inhibition of MK activity or signal pathways downstream of the MK action. Anti-MK antibodies and antisense reagents are obvious candidates of MK inhibitors. Simultaneously, it is necessary to analyze the expression of MK in human pathological specimens to confirm that MK is expressed in a way similar to that observed in animal models.
Acknowledgments
We thank M. Ishihara and K. Aoki for secretarial assistance. This work was supported by grants from the Ministry of Education, Science, Sports and Culture, Japan, including a grant-in-aid for Center of Excellence research.
References
- 1.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz SM, deBlois D, O’Brien ERM. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 3.Ferns GAA, et al. Inhibition of neointima smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 4.Nabel EG, et al. Recombinant platelet-derived growth factor B gene expression in porcine arteries induces intimal hyperplasia in vivo. J Clin Invest. 1993;91:1822–1829. doi: 10.1172/JCI116394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banai S, et al. PDGF-receptor tyrosine kinase blocker AG1295 selectively attenuates smooth muscle cell growth in vitro and reduces neointimal formation after balloon angioplasty in swine. Circulation. 1998;97:1960–1969. doi: 10.1161/01.cir.97.19.1960. [DOI] [PubMed] [Google Scholar]
- 6.Lindner V, Reidy MA. Proliferation of smooth muscle cells after vascular injury is inhibited by an antibody against basic fibroblast growth factor. Proc Natl Acad Sci USA. 1991;88:3739–3743. doi: 10.1073/pnas.88.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boring L, Gosling J, Clearly M, Charo IF. Decreased lesion formation in CCR2–/– mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 8.Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation of atherosclerosis. Biochem Biophys Res Commun. 1988;151:1312–1318. doi: 10.1016/s0006-291x(88)80505-9. [DOI] [PubMed] [Google Scholar]
- 9.Iwasaki W, et al. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997;16:6936–6946. doi: 10.1093/emboj/16.23.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauvala H. An 18-kd heparin-binding protein of developing brain that is distinct from fibroblast growth factors. EMBO J. 1989;8:2933–2941. doi: 10.1002/j.1460-2075.1989.tb08443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merenmies J, Rauvala H. Molecular cloning of the 18-kDa growth-associated protein of developing brain. J Biol Chem. 1990;265:16721–16724. [PubMed] [Google Scholar]
- 12.Li YS, et al. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]
- 13.Kadomatsu K, Huang R-P, Suganuma T, Murata F, Muramatsu T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J Cell Biol. 1990;110:607–616. doi: 10.1083/jcb.110.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng HB, et al. The role of heparin-binding growth-associated molecule (HB-GAM) in the postsynaptic induction in cultured muscle cells. J Neurosci. 1995;15:3027–3038. doi: 10.1523/JNEUROSCI.15-04-03027.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura E, et al. Disruption of the midkine gene (Mdk) resulted in altered expression of a calcium binding protein in the hippocampus of infant mice and their abnormal behavior. Genes Cells. 1998;3:811–822. doi: 10.1046/j.1365-2443.1998.00231.x. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawara A, et al. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer Res. 1995;55:1792–1797. [PubMed] [Google Scholar]
- 17.Czubayko F, Shulte AM, Berchem GJ, Wellstein A. Melanoma angiogenesis and metastasis modulated by ribozyme targeting of the secreted growth factor pleiotrophin. Proc Natl Acad Sci USA. 1996;93:14753–14758. doi: 10.1073/pnas.93.25.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 19.Kadomatsu K, et al. Midkine induces the transformation of NIH3T3 cells. Br J Cancer. 1997;75:354–359. doi: 10.1038/bjc.1997.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 21.Tsutsui J, Uehara K, Kadomatsu K, Matsubara S, Muramatsu T. A new family of heparin-binding factors: strong conservation of midkine (MK) sequences between the human and the mouse. Biochem Biophys Res Commun. 1991;176:792–797. doi: 10.1016/s0006-291x(05)80255-4. [DOI] [PubMed] [Google Scholar]
- 22.Muramatsu H, Muramatsu T. Purification of recombinant midkine and examination of its biological activities: functional comparison of new heparin binding factors. Biochem Biophys Res Commun. 1991;176:792–797. doi: 10.1016/0006-291x(91)91838-4. [DOI] [PubMed] [Google Scholar]
- 23.Take M, et al. Identification of nucleolin as a binding protein for midkine (MK) and heparin-binding growth associated molecule (HB-GAM) J Biochem (Tokyo) 1994;116:1063–1068. doi: 10.1093/oxfordjournals.jbchem.a124628. [DOI] [PubMed] [Google Scholar]
- 24.Ye C, et al. Expression of midkine in the early stages of carcinogenesis in human colorectal cancer. Br J Cancer. 1998;79:179–184. doi: 10.1038/sj.bjc.6690030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yasuhara O, et al. Midkine, a novel neurotrophic factor, is present in senile plaques of Alzheimer disease. Biochem Biophys Res Commun. 1993;192:246–251. doi: 10.1006/bbrc.1993.1406. [DOI] [PubMed] [Google Scholar]
- 26.Xie B, Dong Z, Fidler IJ. Regulatory mechanisms for the expression of type IV collagenases/gelatinases in murine macrophages. J Immunol. 1994;152:3637–3644. [PubMed] [Google Scholar]
- 27.Brown SL, Lundgren CH, Nordt T, Fujii S. Stimulation of migration of human aortic smooth muscle cells by vitronectin: implications for atherosclerosis. Cardiovasc Res. 1994;28:1815–1820. doi: 10.1093/cvr/28.12.1815. [DOI] [PubMed] [Google Scholar]
- 28.Basson CT, Kocher O, Basson MD, Asis A, Madri JA. Differential modulation of vascular cell integrin and extracellular matrix expression in vitro by TGF-β1 correlates with reciprocal effects on cell migration. J Cell Physiol. 1992;153:118–128. doi: 10.1002/jcp.1041530116. [DOI] [PubMed] [Google Scholar]
- 29.Fukazawa H, Nakano S, Mizuno S, Uehara Y. Inhibitors of anchorage-independent growth affect the growth of transformed cells on poly(2-hydroxyethyl methacrylate)-coated surfaces. Int J Cancer. 1996;67:876–882. doi: 10.1002/(SICI)1097-0215(19960917)67:6<876::AID-IJC19>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 30.Kumar A, Hoover JL, Simmons CA, Lindner V, Shebuski RJ. Remodeling and neointimal formation in the carotid artery of normal and P-selectin-deficient mice. Circulation. 1997;96:4333–4342. doi: 10.1161/01.cir.96.12.4333. [DOI] [PubMed] [Google Scholar]
- 31.Takada T, Muramatsu H, Song X-J, Torii S, Muramatsu T. Midkine, a retinoic acid-inducible heparin-biding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J Biochem (Tokyo) 1997;122:453–458. doi: 10.1093/oxfordjournals.jbchem.a021773. [DOI] [PubMed] [Google Scholar]
- 32.Maeda N, Noda M. Involvement of receptor-like protein tyrosine phosphatase ζ/RPTPβ and its ligand pleiotrophin/HB-GAM in neuronal migration. J Cell Biol. 1998;142:203–216. doi: 10.1083/jcb.142.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai S, et al. Osteoblast recruitment and bone formation enhanced by cell matrix-associated heparin-binding growth-associated molecule (HB-GAM) J Cell Biol. 1998;143:1113–1128. doi: 10.1083/jcb.143.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson NE, Chao S, Lindner V, Reidy MA. Intimal smooth muscle cell proliferation after balloon catheter injury. The role of basic fibroblast growth factor. Am J Pathol. 1992;140:1017–1023. [PMC free article] [PubMed] [Google Scholar]
- 35.Ohyama Y, Miyamoto K, Minamino N, Matsuo H. Isolation and identification of midkine and pleiotrophin in bovine follicular fluid. Mol Cell Endocrinol. 1994;105:203–208. doi: 10.1016/0303-7207(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 36.Ellis SG, Roubin GS, Wilentz J, Douglas JS, Jr, King SB. Effect of 18- to 24-hour heparin administration for prevention of restenosis after uncomplicated coronary angioplasty. Am Heart J. 1989;117:777–782. doi: 10.1016/0002-8703(89)90612-1. [DOI] [PubMed] [Google Scholar]
- 37.Emanuelsson H, et al. Long-term effects of angiopeptin treatment in coronary angioplasty. Reduction of clinical events but not angiographic restenosis. Circulation. 1995;91:1689–1696. doi: 10.1161/01.cir.91.6.1689. [DOI] [PubMed] [Google Scholar]
- 38.The Multicenter European Research Trial with Cilazapril after Angioplasty to Prevent Transluminal Coronary ObstructionRestenosis (MERCATOR) Study Group. Does the new angiotensin converting enzyme inhibitor cilazapril prevent restenosis after percutaneous transluminal coronary angioplasty? Results of the MERCATOR study: a multicenter, randomized, double-blind placebo-controlled trial. Circulation. 1992;86:100–110. doi: 10.1161/01.cir.86.1.100. [DOI] [PubMed] [Google Scholar]
- 39.Kaneda N, Talukder AH, Nishiyama H, Koizumi S, Muramatsu T. Midkine, a heparin-binding growth/differentiation factor, exhibits nerve cell adhesion and guidance activity for neurite outgrowth in vitro. J Biochem (Tokyo) 1996;119:1150–1156. doi: 10.1093/oxfordjournals.jbchem.a021361. [DOI] [PubMed] [Google Scholar]