

Abstract
Adenoviral transduction with CD40L and poxviral transduction with B7-1, ICAM-1, and LFA-3 (TRICOM) have been used to enhance the antigen-presenting capacity of chronic lymphocytic leukemia (CLL) cells. This study compares the same vector (modified vaccinia virus strain Ankara (MVA)) encoding CD40L or TRICOM for ability to enhance the immunogenicity of CLL cells. CLL cells from some patients showed differential responses to each vector in terms of induction of autologous T-cell responses. This study supports the rationale for the use of CLL cells modified ex vivo with pre-specified recombinant MVA vectors as a whole tumor-cell vaccine for immunotherapy in CLL patients.
Keywords: immunotherapy, CLL, TRICOM, CD40L, vaccine
1. Introduction
Chronic lymphocytic leukemia (CLL) is a malignancy characterized by the accumulation of clonal CD5+ B lymphocytes [1]. With current treatments consisting of combinations of various cytotoxic drugs, CLL remains an incurable disease; resistance to chemotherapy develops in the majority of patients, and significant toxicities are seen [2]. A better understanding of the molecular and cellular mechanisms of CLL is leading to the development of alternative forms of therapy that might efficiently work against the disease. One such therapy involves ex-vivo modified CLL cells which can be used as a whole tumor-cell vaccine to stimulate an anti-tumor immune response in CLL patients [3]. CLL cells express tumor-associated antigens and major histocompatibility complex (MHC) molecules, but have been shown to be inefficient at antigen presentation due to a lack of expression of T-cell costimulatory molecules on the cell surface [4, 5]. Therefore, strategies are being investigated in an effort to increase the immunogenicity of CLL cells for use as a whole tumor-cell vaccine. CD40 activation has been used as one means to increase expression of various costimulatory molecules on the surface of CLL cells and thus enhance their antigen-presenting ability. CD40 ligand (CD40L)-induced signaling has been achieved via coculture of CLL cells with CD40L+ feeder cells [4, 6, 7], as well as direct gene transfer of CD40L into CLL cells by adenoviral vectors [8, 9]. A phase I clinical trial in which adenovirally-CD40L-transduced autologous CLL cells were intravenously infused to patients with CLL showed increases in the number of leukemia-specific T cells following treatment, as well as reductions in leukemia cell counts [9]. Further, combined expression of CD40L and IL-2 or OX40L by CLL cells, via direct or indirect adenoviral transduction, was shown to augment the T-cell activation induced by CD40L alone [10, 11].
Replication-defective poxviral vectors encoding for the costimulatory molecules B7-1, ICAM-1, and LFA-3 (designated TRICOM) constitute another strategy to increase the immunogenicity of CLL cells. We have previously shown that both murine and human normal B cells could be infected with a nonreplicative fowlpox vector encoding TRICOM to enhance their APC potency [12, 13]. In animal studies, B lymphoma cells transduced with a recombinant fowlpox vector encoding TRICOM induced anti-tumor responses more effectively than non-transduced lymphoma cells [14]. In human cells in vitro, we previously compared the ability of replication-defective poxviral vectors encoding TRICOM to infect CLL cells and showed that a recombinant modified vaccinia virus strain Ankara (MVA)-TRICOM vector, but not recombinant fowlpox-TRICOM, was most efficient at enhancing the antigen-presenting capacity of CLL cells [15]. MVA-TRICOM-infected CLL cells induced proliferation of autologous T cells and generated cytotoxic T lymphocytes with reactivity against unmodified CLL cells [15].
In light of the positive results from the phase I clinical trial using CD40L-transduced CLL cells and our results with MVA-TRICOM-modified CLL cells in vitro, here we evaluated for the first time an MVA vector platform for delivery of CD40L to CLL cells (MVA-CD40L) and compared it to MVA-TRICOM on their ability to enhance the immunogenicity of CLL cells in vitro. Our results demonstrated that the novel MVA-CD40L vector is very efficient, even at low multiplicity of infection (MOI), at inducing CD40L-expression on the surface of CLL cells. Moreover, MVA-TRICOM-infected and MVA-CD40L-infected CLL cells appeared equally potent at inducing autologous T-cell proliferation among the patients assayed, although a level of disparity was observed among patient T-cell responses to either vector-modified CLL cells. Therefore, the results from this study further support the rationale for the use of CLL cells modified ex vivo with recombinant MVA as a whole tumor-cell vaccine for the immunotherapy of CLL, either by modification with the MVA-CD40L or MVA-TRICOM vector. The choice of vector could potentially be predetermined by in-vitro analyses prior to therapy.
2. Materials and methods
2.1. PBMCs from CLL patients and healthy donors
Peripheral blood was collected at the University of Pittsburgh Cancer Institute from patients diagnosed with CLL, after informed consent was obtained and following approval by the University of Pittsburgh Institutional Review Board. Demographics of patients included in this study are presented in Supplemental Table 1. Peripheral blood was collected at NIH from healthy donors, after informed consent was obtained and following approval by the NIH Institutional Review Board. Peripheral blood mononuclear cells (PBMCs) were isolated as previously described [15]. Unless otherwise noted, cells were cultured in RPMI 1640 medium (Mediatech, Inc., Herndon, VA) supplemented with 2 mM glutamine, 1× antibiotic/antimycotic solution (Mediatech, Inc.), and 10% human AB serum (Gemini Bio-Products, West Sacramento, CA).
2.2. Recombinant MVA
Recombinant MVA virus expressing genes encoding for the human B7-1, ICAM-1, and LFA-3 costimulatory molecules (designated MVA-TRICOM) has previously been described [15]. MVA-CD40L contains an 870 bp DNA fragment with the open reading frame of human CD40L under the control of the vaccinia virus 40K promoter [16]. Wild-type MVA virus (designated MVA-WT) was used as a control vector. All viruses were obtained as part of a CRADA with Therion Biologics (Cambridge, MA).
2.3. Infection of CLL cells with recombinant MVA
CLL cells were resuspended at 4 × 106 cells/mL in Opti-MEM (Invitrogen, Carlsbad, CA), plated at 2 × 106 cells/well (in 0.5 mL) on a 24-well plate, and infected with MVA virus for 1 h at 37°C. Following infection, 1.5 mL of prewarmed medium containing 10% human AB serum was added to the cells, and cells were cultured for an additional 24 h. At 24 hours post-infection, the time at which cells were harvested for analysis of expression of costimulatory molecules or for setup of proliferation assay, no significant decrease in cell viability was observed for MVA-infected CLL cells vs. uninfected control cells. However, MVA is cytopathic and apoptosis of infected cells has been shown at 48-96 hours post-infection in other systems [17, 18]. For antibody blocking experiments, CLL cells were pre-treated with purified antibody to human CD40L (Clone 24-31; eBioscience, San Diego, CA) or control mouse IgG1 (Serotec, Inc., Raleigh, NC) at a concentration of 10 μg/mL for 1 h at 37°C, then infected with MVA as above in the presence of the same concentration of antibody.
2.4. Flow cytometry
Twenty-four hours after infection, CLL cells were analyzed by flow cytometry for expression of costimulatory molecules. CD19-FITC, CD19-PE-Cy5, CD40L-FITC, CD40L-PE, B7-1-PE, ICAM-1-PE, and LFA-3-PE were purchased from BD Biosciences Pharmingen (San Diego, CA). To determine whether upregulation of costimulatory molecules occurred following infection with recombinant MVA vectors, values were compared to uninfected CLL cells from the same patient. For expression of CD40L and B7-1, which exhibit negative/low expression on uninfected CLL cells, samples which showed a total expression of 10% or less following infection were considered to be negative for upregulation; for samples with a percentage expression of more than 10%, upregulation was defined as an increase of greater than 50% in percentage expression and/or an increase of 2-fold or greater in mean fluorescence intensity (MFI) of expression compared to the uninfected control cells. For expression of ICAM-1 and LFA-3, which exhibit significant expression on most uninfected CLL cells, upregulation was defined as an increase of greater than 20% in percentage expression and/or an increase of 2-fold or greater in MFI of expression compared to the uninfected control cells.
2.5. T-cell proliferation assays
Allogeneic mixed lymphocyte reaction (MLR) and autologous lymphocyte proliferation assays were conducted as previously described [15].
3. Results
3.1. Phenotypic and functional modification of CLL cells following infection with MVA-TRICOM or MVA-CD40L
We previously compared MVA and other replication-defective poxviral vectors encoding for TRICOM molecules and showed that MVA-TRICOM was the most efficient at augmenting the antigen-presenting ability of CLL cells [15]. Here MVA-TRICOM and MVA-CD40L were compared for their ability (a) to increase expression of the costimulatory molecules CD40L, B7-1, ICAM-1, and LFA-3 on the surface of CLL cells and (b) to enhance antigen-presenting capacity of the CLL cells. As shown in Tables 1 and 2, at a multiplicity of infection (MOI) of 5, the optimal dose determined by titration, MVA-CD40L increased expression of CD40L on the surface of the CLL cells, by percentage and/or MFI, in 12 of 12 patients analyzed, while MVA-TRICOM was able to enhance the expression of CD40L on the surface of CLL cells in only 5 of 12 patients analyzed. The expression of B7-1, normally very low among uninfected CLL cells, was enhanced by both vectors in 10 out of 12 samples (Table 2), with 2 additional samples showing upregulation of B7-1 expression only in response to infection with MVA-TRICOM. The pattern of upregulation of ICAM-1 was similar to that of B7-1 in that both vectors upregulated expression of ICAM-1 in 8 of 12 samples, with 4 additional samples responding only to MVA-TRICOM (Table 2). LFA-3 upregulation was more efficient after infection with MVA-TRICOM than with MVA-CD40L, with 11 out of 12 samples showing upregulation in response to MVA-TRICOM and only 4 out of 12 samples showing upregulation in response to MVA-CD40L (Table 2). In general, the MFI of B7-1, ICAM-1, and LFA-3 expression was markedly increased by MVA-TRICOM compared to MVA-CD40L (Table 1).
Table 1. Expression of CD40L and TRICOM molecules following infection of CLL cells with MVA-TRICOM or MVA-CD40L.
| CD40L % (MFI) | B7-1 % (MFI) | ICAM-1 % (MFI) | LFA-3 % (MFI) | |
|---|---|---|---|---|
| CLL 1 | ||||
| Uninfected | 10.3 (75) | 35.1 (49) | 88.9 (99) | 95.0 (243) |
| MVA-WT | 5.8 (58) | 13.2 (51) | 83.2 (105) | 93.6 (225) |
| MVA-TRICOM | 7.7 (156) | 83.1 (1712) | 91.7 (3432) | 90.7 (3063) |
| MVA-CD40L | 89.6 (3445) | 29.3 (72) | 82.6 (272) | 91.4 (339) |
| CLL 2 | ||||
| Uninfected | 15.3 (19) | 5.7 (19) | 76.2 (37) | 52.3 (47) |
| MVA-WT | 9.6 (25) | 5.2 (27) | 83.7 (33) | 68.6 (47) |
| MVA-TRICOM | 29.3 (26) | 64.8 (487) | 84.7 (798) | 80.6 (758) |
| MVA-CD40L | 54.1 (29) | 19.4 (25) | 72.0 (231) | 65.5 (85) |
| CLL 3 | ||||
| Uninfected | 3.6 (20) | 3.5 (20) | 21.6 (17) | 75.2 (38) |
| MVA-WT | 2.7 (12) | 3.4 (12) | 29.2 (11) | 79.8 (36) |
| MVA-TRICOM | 2.1 (70) | 75.8 (1285) | 72.0 (1059) | 83.2 (683) |
| MVA-CD40L | 81.6 (930) | 14.1 (45) | 36.1 (63) | 70.1 (53) |
| CLL 4 | ||||
| Uninfected | 4.6 (95) | 3.4 (21) | 38.6 (17) | 70.5 (49) |
| MVA-WT | 8.6 (15) | 8.3 (9) | 49.9 (14) | 83.5 (37) |
| MVA-TRICOM | 10.0 (11) | 86.4 (778) | 89.7 (1269) | 92.3 (945) |
| MVA-CD40L | 89.5 (774) | 25.1 (28) | 65.7 (53) | 72.8 (75) |
| CLL 5 | ||||
| Uninfected | 11.2 (71) | 5.5 (67) | 62.7 (127) | 79.5 (170) |
| MVA-WT | 12.8 (72) | 7.5 (57) | 41.6 (74) | 84.3 (152) |
| MVA-TRICOM | 13.0 (141) | 86.6 (1378) | 83.3 (3143) | 90.2 (2215) |
| MVA-CD40L | 89.9 (2010) | 16.1 (226) | 56.9 (228) | 86.6 (233) |
| CLL 6 | ||||
| Uninfected | 13.9 (25) | 17.8 (30) | 52.0 (69) | 85.7 (80) |
| MVA-WT | 21.5 (42) | 16.9 (42) | 62.4 (69) | 86.3 (70) |
| MVA-TRICOM | 28.3 (32) | 36.3 (674) | 86.6 (570) | 53.4 (40) |
| MVA-CD40L | 24.0 (50) | 25.8 (88) | 63.2 (165) | 88.9 (102) |
| CLL 7 | ||||
| Uninfected | 4.6 (60) | 4.2 (52) | 67.5 (20) | 67.4 (24) |
| MVA-WT | 5.2 (100) | 2.9 (40) | 71.3 (35) | 79.3 (29) |
| MVA-TRICOM | 3.3 (144) | 37.3 (868) | 67.3 (555) | 74.5 (250) |
| MVA-CD40L | 38.4 (79) | 15.3 (41) | 74.3 (24) | 76.1 (29) |
| CLL 8 | ||||
| Uninfected | 7.0 (45) | 1.7 (36) | 93.1 (89) | 93.3 (102) |
| MVA-WT | 1.4 (68) | 2.6 (55) | 87.8 (92) | 87.2 (88) |
| MVA-TRICOM | 0.5 (54) | 68.7 (1133) | 85.7 (3474) | 82.7 (2428) |
| MVA-CD40L | 91.7 (1193) | 91.4 (482) | 92.9 (1196) | 89.2 (687) |
| CLL 9 | ||||
| Uninfected | 4.5 (19) | 4.6 (20) | 86.3 (50) | 81.4 (73) |
| MVA-WT | 5.1 (38) | 4.5 (20) | 84.4 (51) | 61.9 (60) |
| MVA-TRICOM | 16.3 (21) | 74.3 (454) | 92.2 (1443) | 88.9 (1002) |
| MVA-CD40L | 69.3 (141) | 31.1 (122) | 85.3 (226) | 69.4 (176) |
| CLL 10 | ||||
| Uninfected | 7.2 (53) | 2.6 (53) | 47.8 (26) | 57.6 (39) |
| MVA-WT | 11.8 (71) | 3.2 (47) | 59.3 (32) | 68.6 (40) |
| MVA-TRICOM | 12.4 (51) | 48.3 (296) | 77.2 (572) | 77.1 (486) |
| MVA-CD40L | 32.8 (35) | 37.1 (47) | 85.6 (124) | 86.8 (97) |
| CLL 11 | ||||
| Uninfected | 12.3 (32) | 8.4 (20) | 56.0 (39) | 74.2 (48) |
| MVA-WT | 15.0 (83) | 9.3 (27) | 60.5 (40) | 67.4 (51) |
| MVA-TRICOM | 22.0 (74) | 28.6 (209) | 59.0 (242) | 64.9 (210) |
| MVA-CD40L | 16.9 (136) | 9.1 (53) | 43.2 (79) | 51.3 (66) |
| CLL 12 | ||||
| Uninfected | 9.9 (37) | 9.8 (17) | 70.9 (22) | 81.9 (44) |
| MVA-WT | 6.5 (21) | 44.2 (14) | 64.6 (25) | 85.3 (31) |
| MVA-TRICOM | 7.7 (26) | 79.0 (263) | 96.4 (667) | 85.6 (418) |
| MVA-CD40L | 78.1 (352) | 12.4 (40) | 46.5 (50) | 80.3 (40) |
CLL cells were infected with the indicated MVA vectors. Flow cytometry analysis was performed 24 hours following infection to determine expression of CD40L and TRICOM molecules by the CLL cells. The percentage and MFI of expression of these molecules by CD19+ CLL cells is shown.
Table 2. Comparison of number of CLL patient samples exhibiting upregulation of costimulatory molecules following infection with MVA-TRICOM or MVA-CD40L.
| CD40L | B7-1 | ICAM-1 | LFA-3 | |
|---|---|---|---|---|
| MVA-TRICOM+ | 5/12 | 12/12 | 12/12 | 11/12 |
| MVA-CD40L+ | 12/12 | 10/12 | 8/12 | 4/12 |
| MVA-TRICOM+/MVA-CD40L+ | 5/12 | 10/12 | 8/12 | 4/12 |
| MVA-TRICOM+/MVA-CD40L- | 0/12 | 2/12 | 4/12 | 7/12 |
| MVA-TRICOM-/MVA-CD40L+ | 7/12 | 0/12 | 0/12 | 0/12 |
| MVA-TRICOM-/MVA-CD40L- | 0/12 | 0/12 | 0/12 | 1/12 |
CLL cells were infected with either MVA-TRICOM or MVA-CD40L. Flow cytometry analysis was performed 24 hours following infection to determine expression of CD40L and TRICOM molecules by the CLL cells. The number of patient samples exhibiting upregulation of each costimulatory molecule following infection with MVA-TRICOM (MVA-TRICOM+) or MVA-CD40L (MVA-CD40L+) is shown. Samples which did not exhibit upregulation of each costimulatory molecule following infection with MVA-TRICOM or MVA-CD40L are indicated by MVA-TRICOM- and MVA-CD40L-, respectively. The criteria for determining upregulation are explained in “Materials and methods.”
Despite differences in upregulation of costimulatory molecules on CLL cells, both MVA-TRICOM and MVA-CD40L increased the immunogenicity of CLL cells, as measured by their ability to induce proliferation of allogeneic CD3+ T cells from healthy donors in an MLR (Figure 1). In 4 patients assayed, infection of CLL cells with either MVA-TRICOM or MVA-CD40L enhanced the proliferation of healthy donor T cells above the levels induced by uninfected CLL cells or CLL cells infected with control vector. MVA-TRICOM infection was greater in enhancing immunogenicity of the CLL cells in 1 of 4 patients analyzed (patient 7, Figure 1) and slightly inferior to MVA-CD40L in 1 of 4 patients (patient 8, Figure 1). MVA-TRICOM-infected and MVA-CD40L-infected CLL cells induced similar levels of allogeneic T-cell proliferation in the remaining 2 patients (patients 3 and 5, Figure 1). CLL cells were subsequently infected with MVA-CD40L at increasing MOI (10 or 20) to determine if the immunogenicity of CLL cells could be further increased. While increased MOI resulted in greater expression of CD40L, it did not correlate with enhanced expression of B7-1, ICAM-1, and LFA-3 on the CLL cells (data not shown). Further, infection with MVA-CD40L at increasing MOI resulted in a decreased antigen-presenting capacity of the CLL cells in an allogeneic MLR (data not shown). We concluded that, at an optimal MOI of 5, both MVA-TRICOM and MVA-CD40L could upregulate costimulatory molecules on CLL cells and thereby induce proliferation of allogeneic T cells from healthy donors.
Figure 1. Proliferation of allogeneic normal donor T cells in response to CLL cells infected with MVA-TRICOM or MVA-CD40L.

CLL cells from 4 patients were infected with MVA-WT, MVA-TRICOM, or MVA-CD40L. Following 24 hours of infection, CLL cells were washed, irradiated, and cocultured with allogeneic CD3+ T cells from a healthy donor at a ratio of effector to stimulator cells of 10:1. Error bars indicate the standard deviation of replicate measurements. Note that, due to variability in responses, different scales were used on the graphs.
To further characterize the upregulation of costimulatory molecules on CLL cells by MVA-CD40L, infected CLL cells were simultaneously analyzed for CD40L and B7-1, ICAM-1, or LFA-3. The data in Figure 2A demonstrate that there were 3 populations of CLL cells following infection with MVA-CD40L, expressing negative, low, or high levels of CD40L, designated as “negative” (N), “low MFI” (L) and “high MFI” (H) populations, respectively. The “high MFI” population exhibited expression of B7-1, ICAM-1, and LFA-3 similar to that of MVA-WT-infected CLL cells. In contrast, both the “negative” and “low MFI” populations showed upregulation of B7-1, ICAM-1, and LFA-3 compared to MVA-WT-infected cells. We hypothesized that, after MVA-CD40L infection, CLL cells with efficient expression of the CD40L transgene may engage and signal to other CLL cells via CD40L-CD40 interactions, thus inducing upregulation of the costimulatory molecules B7-1, ICAM-1, and LFA-3. To test this hypothesis, we treated CLL cells with blocking antibody to CD40L before and during infection with MVA-CD40L. The results in Figure 2B demonstrate that cell populations expressing negative or low levels of CD40L (“N” and “L”, respectively) do not upregulate B7-1, ICAM-1, and LFA-3 in presence of anti-CD40L blocking antibody compared to the isotype control. Further, the cell population with low levels of CD40L (“L”) largely disappears in presence of blocking antibody to CD40L, representing approximately 5% of cells compared to 25% in the isotype control. These results supported the hypothesis that, following MVA-CD40L infection of CLL cells, signaling via CD40L induces upregulation of the costimulatory molecules B7-1, ICAM-1, LFA-3, and CD40L on other CLL cells.
Figure 2. Upregulation of costimulatory molecules by CLL cells following MVA-CD40L infection.
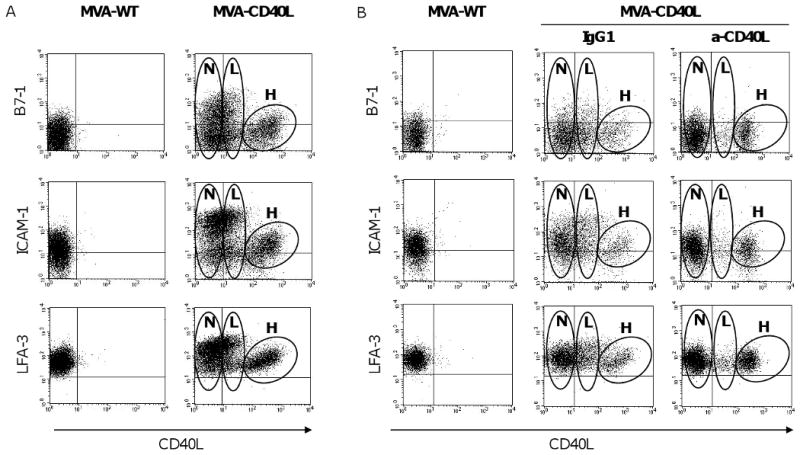
(A) CLL cells were infected with MVA-WT or MVA-CD40L. Following 24 hours of infection, CLL cells were analyzed by flow cytometry for expression of B7-1, ICAM-1, and LFA-3 by CD40L-expressing and non-expressing populations. The plots shown were gated on CD19+ B cells. After infection with MVA-CD40L, 3 populations of CLL cells that expressed negative, low, or high levels of CD40L were observed; these are designated as “negative” (N), “low MFI” (L), and “high MFI” (H) populations on the plots shown. (B) CLL cells were treated with blocking antibody (anti-CD40L or isotype-control mouse IgG1) before and during infection with MVA-CD40L.
3.2. Comparison of CLL cells modified with MVA-TRICOM and MVA-CD40L for ability to stimulate autologous T cells
MVA-TRICOM and MVA-CD40L were further compared for their ability to augment the antigen-presenting ability of CLL cells in autologous T-cell proliferation assays. Both MVA-TRICOM-infected and MVA-CD40L-infected CLL cells induced a greater than two-fold increase in autologous T-cell proliferation compared to control vector in 4 of 6 patients assayed (patients 2, 3, 4, and 5 for MVA-TRICOM; patients 1, 3, 4, and 5 for MVA-CD40L) (Figure 3). Responses were variable among patients. MVA-TRICOM infection was inferior to MVA-CD40L in enhancing the immunogenicity of the CLL cells in 1 of 6 patients analyzed (patient 1), superior to MVA-CD40L in 3 of 6 patients (patients 2, 3, and 4), and equivalent to MVA-CD40L in 1 of 6 patients (patient 5); T cells from one patient showed poor proliferation to autologous CLL cells infected with either vector (patient 6) (Figure 3). CLL cells from the same 6 patients were also analyzed for costimulatory molecule expression following infection (Table 1) to determine whether expression of costimulatory molecules was predictive of response in the proliferation assay. There did not appear to be a direct correlation between magnitude of upregulation of any of the costimulatory molecules analyzed and immunogenicity in the autologous proliferation assay.
Figure 3. Proliferation of autologous patient T cells in response to irradiated CLL cells infected with MVA-TRICOM or MVA-CD40L.

CLL cells from 6 patients were infected with MVA-WT, MVA-TRICOM, or MVA-CD40L. Following 24 hours of infection, CLL cells were washed, irradiated, and cocultured with autologous CD3+ T cells at a ratio of effector to stimulator cells of 2.5:1. Error bars indicate the standard deviation of replicate measurements. Note that, due to variability in responses, different scales were used on the graphs. Costimulatory molecule expression by the CLL cells following infection is shown in Table 1.
Because MVA-CD40L-infected CLL cells appear to engage and signal to uninfected CLL cells, inducing upregulation of costimulatory molecules, we examined whether the use of non-irradiated cells in proliferation assays would increase ‘crosstalk’ with uninfected cells and, consequently, immunogenicity of CLL cells in the assay. Utilizing non-irradiated CLL cells, combined proliferation of T cells and CLL cells was higher with MVA-CD40L-infected cells than MVA-TRICOM-infected cells in 3 of 4 patients assayed (patients 5, 7, and 8; black bars, Figure 4). However, while CLL cells alone exhibited low proliferation following infection with MVA-WT or MVA-TRICOM, CLL cells from 2 of the patients showed significant proliferation after MVA-CD40L infection (patients 5 and 7; white bars, Figure 4); proliferation of the non-irradiated, MVA-CD40L-modified CLL cells accounted for the enhanced activity of MVA-CD40L versus MVA-TRICOM-infected CLL cells in these two patients.
Figure 4. Proliferation of autologous patient T cells in response to non-irradiated CLL cells infected with MVA-TRICOM or MVA-CD40L.

CLL cells from 4 patients were infected with MVA-WT, MVA-TRICOM, or MVA-CD40L. Following 24 hours of infection, CLL cells were washed and, without irradiation, cocultured with autologous CD3+ T cells at a ratio of effector to stimulator cells of 2.5:1. Proliferation by the CLL cells alone is indicated by white bars; proliferation by T cells and CLL cells together is indicated by black bars. Error bars indicate the standard deviation of replicate measurements. Note that, due to variability in responses, different scales were used on the graphs.
Altogether, our results demonstrated that MVA-TRICOM and MVA-CD40L are each effective at enhancing the immunogenicity of CLL cells in vitro. However, there was a degree of variability among various patients' T-cell responses to CLL cells modified via infection with the MVA-TRICOM or MVA-CD40L vector.
4. Discussion
CLL B cells are resistant to transduction with most of the currently available vector systems [19]. Adenovirus has been extensively used, but high multiplicities of infection (MOI from 200 to 2000) are required to achieve sufficient transgene expression in CLL cells [8, 9, 20]. We report here on the ability of MVA vectors encoding for human CD40L or TRICOM to efficiently infect CLL cells in vitro and to enhance their antigen-presenting capacity. MVA is an efficient vector for gene transfer to CLL cells, capable of inducing high-level gene expression at an MOI of 5. While both MVA-CD40L and MVA-TRICOM were able to infect all CLL samples analyzed, the efficiency of infection, measured as the level of expression of the encoded transgene(s), varied among patients. MVA-CD40L, for example, induced CD40L expression in 17-92% of CLL cells; similarly, MVA-TRICOM induced expression of B7-1, normally very low in most CLL cells, in 29-87% of CLL cells. The reason for this variability observed across all patients analyzed is not known at this time, but may be a consequence of the biologically heterogeneous nature of CLL.
The safety profile of MVA makes it potentially useful in the immunotherapy of patients with CLL. MVA is a highly attenuated strain of vaccinia virus [21, 22] that was used in the final stages of the smallpox eradication campaign to immunize more than 120,000 individuals in Germany and Turkey with little significant side effects, including in high-risk groups such as the young, old, or immunosuppressed [21, 23]. Its safety profile has been confirmed in clinical trials for vaccination of individuals infected with HIV-1 [24, 25]. MVA-derived vaccines have been shown to be as effective or more effective than live vaccinia virus vaccines [26-29]. Unlike vaccinia and most orthopoxviruses, MVA has been demonstrated to induce NF-κB activation, leading to increased production of pro-inflammatory cytokines such as interleukin-6 and tumor necrosis factor-α [29-31]. In animal studies, the local immunostimulatory environment following vaccination with MVA-TRICOM led to enhanced tumor-specific immune responses and anti-tumor activity compared to priming with a replication-competent vaccinia vector [32].
We show here that, following infection with MVA-CD40L, 3 populations of CLL cells appeared. The population of cells characterized by high expression of CD40L, designated here as “H”, can be considered the result of MVA-CD40L infection followed by efficient expression of the encoded transgene. This population of cells failed to upregulate additional costimulatory molecules, B7-1, ICAM-1, and LFA-3. In contrast, cells that are negative for CD40L, designated here as “N”, have upregulated the surface expression of B7-1, ICAM-1, and LFA-3, compared to MVA-WT-infected cells. We have speculated that infected CLL cells with high levels of the encoded transgene (i.e., CD40L) may engage and signal to other CLL cells via CD40L-CD40 interactions, thus inducing upregulation of the costimulatory molecules B7-1, ICAM-1, and LFA-3. Supporting this hypothesis, infection of CLL cells with MVA-CD40L in presence of blocking antibody to CD40L abrogated the upregulation of costimulatory molecules by the CD40L-negative population. Upregulation of costimulatory molecules by ‘bystander’ CLL cells following infusion of CD40L-transduced CLL cells was previously demonstrated by Wierda et al. [9]. There is also an intermediate population of cells with low levels of CD40L that appeared following MVA-CD40L infection, designated here as “L”. The emergence of this population could be attributed to the endogenous upregulation of CD40L, B7-1, ICAM-1, and LFA-3 following crosstalk with the high-CD40L population and, in small part, to low efficiency infection by MVA-CD40L.
CD40L-induced signaling has been used previously to increase expression of costimulatory molecules on CLL cells and thus their ability to present antigen to T cells [4, 6-9]. Supporting the utility of this strategy, a phase I clinical trial in which adenovirally-CD40L-transduced autologous CLL cells were intravenously infused to patients with CLL showed increases in the number of leukemia-specific T cells following treatment, as well as reductions in leukemia cell counts [9]. MVA-TRICOM also was used previously to increase the immunogenicity of CLL cells, inducing cytotoxic T lymphocytes with reactivity against unmodified CLL cells in vitro [15]. This study shows for the first time a direct comparison of MVA vectors encoding CD40L or TRICOM for their ability to enhance the antigen-presenting capacity of CLL cells in vitro. In the studies shown here, MVA-TRICOM-infected and MVA-CD40L-infected CLL cells both showed induction of autologous T-cell proliferation among the patients assayed; there was, however, a degree of variability in response among patients, with some patients responding better to MVA-TRICOM or MVA-CD40L. The variability in autologous T-cell responses among patient samples did not appear to correlate with the magnitude of upregulation of any of the costimulatory molecules analyzed (CD40L, B7-1, ICAM-1, LFA-3). We thus hypothesize that signaling through CD40L or TRICOM molecules on CLL cells could be inducing additional molecules not tested in this study, which might be of relevance for enhancing the APC capacity of CLL cells. Because of the disparity among patient T-cell responses to either MVA-TRICOM- or MVA-CD40L-modified CLL cells, we cannot conclude here which vector will be superior and should be preferred in the setting of a clinical trial for the treatment of CLL. We postulate that screening of patients for in-vitro T-cell responses against autologous MVA-TRICOM- versus MVA-CD40L-modified CLL cells might be useful to select those patients who might benefit from receiving a vaccine based on the use of MVA-TRICOM, MVA-CD40L, or a combination of both vectors for modification of CLL cells. This screening could also be useful to exclude patients who might not benefit from this vaccine approach, by identifying those cases where no autologous T-cell proliferation is detected in response to either vector-modified CLL cells.
In a previous clinical trial in which adenovirally-CD40L-transduced autologous CLL cells were intravenously infused to patients with CLL, non-irradiated cells were utilized [9]. Here we demonstrated that non-irradiated, MVA-CD40L-infected CLL cells exhibit significant proliferation in some patient samples, compared to low rates of proliferation following infection with MVA-TRICOM (or control MVA-WT vector). Thus, while CD40L transduction increases the antigen-presenting capacity of CLL cells and the susceptibility of CLL cells to T-cell-mediated cytotoxicity [8], CD40L-induced signaling has a potential drawback in that it may increase proliferation of CLL cells [33, 34]. Moreover, it has been previously shown that CD40L signaling could also increase resistance of CLL cells to drug-induced apoptosis as in response to the nucleoside analog fludarabine, one of the current therapies for CLL [35-37]. The enhanced proliferation of CLL cells following MVA-CD40L infection in some patients suggests that any future immunotherapy with CLL cells modified ex vivo with MVA-CD40L should be performed with irradiated CLL cells.
In conclusion, this study shows the potential of the MVA vector platform for efficient delivery, at very low multiplicity of infection, of either CD40L or the TRICOM molecules to CLL cells, and it further supports the rationale for the use of CLL cells modified ex vivo with recombinant MVA-CD40L and/or MVA-TRICOM as a whole tumor-cell vaccine for the immunotherapy of CLL.
Supplementary Material
Acknowledgments
We acknowledge the technical assistance of Margie Duberstein and the editorial assistance of Debra Weingarten in the preparation of this manuscript. We thank Dr. James Hodge and Dr. Ravi Madan for critical reading of the manuscript. This research was supported by the NIH Intramural Research Program, Center for Cancer Research, National Cancer Institute.
Footnotes
Author contributions: MTL designed and performed research; analyzed data; wrote the paper; KAF designed research, contributed vital reagents; K-YT designed and performed research; JS designed research, analyzed data, wrote the paper and CP designed and performed research, analyzed data, wrote the paper
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wierda WG, Kipps TJ. Chronic lymphocytic leukemia. Curr Opin Hematol. 1999;6:253–61. doi: 10.1097/00062752-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Tsimberidou AM, Keating MJ. Treatment of fludarabine-refractory chronic lymphocytic leukemia. Cancer. 2009;115:2824–36. doi: 10.1002/cncr.24329. [DOI] [PubMed] [Google Scholar]
- 3.Pleyer L, Egle A, Hartmann TN, Greil R. Molecular and cellular mechanisms of CLL: novel therapeutic approaches. Nat Rev Clin Oncol. 2009;6:405–18. doi: 10.1038/nrclinonc.2009.72. Epub 2009 Jun 2. [DOI] [PubMed] [Google Scholar]
- 4.Buhmann R, Nolte A, Westhaus D, Emmerich B, Hallek M. CD40-activated B-cell chronic lymphocytic leukemia cells for tumor immunotherapy: stimulation of allogeneic versus autologous T cells generates different types of effector cells. Blood. 1999;93:1992–2002. [PubMed] [Google Scholar]
- 5.Rezvany MR, Jeddi-Tehrani M, Rabbani H, Lewin N, Avila-Carino J, Osterborg A, et al. Autologous T lymphocytes may specifically recognize leukaemic B cells in patients with chronic lymphocytic leukaemia. Br J Haematol. 2000;111:608–17. doi: 10.1046/j.1365-2141.2000.02383.x. [DOI] [PubMed] [Google Scholar]
- 6.Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med. 1993;177:925–35. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van den Hove LE, Van Gool SW, Vandenberghe P, Bakkus M, Thielemans K, Boogaerts MA, et al. CD40 triggering of chronic lymphocytic leukemia B cells results in efficient alloantigen presentation and cytotoxic T lymphocyte induction by up-regulation of CD80 and CD86 costimulatory molecules. Leukemia. 1997;11:572–80. doi: 10.1038/sj.leu.2400598. [DOI] [PubMed] [Google Scholar]
- 8.Kato K, Cantwell MJ, Sharma S, Kipps TJ. Gene transfer of CD40-ligand induces autologous immune recognition of chronic lymphocytic leukemia B cells. J Clin Invest. 1998;101:1133–41. doi: 10.1172/JCI1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wierda WG, Cantwell MJ, Woods SJ, Rassenti LZ, Prussak CE, Kipps TJ. CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood. 2000;96:2917–24. [PubMed] [Google Scholar]
- 10.Takahashi S, Rousseau RF, Yotnda P, Mei Z, Dotti G, Rill D, et al. Autologous antileukemic immune response induced by chronic lymphocytic leukemia B cells expressing the CD40 ligand and interleukin 2 transgenes. Hum Gene Ther. 2001;12:659–70. doi: 10.1089/104303401300057360. [DOI] [PubMed] [Google Scholar]
- 11.Biagi E, Dotti G, Yvon E, Lee E, Pule M, Vigouroux S, et al. Molecular transfer of CD40 and OX40 ligands to leukemic human B cells induces expansion of autologous tumor-reactive cytotoxic T lymphocytes. Blood. 2005;105:2436–42. doi: 10.1182/blood-2004-07-2556. Epub 2004 Nov 9. [DOI] [PubMed] [Google Scholar]
- 12.Hodge JW, Grosenbach DW, Rad AN, Giuliano M, Sabzevari H, Schlom J. Enhancing the potency of peptide-pulsed antigen presenting cells by vector-driven hyperexpression of a triad of costimulatory molecules. Vaccine. 2001;19:3552–67. doi: 10.1016/s0264-410x(01)00062-7. [DOI] [PubMed] [Google Scholar]
- 13.Palena C, Zhu M, Schlom J, Tsang KY. Human B cells that hyperexpress a triad of costimulatory molecules via avipox-vector infection: an alternative source of efficient antigen-presenting cells. Blood. 2004;104:192–9. doi: 10.1182/blood-2003-09-3211. Epub 2004 Mar 9. [DOI] [PubMed] [Google Scholar]
- 14.Briones J, Timmerman JM, Panicalli DL, Levy R. Antitumor immunity after vaccination with B lymphoma cells overexpressing a triad of costimulatory molecules. J Natl Cancer Inst. 2003;95:548–55. doi: 10.1093/jnci/95.7.548. [DOI] [PubMed] [Google Scholar]
- 15.Palena C, Foon KA, Panicali D, Yafal AG, Chinsangaram J, Hodge JW, et al. Potential approach to immunotherapy of chronic lymphocytic leukemia (CLL): enhanced immunogenicity of CLL cells via infection with vectors encoding for multiple costimulatory molecules. Blood. 2005;106:3515–23. doi: 10.1182/blood-2005-03-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gritz L, Destree A, Cormier N, Day E, Stallard V, Caiazzo T, et al. Generation of hybrid genes and proteins by vaccinia virus-mediated recombination: application to human immunodeficiency virus type 1 env. J Virol. 1990;64:5948–57. doi: 10.1128/jvi.64.12.5948-5957.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chahroudi A, Garber DA, Reeves P, Liu L, Kalman D, Feinberg MB. Differences and similarities in viral life cycle progression and host cell physiology after infection of human dendritic cells with modified vaccinia virus Ankara and vaccinia virus. J Virol. 2006;80:8469–81. doi: 10.1128/JVI.02749-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greiner S, Humrich JY, Thuman P, Sauter B, Schuler G, Jenne L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin Exp Immunol. 2006;146:344–53. doi: 10.1111/j.1365-2249.2006.03177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendtner CM, Kofler DM, Mayr C, Bund D, Hallek M. The potential of gene transfer into primary B-CLL cells using recombinant virus vectors. Leuk Lymphoma. 2004;45:897–904. doi: 10.1080/10428190310001638896. [DOI] [PubMed] [Google Scholar]
- 20.Cantwell MJ, Sharma S, Friedmann T, Kipps TJ. Adenovirus vector infection of chronic lymphocytic leukemia B cells. Blood. 1996;88:4676–83. [PubMed] [Google Scholar]
- 21.Blanchard TJ, Alcami A, Andrea P, Smith GL. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol. 1998;79:1159–67. doi: 10.1099/0022-1317-79-5-1159. [DOI] [PubMed] [Google Scholar]
- 22.Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol. 1998;79:347–52. doi: 10.1099/0022-1317-79-2-347. [DOI] [PubMed] [Google Scholar]
- 23.Mayr A, Danner K. Vaccination against pox diseases under immunosuppressive conditions. Dev Biol Stand. 1978;41:225–34. [PubMed] [Google Scholar]
- 24.Cosma A, Nagaraj R, Buhler S, Hinkula J, Busch DH, Sutter G, et al. Therapeutic vaccination with MVA-HIV-1 nef elicits Nef-specific T-helper cell responses in chronically HIV-1 infected individuals. Vaccine. 2003;22:21–9. doi: 10.1016/s0264-410x(03)00538-3. [DOI] [PubMed] [Google Scholar]
- 25.Harrer E, Bauerle M, Ferstl B, Chaplin P, Petzold B, Mateo L, et al. Therapeutic vaccination of HIV-1-infected patients on HAART with a recombinant HIV-1 nef-expressing MVA: safety, immunogenicity and influence on viral load during treatment interruption. Antivir Ther. 2005;10:285–300. [PubMed] [Google Scholar]
- 26.Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12:1032–40. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 27.Carroll MW, Overwijk WW, Chamberlain RS, Rosenberg SA, Moss B, Restifo NP. Highly attenuated modified vaccinia virus Ankara (MVA) as an effective recombinant vector: a murine tumor model. Vaccine. 1997;15:387–94. doi: 10.1016/s0264-410x(96)00195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belyakov IM, Wyatt LS, Ahlers JD, Earl P, Pendleton CD, Kelsall BL, et al. Induction of a mucosal cytotoxic T-lymphocyte response by intrarectal immunization with a replication-deficient recombinant vaccinia virus expressing human immunodeficiency virus 89.6 envelope protein. J Virol. 1998;72:8264–72. doi: 10.1128/jvi.72.10.8264-8272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramirez JC, Gherardi MM, Esteban M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol. 2000;74:923–33. doi: 10.1128/jvi.74.2.923-933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oie KL, Pickup DJ. Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-kappaB activation. Virology. 2001;288:175–87. doi: 10.1006/viro.2001.1090. [DOI] [PubMed] [Google Scholar]
- 31.Guerra S, Lopez-Fernandez LA, Conde R, Pascual-Montano A, Harshman K, Esteban M. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J Virol. 2004;78:5820–34. doi: 10.1128/JVI.78.11.5820-5834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodge JW, Higgins J, Schlom J. Harnessing the unique local immunostimulatory properties of modified vaccinia Ankara (MVA) virus to generate superior tumor-specific immune responses and antitumor activity in a diversified prime and boost vaccine regimen. Vaccine. 2009;27:4475–82. doi: 10.1016/j.vaccine.2009.05.017. Epub 2009 May 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ledbetter JA, Shu G, Gallagher M, Clark EA. Augmentation of normal and malignant B cell proliferation by monoclonal antibody to the B cell-specific antigen BP50 (CDW40) J Immunol. 1987;138:788–94. [PubMed] [Google Scholar]
- 34.Crawford DH, Catovsky D. In vitro activation of leukaemic B cells by interleukin-4 and antibodies to CD40. Immunology. 1993;80:40–4. [PMC free article] [PubMed] [Google Scholar]
- 35.Romano MF, Lamberti A, Tassone P, Alfinito F, Costantini S, Chiurazzi F, et al. Triggering of CD40 antigen inhibits fludarabine-induced apoptosis in B chronic lymphocytic leukemia cells. Blood. 1998;92:990–5. [PubMed] [Google Scholar]
- 36.Kitada S, Zapata JM, Andreeff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;106:995–1004. doi: 10.1046/j.1365-2141.1999.01642.x. [DOI] [PubMed] [Google Scholar]
- 37.Kater AP, Evers LM, Remmerswaal EB, Jaspers A, Oosterwijk MF, van Lier RA, et al. CD40 stimulation of B-cell chronic lymphocytic leukaemia cells enhances the anti-apoptotic profile, but also Bid expression and cells remain susceptible to autologous cytotoxic T-lymphocyte attack. Br J Haematol. 2004;127:404–15. doi: 10.1111/j.1365-2141.2004.05225.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.