

Abstract
Background
TOR1A encodes a chaperone-like AAA-ATPase whose ΔGAG (ΔE) mutation is responsible for an early-onset, generalized dystonia syndrome. Because of the established role of the TOR1A gene in heritable generalized dystonia (DYT1), a potential genetic contribution of TOR1A to the more prevalent and diverse presentations of late-onset, focal dystonia has been suggested.
Results
A novel TOR1A missense mutation (c.613T>A, p.F205I) in a patient with late-onset, focal dystonia is reported. The mutation occurs in a highly evolutionarily conserved region encoding the AAA-ATPase domain. Expression assays revealed that expression of F205I or ΔE, but not wildtype TOR1A produced frequent intracellular inclusions.
Conclusions
We have identified a novel, rare TOR1A variant in an individual with late-onset, focal dystonia and provided evidence that the mutation impairs TOR1A function. Together these findings raise the possibility that this novel TOR1A variant may contribute to the expression of dystonia. In light of these findings, a more comprehensive genetic effort is warranted to identify the role of this and other rare TOR1A variants in the expression of late-onset, focal dystonia.
Keywords: dystonia, TOR1A, sequence variant, depression, torsinA
INTRODUCTION
Dystonia is a movement disorder characterized by sustained, involuntary postures. As is the case for many neurological disorders, genetic causes for some relatively rare forms are known, whereas insight to the causes of the more prevalent late-onset, “idiopathic” forms of the disorder is far more limited. While a genetic basis for late-onset, focal dystonias has been hypothesized, a genetic cause for this more widely prevalent form of dystonia has been elusive1. The ΔGAG mutation of the TOR1A gene (DYT1) is responsible for an autosomal dominant, early-onset generalized dystonia 2. Because of this association, other studies have explored the possibility that ΔGAG or other mutations of the TOR1A gene may contribute to the expression of idiopathic focal dystonia 3–12. For the most part, no single genetic association has been found to explain a major fraction of late-onset, focal dystonias; suggesting that if a genetic contribution were present, it may involve heterogeneous genetic contributions and/or complex gene-environment interactions. Here, we identify a novel sequence variant of TOR1A in a patient with late-onset, focal dystonia and study the functional consequences of this mutation.
PATIENT DETAILS
The patient was in his fifth decade of life when he reported involuntary jaw movements and grimacing beginning 3 years prior. The movements could not be suppressed voluntarily and were not associated with an urge to perform them. The patient had a history of remote exposure (greater than 10 years prior) to dopamine receptor-blocking agents and quetiapine. The patient has been treated with lithium monotherapy for bipolar disorder for more than 10 years with serum levels documented in the normal therapeutic range. No other history of potential inciting exposures, bruxism or trauma was identified. The patient’s medical history was unremarkable for other neurological conditions, but did include a history of bipolar disorder. The patient’s family history was negative for a history of dystonia, but did include tremor and depression. Neurological examination revealed an orofacial movement, primarily involving jaw retraction, consistent with an oromandibular dystonia. Repetitive chewing movements were not observed. Cogwheel tone without rigidity and a mild tremor present with action and posture but not rest was present in the upper extremities. Deep tendon reflexes at the Achilles tendon were absent bilaterally. No other neurological abnormalities were present. Diagnostic evaluations included normal serum electrolytes, complete blood count, peripheral blood smear, erythrocyte sedimentation rate, prothrombin and partial thromboplastin time, ceruloplasmin and MRI of the brain. The facial movements responded well to treatment with anticholinergic agents.
METHODS
Subjects
All subjects were consented and enrolled as part of three NIMH-supported mood disorder study databases at Duke University: the Mental Health Clinical Research Center for the Study of Depression in Late Life, the Neurocognitive Outcomes of Depression in the Elderly study, and Bipolar Disorder in Late Life study. Individuals were recruited for those studies based on the presence of a psychiatric condition (depression or bipolar disorder). Control subjects were individuals without the psychiatric conditions of interest. All studies, including this genetic analysis, were approved by the Duke Institutional Review Board.
Sequencing and genotype analysis
Exonic sequences of TOR1A were sequenced in their entirety in the index patient. PCR primers were: Exon 1 5′-GGAAGCGTGGGTCTGGC/5′-TTCAGCCCTAGTGCCATCG; Exon 2 5′-TTTCTTATGGGCTGTAAATGTGG/5′-AACAAAGAGACCCCAAACCC; Exons 3 and 4 5′-AGAAGGAGCTGATTGATGGC/5′-ACACTTAGGGTGCAGGATTAGG; Exon 5 5′-GTCTATAGGGCAGGTGGGTG/5′-GTGGAAGTGTGGAAGGACTG.
Sequencing was performed on ABI 3730 according to standard protocols. Analysis was performed with Sequencher software (GeneCodes Inc.). Genotyping was performed by applying the Taqman platform to custom assays. Eight hundred de-identified population controls were genotyped for the c.613T>A change using the identified mutant chromosome as positive control.
Transfection and immunocytochemistry
Mammalian expression plasmids containing human wildtype and ΔGAG TOR1A cDNA with EGFP fused at the C-terminus were kindly provided by Dr. William Dauer (University of Michigan). Quickchange Site-Directed Mutagenesis Kit (Stratagene) was used to introduce the F205I mutation into the TOR1A protein using the primers, 5′-CCAGAAAGCCATGTTCATAATTCTCAGCAATGCTGGAGC-3′ and 5′-GCTCCAGCATTGCTGAGAATTATGAACATGGCTTTCTGG-3′. BHK21 cells grown in 24 well plates containing DMEM with 5% FBS were transfected with 0.25 micrograms of plasmid DNA using GeneJuice Transfection Reagent (Novagen) and examined 48 h post-transfection. Cells were fixed in 4% paraformaldehyde and immunostained with the following primary antibodies: for nuclear envelope, rabbit anti-LaminB1 (Santa Cruz Biotechnology); for endoplasmic reticulum (ER), mouse anti-KDEL (StressGen Biotechnologies); for autophagosome, rabbit anti-MAP1LC3B (Abgent); and for lysosome, rat anti-Lamp1 (Abcam). Secondary antibodies were Alexa Fluor 568-conjugated goat anti-rat, rabbit, or mouse (Molecular Probes). For counting percentage of cells with fluorescent inclusions, cells were visualized using a Zeiss Axioskop fluorescent microscope and 100 cells/experiment were counted from triplicate coverslips for each transfected construct. Inclusions were defined by the presence of focal punctate concentration of EGFP fluorescence that was easily distinguishable from the diffuse, reticular fluorescence typically seen in wildtype TOR1A-expressing cells. Quantification of inclusion size was performed using the morphometry analysis function in Metamorph software (Molecular Devices). For immunolocalization, images were obtained using a Leica SP5 confocal microscope. Images were obtained under identical conditions across constructs for all experiments. No appreciable difference in expression level was detectable among individual cells because optimal exposure conditions were identical. Western immunoblotting of lysates from transfected cultures showed similar expression levels of TOR1A by all constructs (Supplemental Figure 1).
RESULTS
This case report describes an individual with a novel mutation in exon 3 of TOR1A that changed a phenylalanine to isoleucine (c.613T>A, p.F205I). This variant was not identified in 800 anonymized population controls (1600 chromosomes). Because of the presence of dystonia in an individual with a rare, novel TOR1A mutation and the known association of the ΔGAG TOR1A mutation with generalized dystonia2, the pathological significance of this mutation was further explored.
The mutation affects a residue in the AAA domain of the ATPase homologous region of TOR1A 13, 14 (Figure 1A). The phenylalanine at position 205 is highly conserved across species and is part of a highly conserved beta strand motif in the AAA domain 14 (Figure 1B). SIFT software analysis (http://sift.jcvi.org/)15 was performed to predict the probability that this substitution produces deleterious effects on protein function. Scores <0.05 support the hypothesis that the mutation may be pathogenic. Such an analysis of the F205I mutation yielded a score of 0.03 indicating likely pathogenicity.
Figure 1.
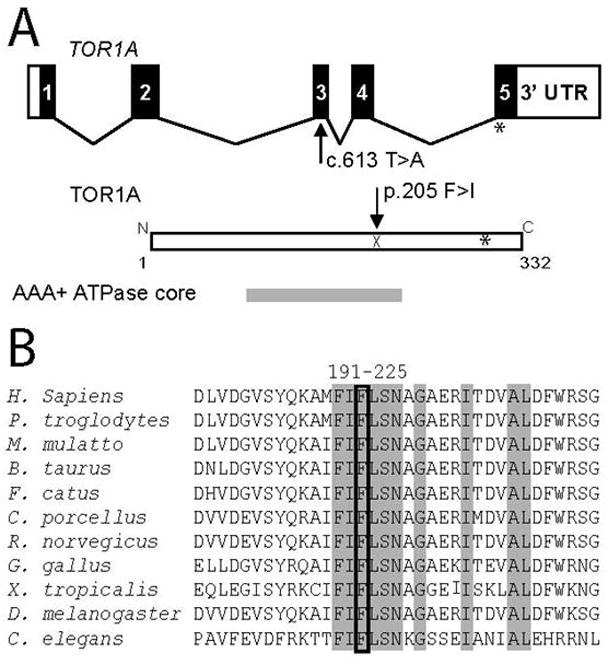
(A) Genomic structure of TOR1A and relative location of novel mutation. Exons are represented by rectangles and coding exonic regions are shown in black. Below is a representation of TOR1A protein with location of ATPase functional domain relative to full length protein shown. For reference, the location of the ΔE(302/303) mutation is shown with an asterisk. (B) Alignment of TOR1A amino acid sequences with residues 191–225 of the human protein. Mutated residue is boxed and surrounding identical residues are shaded grey. Alignments are among Torsin family 1, member A predicted proteins, except for C. elegans (tor-1) and D. melanogaster (torp4a).
To obtain direct experimental evidence for the effect of the mutation on TOR1A function, we tested whether cDNA encoding F205I TOR1A, as a fusion protein with EGFP to aid visualization, produced a similar phenotype as the ΔE mutant TOR1A-EGFP fusion protein when expressed in cultured cells. This assay has been previously used to identify cellular abnormalities characteristic of the ΔE mutation 6, 16–20. In those studies, the ΔE mutant produced TOR1A inclusions that largely colocalized with the nuclear envelope protein, LaminB1. The inclusions are thought to reflect a disruption of normal membrane trafficking as evidenced by ultrastructural studies 18, 19, 21. These abnormalities were not significantly caused by expression of wildtype TOR1A under similar conditions. However, because of recent evidence that wildtype TOR1A may also produce cellular pathology at high expression levels 22, for all experiments, we compared the effects of a cDNA encoding the novel mutation to both a positive control (ΔE mutation) and negative control (wildtype).
Following transient transfection of BHK cells, we found that, consistent with previous reports6, 16–20, expression of ΔE-TOR1A-EGFP frequently produced inclusion bodies in transfected cells (79% of transfected cells) whereas, expression of wildtype TOR1A-EGFP only rarely resulted in cells with detectable inclusion bodies (10%) (Figure 2A). When F205I-TOR1A-EGFP was expressed, cellular pathology that was readily distinguishable from cells expressing wildtype TOR1A-EGFP was observed. In contrast to wildtype TOR1A-EGFP, F205I-TOR1A-EGFP frequently produced inclusions in transfected cells (44% F205I vs. 10% wildtype; p<0.05). While both the F205I mutant and ΔE mutant TOR1A proteins frequently produced inclusions, the ΔE mutant TOR1A produced inclusions in a significantly greater fraction of transfected cells (79% ΔE vs. 44% F205I; p<0.05); an observation that suggests the novel mutant TOR1A may not cause identical pathophysiology as the ΔE mutant.
Figure 2.
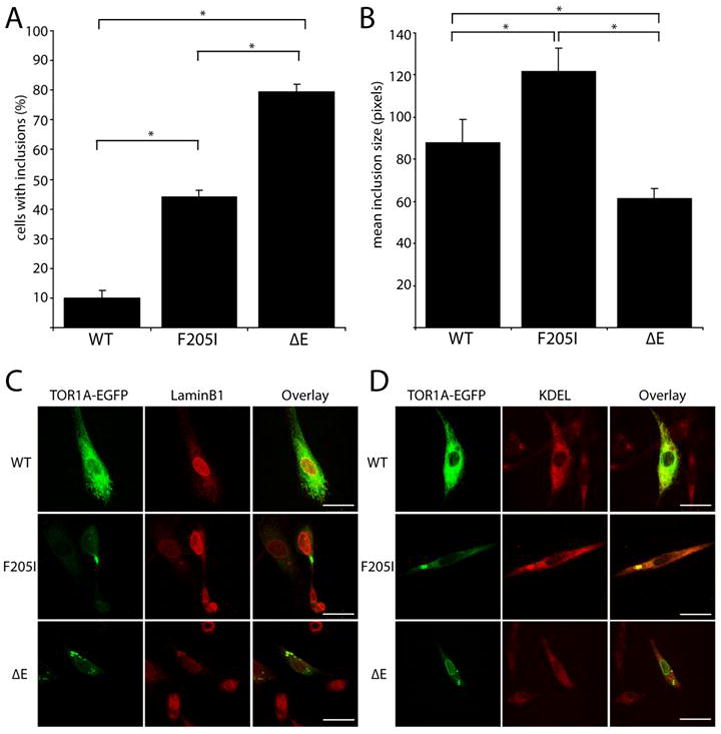
Novel F205I mutation produced inclusions in BHK cell expression studies. (A) Under identical transfection conditions, BHK cells expressing TOR1A-EGFP exhibited fluorescent inclusions in 44% of F205I, 10% of wildtype and 79% of ΔE (*p< 0.05, n= 200 cells per construct from a minimum of two experimental replications). (B) Mean pixel size of TOR1A-EGFP inclusions (* p < 0.05, n= 60 WT, 107 F205I, and 187 ΔE puncta from a minimum of 3 experimental replications). (C, D) Representative images of confocal immunocolocalization of TOR1A-EGFP in BHK cells. LaminB1 and KDEL are nuclear envelope and endoplasmic reticulum markers, respectively. Scale bar indicates 25 microns.
Further analysis of the F205I inclusions revealed that on average these inclusions were larger than those in the ΔE or wildtype conditions (Figure 2B, C, D) raising the possibility that their cellular origin may be different. To investigate this, colocalization of EGFP fluorescence with cellular markers for the nuclear envelope (LaminB1), endoplasmic reticulum (KDEL), lysosomes (Lamp1), and autophagosomes (MAP1LC3B) was examined (Figure 2C,D). As has been previously reported, we found that wildtype TOR1A colocalized with the endoplasmic reticulum marker, KDEL (100%, n=52 cells) but not the nuclear envelope marker, LaminB1 (6%, n=48 cells) whereas the majority of cells expressing ΔE mutant TOR1A had EGFP fluorescence that colocalized with LaminB1 (89%, n=79 cells) and not KDEL (12%, n=68 cells). F205I mutant TOR1A, in contrast to the pronounced colocalization of ΔE TOR1A with nuclear envelope markers, preferentially colocalized with the ER marker, KDEL (96%, n=67 cells) relative to Lamin B1 (19%, n=41 cells). No colocalization was observed with lysosomal or autophagosomal markers (data not shown). Together, these findings demonstrate that both F205I and ΔE TOR1A produce intracellular inclusions frequently in contrast to wildtype protein, but that the subcellular nature of the inclusions is not identical.
DISCUSSION
In this study, we have identified an individual with late-onset, focal dystonia and a novel missense mutation in TOR1A(c.613T>A, p.F205I). It is noteworthy that the index patient and his family had a history of psychiatric illness since an increased incidence of depression has been reported among carriers of the ΔGAG TOR1A mutation independent of the presence of dystonia 23, 24. Given the clinical context of this individual’s rare sequence variant, we further investigated the potential significance of this mutation on TOR1A function.
The c.613T>A, p.F205I TOR1A mutation involves a highly conserved amino acid residue that lies within an important functional domain of the protein and is predicted to have a pathogenic effect by protein function prediction algorithms. Direct experimental evidence that F205I mutant TOR1A was functionally impaired was obtained using cell culture expression studies. We found that the F205I mutation produces frequent inclusions when expressed in cultured cells, a phenotype shared by the ΔE mutant TOR1A, but not wildtype protein. Although both F205I and ΔE TOR1A frequently produced cellular pathology, we also identified differences suggesting that the pathophysiology of F205I mutant TOR1A is unlikely to be identical to that caused by the ΔE mutant TOR1A; an observation possibly reflective of the milder clinical phenotype in which the F205I mutation was identified. Together, these observations raise the possibility that the c.613T>A, p.F205I variant of TOR1A may contribute to the presentation of late-onset, focal dystonia by altering the normal activity of TOR1A.
To date, an understanding of the genetic basis for focal dystonia has been limited 3–12, 25. The potential for TOR1A to contribute to the expression of focal dystonias is suggested by the observation that in some cases, the ΔGAG mutation which normally causes generalized dystonia may produce a focal dystonia25. However, evidence that other mutations of TOR1A are involved in dystonia is much sparser3, 25. One limitation of prior studies is that most involve screening only for known dystonia mutations that are usually associated with generalized forms of the disorder. In these cases, contribution of novel rare sequence variants could be missed. Recently, the most common TOR1A variant identified to date, p.D216H, which is estimated to be present in approximately 12% of the population, has been implicated in the expression of focal dystonia and as a genetic modifier for the expression of generalized dystonia due to theΔGAG TOR1A mutation 12, 26. Additionally, an individual with early-onset, generalized dystonia and a novel p.R288Q TOR1A mutation has been described 27. Interestingly, both of these mutations produce cellular abnormalities in vitro, strengthening the potential association between in vitro phenotype and pathogenicity6, 27.
In summary, we have identified an individual with late-onset, focal dystonia and a novel TOR1A mutation (c.613T>A, p.F205I) and presented evidence that the mutation impairs TOR1A function. We suggest that, in light of these findings, a more comprehensive genetic evaluation is warranted to investigate the role of this and other rare sequence variants of TOR1A in late-onset, focal dystonia.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by the Institute for Genome Sciences and Policy, Duke University (N.C.), the Jake’s Ride Grant from the Bachmann-Strauss Dystonia and Parkinson Foundation (N.C.), National Institute of Mental Health Grants R01-MH57027 (J.L.B.), P50 MH60451, R01 MH54846 and K24 MH70027 (D.C.S.). We thank the patients and volunteers who kindly agreed to participate in this study.
Footnotes
COMPETING INTERESTS: The authors have no financial conflicts to disclose.
References
- 1.Defazio G, Berardelli A, Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain. 2007;130(Pt 5):1183–93. doi: 10.1093/brain/awl355. [DOI] [PubMed] [Google Scholar]
- 2.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17(1):40–8. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 3.Clarimon J, Asgeirsson H, Singleton A, Jakobsson F, Hjaltason H, Hardy J, Sveinbjornsdottir S. Torsin A haplotype predisposes to idiopathic dystonia. Ann Neurol. 2005;57(5):765–7. doi: 10.1002/ana.20485. [DOI] [PubMed] [Google Scholar]
- 4.Clarimon J, Brancati F, Peckham E, Valente EM, Dallapiccola B, Abruzzese G, Girlanda P, Defazio G, Berardelli A, Hallett M, Singleton AB. Assessing the role of DRD5 and DYT1 in two different case-control series with primary blepharospasm. Mov Disord. 2007;22(2):162–6. doi: 10.1002/mds.21182. [DOI] [PubMed] [Google Scholar]
- 5.Kamm C, Asmus F, Mueller J, Mayer P, Sharma M, Muller UJ, Beckert S, Ehling R, Illig T, Wichmann HE, Poewe W, Mueller JC, Gasser T. Strong genetic evidence for association of TOR1A/TOR1B with idiopathic dystonia. Neurology. 2006;67(10):1857–9. doi: 10.1212/01.wnl.0000244423.63406.17. [DOI] [PubMed] [Google Scholar]
- 6.Kock N, Naismith TV, Boston HE, Ozelius LJ, Corey DP, Breakefield XO, Hanson PI. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum Mol Genet. 2006;15(8):1355–64. doi: 10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- 7.Sibbing D, Asmus F, Konig IR, Tezenas du Montcel S, Vidailhet M, Sangla S, Oertel WH, Brice A, Ziegler A, Gasser T, Bandmann O. Candidate gene studies in focal dystonia. Neurology. 2003;61(8):1097–101. doi: 10.1212/01.wnl.0000090560.20641.ab. [DOI] [PubMed] [Google Scholar]
- 8.Kabakci K, Hedrich K, Leung JC, Mitterer M, Vieregge P, Lencer R, Hagenah J, Garrels J, Witt K, Klostermann F, Svetel M, Friedman J, Kostic V, Bressman SB, Breakefield XO, Ozelius LJ, Pramstaller PP, Klein C. Mutations in DYT1: extension of the phenotypic and mutational spectrum. Neurology. 2004;62(3):395–400. doi: 10.1212/01.wnl.0000113024.84178.f7. [DOI] [PubMed] [Google Scholar]
- 9.van den Bos M, Marotta R, Goldup S, Chataway T, Firgaira F, Thyagarajan D. Writer’s cramp in an Australian pedigree with DYT1 dystonia. J Clin Neurosci. 2004;11(5):537–9. doi: 10.1016/S0967-5868(03)00226-1. [DOI] [PubMed] [Google Scholar]
- 10.Hague S, Klaffke S, Clarimon J, Hemmer B, Singleton A, Kupsch A, Bandmann O. Lack of association with TorsinA haplotype in German patients with sporadic dystonia. Neurology. 2006;66(6):951–2. doi: 10.1212/01.wnl.0000203344.43342.18. [DOI] [PubMed] [Google Scholar]
- 11.Defazio G, Matarin M, Peckham EL, Martino D, Valente EM, Singleton A, Crawley A, Aniello MS, Brancati F, Abbruzzese G, Girlanda P, Livrea P, Hallett M, Berardelli A. The TOR1A polymorphism rs1182 and the risk of spread in primary blepharospasm. Mov Disord. 2009;24(4):613–6. doi: 10.1002/mds.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruggemann N, Kock N, Lohmann K, Konig IR, Rakovic A, Hagenah J, Schmidt A, Ziegler A, Jabusch HC, Siebner H, Altenmuller E, Munchau A, Klein C. The D216H variant in the DYT1 gene: a susceptibility factor for dystonia in familial cases? Neurology. 2009;72(16):1441–3. doi: 10.1212/WNL.0b013e3181a1861e. [DOI] [PubMed] [Google Scholar]
- 13.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Jacoby D, Penney J, Risch NJ, Fahn S, Gusella JF, Breakefield XO. The gene (DYT1) for early-onset torsion dystonia encodes a novel protein related to the Clp protease/heat shock family. Adv Neurol. 1998;78:93–105. [PubMed] [Google Scholar]
- 14.Zhu L, Wrabl JO, Hayashi AP, Rose LS, Thomas PJ. The torsin-family AAA+ protein OOC-5 contains a critical disulfide adjacent to Sensor-II that couples redox state to nucleotide binding. Mol Biol Cell. 2008;19(8):3599–612. doi: 10.1091/mbc.E08-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 16.Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000;9(9):1403–13. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- 17.Kustedjo K, Bracey MH, Cravatt BF. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem. 2000;275(36):27933–9. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- 18.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci U S A. 2004;101(3):847–52. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24(11):2593–601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bragg DC, Camp SM, Kaufman CA, Wilbur JD, Boston H, Schuback DE, Hanson PI, Sena-Esteves M, Breakefield XO. Perinuclear biogenesis of mutant torsin-A inclusions in cultured cells infected with tetracycline-regulated herpes simplex virus type 1 amplicon vectors. Neuroscience. 2004;125(3):651–61. doi: 10.1016/j.neuroscience.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 21.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48(6):923–32. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Grundmann K, Reischmann B, Vanhoutte G, Hubener J, Teismann P, Hauser TK, Bonin M, Wilbertz J, Horn S, Nguyen HP, Kuhn M, Chanarat S, Wolburg H, Van der Linden A, Riess O. Overexpression of human wildtype torsinA and human DeltaGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol Dis. 2007;27(2):190–206. doi: 10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Jahanshahi M. Behavioral and psychiatric manifestations in dystonia. Adv Neurol. 2005;96:291–319. [PubMed] [Google Scholar]
- 24.Heiman GA, Ottman R, Saunders-Pullman RJ, Ozelius LJ, Risch NJ, Bressman SB. Increased risk for recurrent major depression in DYT1 dystonia mutation carriers. Neurology. 2004;63(4):631–7. doi: 10.1212/01.wnl.0000137113.39225.fa. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz CS, Bressman SB. Genetics and treatment of dystonia. Neurol Clin. 2009;27(3):697–718. vi. doi: 10.1016/j.ncl.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Risch NJ, Bressman SB, Senthil G, Ozelius LJ. Intragenic Cis and Trans modification of genetic susceptibility in DYT1 torsion dystonia. Am J Hum Genet. 2007;80(6):1188–93. doi: 10.1086/518427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zirn B, Grundmann K, Huppke P, Puthenparampil J, Wolburg H, Riess O, Muller U. Novel TOR1A mutation p.Arg288Gln in early-onset dystonia (DYT 1) J Neurol Neurosurg Psychiatry. 2008 doi: 10.1136/jnnp.2008.148270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.