

Abstract
Only rarely do corticotroph pituitary tumors become invasive leading to symptoms caused by compression of cranial nerves and other local structures. When aggressive pituitary neuroendocrine tumors do develop, conventional treatment options are of limited success. A 50-year-old man developed a giant invasive corticotroph pituitary tumor 2 years after initial presentation. His tumor and symptoms failed to respond to maximal surgical, radio-surgical, radiation and medical therapy and a bilateral adrenalectomy was done. He subsequently developed rapid growth of his tumor leading to multiple cranial nerve deficits. He was administered salvage chemotherapy with capecitabine and temozolomide (CAPTEM), a novel oral chemotherapy regimen developed at our institution for treatment of neuroendocrine tumors. After two cycles of CAPTEM, his tumor markedly decreased in size and ACTH levels fell by almost 90%. Despite further decreases in ACTH levels, his tumor recurred after 5 months with increased avidity on PET scan suggesting a transformation to a more aggressive phenotype. Temozolomide had been reported to be effective against other pituitary tumors and this case adds to this literature demonstrating its use along with capecitabine (CAPTEM) against a corticotroph tumor. Further evaluation of the CAPTEM regimen in patients with pituitary neuroendocrine tumors which fail to respond to classic treatments is warranted.
Introduction
Pituitary corticotroph adenomas rarely develop into aggressive and invasive tumors [1]. Many of these occur in the setting of Nelson's syndrome, defined as clinically significant enlargement of a corticotroph adenoma after bilateral adrenalectomy, and growth typically occurs over months to years. Standard treatment options include surgical debulking and radiotherapy [2]. When conventional treatments fail to control aggressive corticotroph tumors, few options remain.
Eleven cases of the successful use of temozolomide alone to treat pituitary tumors have been reported [3–9]. We describe a patient with a corticotroph adenoma who failed standard treatment and developed rapid tumor growth with life threatening cranial nerve deficits. We used oral temozolomide and capecitabine (CAPTEM), which led to an initial radiographic and symptomatic response and a correlative decrease in corticotropin (ACTH) levels. This is the first report of the use of the CAPTEM regimen against a pituitary neuroendocrine tumor. Possible pathophysiologic mechanisms underlying the initial response and eventual recurrence of the tumor are discussed. Further, we comment on other reports of temozolomide use, the potential mechanism for CAPTEM's effect and its potential applicability to patients with invasive pituitary neuroendocrine tumors.
Case report
A previously healthy 50-year-old man presented with headaches and the sudden onset of diploplia due to left cranial nerve (CN) VI palsy and was found to have a pituitary mass. Endocrine workup demonstrated hypogonadotropic hypogonadism, a normal prolactin of 6.3 ng/ml, an elevated ACTH of 113 pg/ml (9–52 pg/ml) but a normal 24-hour urinary free cortisol (UFC) of 31 μg/day (normal <50 μg/day). He underwent gross total transsphenoidal resection of the tumor with symptom resolution. Pathology revealed a 1.5 cm adenoma with sheets of ACTH positive cells with a high Ki-67 labeling index (LI) of 31% (Fig. 1) (typical pituitary adenoma <2%). He initially denied symptoms of Cushing's syndrome except reporting that he tanned easily. No physical exam findings suggested Cushing's.
Fig. 1.
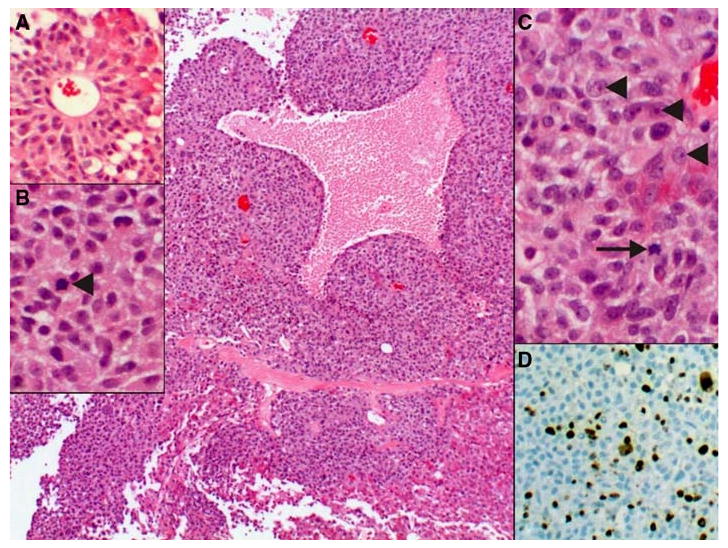
Pituitary tumor, arranged in sheets of pleomorphic epithelial cells embedded in a delicate capillary network. Areas of coagulative necrosis were identified, most likely due to infarction secondary to the large size of the tumor. a In areas, pseudorosette-formation around larger caliber vessels was present. b The cells have a moderate to abundant amount of granular eosinophilic cytoplasm, round to oval nuclei with many mitotic figures (arrowhead). c The neoplastic cells have distinct, eosinophilic nucleoli (arrowheads) and hyperchromatic chromatin. Mitotic figures were prominent (arrow). d The high proliferation rate as indicated by the elevated mitotic count was corroborated by a high Ki67 (mib-1) labeling index, reaching up to 31% of the cells
His CN VI palsy recurred two months later and pituitary magnetic resonance image (MRI) showed recurrence of a 2.8 cm tumor in the left cavernous sinus encasing the left internal carotid artery. Due to the recurrent tumor's location, an 80% partial resection was done with symptomatic improvement. Pathology was similar to the initial resection. His diplopia recurred within weeks. MRI appeared unchanged and the patient underwent gamma knife radio-surgery of the tumor without clinical or radiographic effect.
Six months after his initial presentation, he developed classic signs and symptoms of Cushing's syndrome including central weight gain, a dorsal fat pad, moon facies, easy bruising, hyperpigmentation and hypertension. Laboratory values include an ACTH of 151 pg/ml (9–52 pg/ml), a serum cortisol of 28 μg/dl (5–25 μg/dl) and a urinary free cortisol (UFC) of 498 μg/d (<50 μg/d). Ketoconazole was begun and was titrated to a dose of 400 mg orally three times a day, which controlled his CD symptoms for about 6 months.
One year after presentation, he had a total left CN VI palsy, an ACTH of 767 pg/ml and ketoconazole no longer controlled his symptoms nor UFC levels. MRI showed growth of the tumor by 0.7 cm. He underwent standard conformal external beam radiotherapy (XRT) with 5040 cGy in 28 fractions over 2 months with a decrease in his ACTH to 375 pg/ml but no change in his UFC or MRI. Over the next year, UFC and ACTH levels continued to rise despite addition of metyrapone 250 mg orally twice a day, octreotide 300 μg subcutaneously three times a day and oral cabergoline 0.5 mg twice a week. After initiation of octreotide, imaging indicated relative stability of the tumor. Two years after presentation, he underwent bilateral adrenalectomy because medical therapy failed to control his Cushings symptoms, which included increasing fatigue, proximal muscle weakness, escalating antihypertensive requirements, poor memory and hyperglycemia. Just prior to his bilateral adrenalectomy, his ACTH was 733 pg/ml and UFC was 932 μg/d.
After his adrenalectomy, ketoconazole, metyrapone, octreotide and cabergoline were discontinued and the patient was placed on twice physiologic corticosteroid and mineralocorticoid replacement. Immediately postoperatively, he noted some improvement in symptoms but he began to experience a near constant headache. He was readmitted 2 weeks after his adrenalectomy with dysphagia and aspiration of liquids, new difficulty moving his right eye laterally, numbness over his left jaw and worsening hoarseness. Physical exam revealed hyperpigmentation, bitemporal hemianopsia, anosmia, fixed medial left eye deviation with minimal pupil reactivity, decreased lateral right eye movement, decreased sensation over his left face, uvula deviation to the left and a poor gag reflex. His ACTH was 2,541 pg/ml, a 347% increase from preoperative levels. Levels of alpha subunit, chromogranin A and thyroid function were normal. An HIV test was negative. MRI demonstrated growth of the parasellar tumor further into the left cavernous sinus, the sphenoid sinus and inferiorly to the posterior margin of the clivus and into the basion (Fig. 2b). Because the tumor encased vital structures and was radioresistant, he was deemed not to be a candidate for further neurosurgical or radiation therapy. Although octreotide was restarted immediately and stress dose steroids and rosiglitazone (8 mg/day) were added with an initial reduction in his ACTH by 26%, his symptoms did not improve but worsened such that he required enteral feeding due to his severe dysphagia. An octreotide scan showed octreotide avidity of the tumor and no evidence of extracranial metastases. Octreotide was changed to sandostatin LAR® 30 mg every month.
Fig. 2.

Magnetic Resonance Imaging (MRI) demonstrating changes in the tumor over time. a. Sagittal view of tumor prior to adrenalectomy. b Sagittal and coronal views of the tumor 2 weeks after adrenalectomy demonstrating aggressive growth of tumor anteriorly into the sinuses, inferiorly to invade multiple cranial nerves as well as anteriorly into the suprasellar region. c Sagittal and coronal views of tumor after two cycles of the CAPTEM regimen with marked decrease in the size of the tumor
A trial of salvage chemotherapy with capecitabine (CAP) and temozolomide (TEM) was initiated (CAPTEM) because of our prior experience with this regimen against other neuroendocrine tumors. This protocol, designed in our laboratory to target neuroendocrine tumors, consists of 28 day cycles with oral CAP 1,000 mg twice daily on days 1–14 and oral TEM 100–200 mg/m2/day divided into bid dosing on days 10–14. Because he had no prior chemotherapy he was given 200 mg/m2/day for 5 days on days 10–14 of capecitabine. No chemotherapy is given on days 15–28. The regimen was well tolerated by the patient without any grade 2, 3 or 4 toxicities and no myelotoxicity, diarrhea or hand-foot syndrome. After two cycles, MRI showed a 75% decrease in tumor size with residual in the left sella and cavernous sinus (Fig. 2). Concurrently, his ACTH level decreased to 309 pg/ml from 1,874 pg/ml and his symptoms improved significantly with return of extraocular movements on the right and decreased dysphagia. His performance status improved from three to one. ACTH was felt to be an adequate tumor marker and levels were followed monthly (Fig. 3). After 4 cycles of CAPTEM, ACTH production fell by >95% to 85 pg/ml, his headaches resolved, his CN deficits improved such that he could eat solid foods and his hyperpigmentation decreased.
Fig. 3.
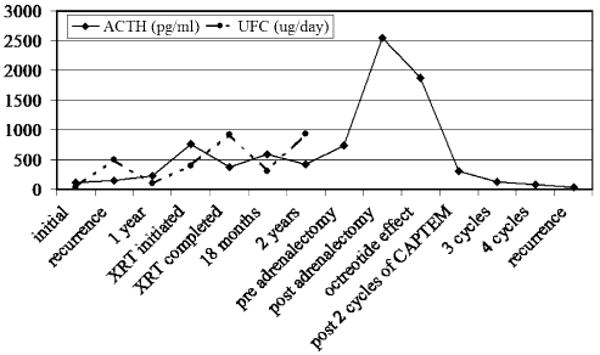
Corticotropin (ACTH) and urinary free cortisol (UFC) trends over the patient's course. A dramatic rise in ACTH is seen post adrenalectomy and levels fall after initiation of the capecitabine and temozolomide (CAPTEM) regimen
However, he returned to the hospital 5 months after initiation of CAPTEM with recurrence of his CN dysfunctions. ACTH level at this time was 36 pg/ml despite MRI evidence of tumor re-growth to its prior dimensions and with further sinus extension. A fluorodeoxyglucose (FDG)-positron emission tomography (PET)-contrast enhanced CT scan of the head demonstrated intense FDG avidity of the mass with a standardized uptake value (SUV) of 14.6/16. The impression was that CAPTEM had eliminated the well-differentiated neuroendocrine corticotroph tumor cells but there had been escape of a population of poorly differentiated small cell neuroendocrine cells and the tumor now behaved as a small cell carcinoma. He noted new back pain and a bone scan was consistent with metastatic disease at the T2 and T3 vertebrae and two ribs but no radiographically positive lesions were seen in this location on X-ray or MRI. He received two cycles of standard etoposide and cisplatin for small cell carcinoma with decreased tumor volume (40%) in the head on MRI. However, he did not tolerate the etoposide/cisplatin regimen hematogically despite reduced doses (50%) and chose to stop all oncologic treatment, enter hospice and he died 1 month later. Autopsy was not performed per the patient's wishes.
Discussion
Our patient had a highly atypical and aggressive corticotroph adenoma that did not meet criteria for pituitary carcinoma, which requires metastases [10], until his final recurrence. At the time of the recurrence, however, his ACTH was normal suggesting that these tumor cells were not ACTH producing cells but rather a more aggressive poorly differentiated small cell type. His initial tumor had an extremely high Ki67 LI which is a measure of tumoral mitotic activity and may have 97% specificity for atypical adenomas when greater than three percent [11]. Despite prior maximal amounts of radiotherapy, our patient's tumor grew rapidly over only 2 weeks after adrenalectomy with concomitant rise in ACTH by 346%. After adrenalectomy for CD, the incidence of Nelson's syndrome may be as high as 39% over 3 years without prior pituitary irradiation [12] and is reduced 50% by prior irradiation [13]. Accelerated growth of ACTH-secreting cells due to the loss of glucocorticoid inhibition typically occurs over months to years. Neither a high Ki67 labeling index nor presence of mitoses are predictive of Nelson's syndrome [12]. In retrospect, octreotide may have contributed to tumor stabilization. Octreotide targets primarily somatostatin receptor subtype 2 (sst2) and less efficiently sst5 and is generally ineffective in lowering ACTH levels in CD as corticotroph adenomas predominantly exhibit sst5 [14]. Clearly, our patient had an unusually aggressive tumor that demonstrated unprecedented growth after adrenalectomy to become life threatening.
Our patient presented with evidence of mass effect of the tumor and without florid CD symptoms. Silent corticotroph adenomas have been described and are reported to be more aggressive and locally invasive than typical corticotroph microadenomas which cause CD [15]. There are reports of patients with large, invasive, silent corticotroph tumors evolving into CD years after initial presentation [16, 17]. This patient's initially normal 24 h urinary free cortisol and lack of clinical signs of Cushing's syndrome despite an elevated plasma ACTH level may have been due to the rapidity of his tumor growth that did not permit signs to become manifest. Alternatively, initial tumoral secretion could have been that of biologically inactive ACTH and precursor of ACTH (POMC) which has been demonstrated in some invasive atypical macroadenomas [18]. The nature of ACTH produced by this patient's tumor may have changed or eventually, as the tumor volume increased, enough biologically active ACTH was produced to lead to Cushing's syndrome.
Due to the severity of his cranial neuropathies and because we had exhausted all conventional treatments, we initiated alternative treatments. We chose rosiglitazone, a peroxisome proliferator-activated receptor gamma (PPAR-gamma) agonist, because PPAR-gamma is expressed on pituitary tumors and because rosiglitazone has been reported to benefit some patients with CD [19] and Nelson's Syndrome [20] and to inhibit growth of pituitary cell lines in vitro [21]. However, it is unlikely that rosiglitazone contributed significantly to his tumor shrinkage as other studies show no benefit of rosiglitazone to patients with Nelson's syndrome [22] and because his ACTH levels did not decrease after initiation of rosiglitazone.
Treatment was also initiated with the chemotherapy regimen CAPTEM, which consists of sequential use of capecitabine and temozolomide and was developed in our lab (RLF). Capecitabine is an oral chemotherapeutic agent which is converted to 5-fluorouracil (5-FU) in vivo by the enzyme, thymidine phosphorylase, which is present in higher concentrations in neoplastic versus non-neoplastic tissue [23]. 5-FU is metabolized to 5-dUMP which produces deficiency of thymidylate by inhibiting thymidylate synthase (TS) which leads to decreased deoxyribonucleic acid (DNA) synthesis and inhibition of cell division by reducing conversion of UMP to dTMP by TS, thus a thymidylate deficiency occurs. Temozolomide is a lipophilic imadozotetrazine derivative which is converted to a methylating alkylator agent that causes DNA damage at any point in the cell cycle through base pair mismatch of O6methyl-guanine with thymidine in the sister chromatid instead of cytosine. The mismatch repair enzymes misread the methylated guanine as an adenosine and place thymidine in the sister chromatid. Thus, the rationale for utilizing capecitabine for 10 days before the addition of temozolamide is the decreased thymidine levels intracellularly accentuates the mismatch repair process such that the deficiency of thymidine leads to a break in the DNA, a potent stimulus for apoptosis. In a carcinoid (BON) neuroendocrine tumor (NET) cell line, the combination is superior for inducing apoptosis by 2–3 fold over temozolamide alone when given in the sequence of capecitabine (5-FU) preceding temozolomide (TMZ) [24, 25] Temozolomide also causes loss of the DNA repair enzyme, methyl-guanine methyl transferase (MGMT), a.k.a O6-alkyl-guanyl-alkyltransferase which is covalently bound and consumed when MGMT binds to the methylated guanine to repair the lesion in DNA. Recent studies have demonstrated that lower levels of MGMT may be predictive of tumor aggression in clinically non-functioning pituitary adenomas [26] and fortunately lower MGMT expression may predict a better response to temozolamide therapy [8, 27]. A recent report by Kulke et. al. demonstrated that well differentiated neuroendocrine tumors outside the pituitary have low endogenous levels of MGMT [28].
Recently temozolomide alone had demonstrated sustained effects in 11 separate cases of pituitary tumors. A malignant prolactinoma had a sustained response to temozolomide for 2 years after temozolomide was discontinued [5]. Three pituitary carcinomas, two prolactin, one GH/Prolactin and one LH secreting, had long term partial responses to temozolomide [3, 4, 7]. In one of these tumors, post-treatment pathology demonstrated decrease in the Ki67 LI from 40 to 5%, as well as, an expected increase in necrosis, fibrosis and hemorrhages and an unexpected increase in neuronal transformation [4]. Another benign but invasive prolactinoma which failed dopamine agonist therapy responded to temozolomide with a greater than 30 fold reduction in serum prolactin and decreased tumor volume [6]. Three additional aggressive prolactinomas [7, 8], and a clinically nonfunctioning pituitary adenoma [7] had significant reductions in tumor mass and hormone levels with temozolamide therapy of variable durations. Two aggressive corticotroph tumors had tumor shrinkage and reductions in hormone levels on temozolamide [9]. Other lipophilic alkylating agents such as carmustine and lomustine may also have temporary anti-proliferative effects against pituitary tumors [5]. Some reports have included cases in which the therapy was unsuccessful [8], but it is unknown what percentage of aggressive pituitary tumors may respond to this chemotherapy as no prospective trials have been conducted.
As this is the first reported case of CAPTEM use for a pituitary tumor and no trials have been conducted it is unknown whether this regimen would be superior to temozolomide alone. However, as previously noted, we have data supporting the concept that pretreatment with capecitabine potentiates the cytotoxicity of temozolomide. Simultaneous capecitabine and temozolomide use has been successfully reported for breast cancer brain metastases [29] but this is the first report of their sequential use against a pituitary tumor. Also, our retrospective trial of 18 patients and prospective ongoing trial in 20 patients with metastatic well and moderately differentiated neuroendocrine carcinomas including carcinoids to the liver who failed high dose sandostatin LAR® (60 mg/month) has shown an average response rate of 67% with CAPTEM, with another 25% achieving stable disease with minor toxicities (unpublished data RLF). The standard of care (streptozocin, doxorubicin) only has a 6% response rate and many grade 3, 4 toxicities [24]. In comparison, a recent report showed a 25% response rate with combined temozolomide and thalidomide therapy in metastatic neuroendocrine tumors [30]. Thus, it is plausible, based upon laboratory and ongoing clinical studies, that the CAPTEM regimen may be superior to temozolomide alone against neuroendocrine cancers and possibly pituitary tumors.
This is the third described case of a temozolomide based regimen used against a corticotroph tumor [9]. This patient's tumor volume initially decreased dramatically by about 75% with an associated reduction in ACTH from 1,874 to 309 pg/ml. Although the ACTH continued to fall and eventually normalized, the patient had tumor re-growth. We hypothesize that the relatively differentiated neuroendocrine corticotroph tumor cells continued to respond to the CAPTEM but that other less differentiated cells in the tumor continued to grow and acted a small cell carcinoma. The 40% response to etoposide and cisplatin also support this hypothesis, as this regimen is highly efficacious for small cell carcinomas. As this patient also had radiotherapy prior to the administration of CAPTEM, we cannot be certain that the latter alone was responsible for the marked tumor shrinkage that initially followed its use. However, radiotherapy to pituitary tumors typically does not produce rapid marked tumor shrinkage as occurred in this patient and thus the time course of tumor shrinkage is more suggestive of an effect of the CAPTEM regimen. The efficacy of temozolamide for corticotroph tumor cells is also supported by a recent report of the effective treatment of a patient with Nelson's syndrome with temozolamide resulting in regression of the tumor and marked reduction in ACTH levels [31].
In conclusion, we report a patient with a highly aggressive, invasive corticotroph tumor that exhibited rapid growth post adrenalectomy. While prior reports have demonstrated durable responses to temozolomide, we report a dramatic but short-term response (5 months), to CAPTEM with ultimate progression probably due to selection of poorly differentiated, small anaplastic cells. Given the prior reports of temozolomide use, as well as our case, the use of CAPTEM should be considered in patients with invasive, aggressive pituitary adenomas or carcinomas who fail conventional medical, surgical and radiation therapies. Additional larger scale studies are needed to further define the role of this chemotherapy regimen in the treatment of aggressive pituitary tumors.
Acknowledgments
Presented at Marie S. Thearle, Pamela U. Freda, Jeffrey N. Bruce, Steven R. Isaacson, Yoomi Lee and Robert L. Fine. The use of Temozolomide (Temodar®) and Capecitabine (Xeloda®) for treatment of an aggressive corticotroph pituitary tumor. 89th Annual Meeting of the Endocrine Society, OR. Toronto, Canada, June, 2007. Supported in part by DK073040 (to PUF).
Contributor Information
Marie S. Thearle, Department of Medicine, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, 650 West 168th St. BB 20-05, New York, NY 10032, USA
Pamela U. Freda, Email: puf1@columbia.edu, Department of Medicine, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, 650 West 168th St. BB 20-05, New York, NY 10032, USA; Department of Medicine, Division of Endocrinology, Columbia University College of Physicians and Surgeons, 650 West 168th St. BB 9-905, New York, NY 10032, USA.
Jeffrey N. Bruce, Department of Neurosurgery, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, New York, NY 10032, USA
Steven R. Isaacson, Department of Radiation Oncology, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, New York, NY 10032, USA
Yoomi Lee, Experimental Therapeutics Program, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, New York, NY 10032, USA.
Robert L. Fine, Email: rlf20@columbia.edu, Department of Medicine, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, 650 West 168th St. BB 20-05, New York, NY 10032, USA; Experimental Therapeutics Program, Division of Medical Oncology, Columbia University College of Physicians and Surgeons, New York, NY 10032, USA.
References
- 1.Lubke D, Saeger W, Ludecke DK. Proliferation markers and EGF in ACTH-secreting adenomas and carcinomas of the pituitary. Endocr Pathol. 1995;6:45–55. doi: 10.1007/BF02914988. [DOI] [PubMed] [Google Scholar]
- 2.Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. 2005;90:3089–3099. doi: 10.1210/jc.2004-2231. [DOI] [PubMed] [Google Scholar]
- 3.Fadul CE, Kominsky AL, Meyer LP, Kingman LS, Kinlaw WB, Rhodes CH, Eskey CJ, Simmons NE. Long-term response of pituitary carcinoma to temozolomide. Report of two cases. J Neurosurg. 2006;105:621–626. doi: 10.3171/jns.2006.105.4.621. [DOI] [PubMed] [Google Scholar]
- 4.Kovacs K, Horvath E, Syro LV, Uribe H, Penagos LC, Ortiz LD, Fadul CE. Temozolomide therapy in a man with an aggressive prolactin-secreting pituitary neoplasm: morphological findings. Hum Pathol. 2007;38:185–189. doi: 10.1016/j.humpath.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Lim S, Shahinian H, Maya MM, Yong W, Heaney AP. Temozolomide: a novel treatment for pituitary carcinoma. Lancet Oncol. 2006;7:518–520. doi: 10.1016/S1470-2045(06)70728-8. [DOI] [PubMed] [Google Scholar]
- 6.Neff LM, Weil M, Cole A, Hedges TR, Shucart W, Lawrence D, Zhu JJ, Tischler AS, Lechan RM. Temozolomide in the treatment of an invasive prolactinoma resistant to dopamine agonists. Pituitary. 2007 doi: 10.1007/s11102-007-0014-1. [DOI] [PubMed] [Google Scholar]
- 7.Hagen C, Schroeder H, Hansen S, Andersen M. Temozolomide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur J Endocrinol. 2009 doi: 10.1530/EJE-09-0389. [DOI] [PubMed] [Google Scholar]
- 8.McCormack AI, McDonald KL, Gill AJ, Clark SJ, Burt MG, Campbell KA, Braund WJ, Little NS, Cook RJ, Grossman AB, Robinson BG, Clifton-Bligh RJ. Low O6-methylguanine-DNA methyltransferase (MGMT) expression and response to temozolomide in aggressive pituitary tumours. Clin Endocrinol (Oxf) 2009;71:226–233. doi: 10.1111/j.1365-2265.2008.03487.x. [DOI] [PubMed] [Google Scholar]
- 9.Mohammed S, Kovacs K, Mason W, Smyth H, Cusimano MD. Use of temozolomide in aggressive pituitary tumors: case report. Neurosurgery. 2009;64:E773–774. doi: 10.1227/01.NEU.0000339115.12803.4E. Discussion E774. [DOI] [PubMed] [Google Scholar]
- 10.Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, Young WF, Jr, Lloyd RV, Davis DH, Guthrie BL, Schoene WC. Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer. 1997;79:804–812. doi: 10.1002/(sici)1097-0142(19970215)79:4<804::aid-cncr18>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 11.Kontogeorgos G. Predictive markers of pituitary adenoma behavior. Neuroendocrinology. 2006;83:179–188. doi: 10.1159/000095526. [DOI] [PubMed] [Google Scholar]
- 12.Assie G, Bahurel H, Coste J, Silvera S, Kujas M, Dugue MA, Karray F, Dousset B, Bertherat J, Legmann P, Bertagna X. Corticotroph tumor progression after adrenalectomy in Cushing's disease: A reappraisal of Nelson's syndrome. J Clin Endocrinol Metab. 2007;92:172–179. doi: 10.1210/jc.2006-1328. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins PJ, Trainer PJ, Plowman PN, Shand WS, Grossman AB, Wass JA, Besser GM. The long-term outcome after adrenalectomy and prophylactic pituitary radiotherapy in adrenocorticotropin-dependent Cushing's syndrome. J Clin Endocrinol Metab. 1995;80:165–171. doi: 10.1210/jcem.80.1.7829606. [DOI] [PubMed] [Google Scholar]
- 14.van der Hoek J, Lamberts SW, Hofland LJ. The role of somatostatin analogs in Cushing's disease. Pituitary. 2004;7:257–264. doi: 10.1007/s11102-005-1404-x. [DOI] [PubMed] [Google Scholar]
- 15.Horvath E, Kovacs K, Killinger DW, Smyth HS, Platts ME, Singer W. Silent corticotropic adenomas of the human pituitary gland: a histologic, immunocytologic, and ultrastructural study. Am J Pathol. 1980;98:617–638. [PMC free article] [PubMed] [Google Scholar]
- 16.Salgado LR, Machado MC, Cukiert A, Liberman B, Kanamura CT, Alves VA. Cushing's disease arising from a clinically nonfunctioning pituitary adenoma. Endocr Pathol. 2006;17:191–199. doi: 10.1385/ep:17:2:191. [DOI] [PubMed] [Google Scholar]
- 17.Tan EU, Ho MS, Rajasoorya CR. Metamorphosis of a non-functioning pituitary adenoma to Cushing's disease. Pituitary. 2000;3:117–122. doi: 10.1023/a:1009961925780. [DOI] [PubMed] [Google Scholar]
- 18.Raffin-Sanson ML, Massias JF, Dumont C, Raux-Demay MC, Proeschel MF, Luton JP, Bertagna X. High plasma proopiomelanocortin in aggressive adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab. 1996;81:4272–4277. doi: 10.1210/jcem.81.12.8954027. [DOI] [PubMed] [Google Scholar]
- 19.Ambrosi B, Dall'Asta C, Cannavo S, Libe R, Vigo T, Epaminonda P, Chiodini I, Ferrero S, Trimarchi F, Arosio M, Beck-Peccoz P. Effects of chronic administration of PPAR-gamma ligand rosiglitazone in Cushing's disease. Eur J Endocrinol. 2004;151:173–178. doi: 10.1530/eje.0.1510173. [DOI] [PubMed] [Google Scholar]
- 20.Andreassen M, Kristensen LO. Rosiglitazone for prevention or adjuvant treatment of Nelson's syndrome after bilateral adrenalectomy. Eur J Endocrinol. 2005;153:503–505. doi: 10.1530/eje.1.01994. [DOI] [PubMed] [Google Scholar]
- 21.Heaney AP, Fernando M, Melmed S. PPAR-gamma receptor ligands: novel therapy for pituitary adenomas. J Clin Invest. 2003;111:1381–1388. doi: 10.1172/JCI16575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullan KR, Leslie H, McCance DR, Sheridan B, Atkinson AB. The PPAR-gamma activator rosiglitazone fails to lower plasma ACTH levels in patients with Nelson's syndrome. Clin Endocrinol (Oxf) 2006;64:519–522. doi: 10.1111/j.1365-2265.2006.02501.x. [DOI] [PubMed] [Google Scholar]
- 23.Amatori F, Di Paolo A, Del Tacca M, Fontanini G, Vannozzi F, Boldrini L, Bocci G, Lastella M, Danesi R. Thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer and normal mucosa in patients. Pharmacogenet Genomics. 2006;16:809–816. doi: 10.1097/01.fpc.0000230410.07899.bc. [DOI] [PubMed] [Google Scholar]
- 24.Isacoff WH, Moss RA, Pecora AL, Fine RL. Temozolomide/capecitabine therapy for metastatic neuroendocrine tumors of the pancreas. A retrospective review. J Clin Oncol, ASCO Annual meeting proceedings part I. 2006;24:14023. [Google Scholar]
- 25.F D, Fine RL, Schreibman SM. Effective treatment of neuroendocrine tumors with temozolomide and capecitabine. J Clin Oncol, ASCO Annual meeting proceedings. 2005;23:4216. [Google Scholar]
- 26.Widhalm G, Wolfsberger S, Preusser M, Woehrer A, Kotter MR, Czech T, Marosi C, Knosp E. O(6)-methylguanine DNA methyltransferase immunoexpression in nonfunctioning pituitary adenomas: are progressive tumors potential candidates for temozolomide treatment? Cancer. 2009;115:1070–1080. doi: 10.1002/cncr.24053. [DOI] [PubMed] [Google Scholar]
- 27.Kovacs K, Scheithauer BW, Lombardero M, McLendon RE, Syro LV, Uribe H, Ortiz LD, Penagos LC. MGMT immunoexpression predicts responsiveness of pituitary tumors to temozolomide therapy. Acta Neuropathol. 2008;115:261–262. doi: 10.1007/s00401-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 28.Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, Meyerhardt JA, Clark JW, Stuart K, Fuchs CS, Redston MS. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338–345. doi: 10.1158/1078-0432.CCR-08-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera E, Meyers C, Groves M, Valero V, Francis D, Arun B, Broglio K, Yin G, Hortobagyi GN, Buchholz T. Phase I study of capecitabine in combination with temozolomide in the treatment of patients with brain metastases from breast carcinoma. Cancer. 2006;107:1348–1354. doi: 10.1002/cncr.22127. [DOI] [PubMed] [Google Scholar]
- 30.Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, Vincitore M, Michelini A, Fuchs CS. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–406. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 31.Moyes VJ, Alusi G, Sabin HI, Evanson J, Berney DM, Kovacs K, Monson JP, Plowman PN, Drake WM. Treatment of Nelson's syndrome with temozolomide. Eur J Endocrinol. 2009;160:115–119. doi: 10.1530/EJE-08-0557. [DOI] [PubMed] [Google Scholar]