

Abstract
Although increased bone marrow fat in age-related bone loss has been associated with lower trabecular mass, the underlying mechanism responsible remains unknown. We hypothesized that marrow adipocytes exert a lipotoxic effect on osteoblast function and survival through the reversible biosynthesis of fatty acids (FA) into the bone marrow microenvironment. We have used a two-chamber system to co-culture normal human osteoblasts (NHOst) with differentiating pre-adipocytes in the absence or presence of an inhibitor of FA synthase (cerulenin) and separated by an insert that allowed unidirectional trafficking of soluble factors only and prevented direct cell–cell contact. Supernatants were assayed for the presence of FA using mass spectophotometry. After 3 weeks in co-culture, NHOst showed significantly lower levels of differentiation and function based on lower mineralization and expression of alkaline phosphatase, osterix, osteocalcin and Runx2. In addition, NHOst survival was affected by the presence of adipocytes as determined by MTS-formazan and TUNEL assays as well as higher activation of caspases 3/7. These toxic effects were inhibited by addition of cerulenin. Furthermore, culture of NHOst with either adipocyte-conditioned media alone in the absence of adipocytes themselves or with the addition of the most predominant FA (stearate or palmitate) produced similar toxic results. Finally, Runx2 nuclear binding was affected by addition of either adipocyte conditioned media or FA into the osteogenic media. We conclude that the presence of FA within the marrow milieu can contribute to the age-related changes in bone mass and can be prevented by the inhibition of FA synthase.
Keywords: fatty acids; osteoblastogenesis; osteoporosis; lipotoxicity, apoptosis
Introduction
The redistribution of body fat is a prevalent feature of the aging process [1, 2]. With aging, body fat appears to accumulate in depots where it should not, infiltrating normal tissues such as muscle and pancreas [3]. Bone marrow is no exception since fat content increases within this haematopoietic and osteogenic tissue, making it an ectopic fat depot in older aged individuals [1, 4, 5].
Various theories have been proposed for the role and the cause of the increased bone marrow fat [4–6]. It has been proposed that the conversion of osteoblast progenitors into adipocyte-like cells accounts for the increased bone marrow fat at the expense of bone forming cells [4, 5]. It has also been suggested that an inverse relationship exists in the marrow cavity between bone and fat where fat would occupy the space left empty by age-related bone loss or altered haematopoietic capacity [7, 8].
The accumulation of reactive lipids in metabolically active tissues such as pancreatic islets, liver, heart and skeletal muscle leads to lipotoxicity [3, 9]. Lipotoxicity has been shown to contribute to the pathophysiology of insulin resistance, type 2 diabetes, steatotic liver disease and heart failure [3, 9]. In the case of aging bone marrow, similar lipotoxity could impact mature osteoblasts negatively due to their close proximity to marrow adipocytes [10].
In fact, there is evidence suggesting that bone marrow fat plays a toxic role similar to that seen in other organs, resulting in the further loss of mature osteoblasts. In vitro studies have found that co-culture of osteoblasts with adipocytes results in a decrease of osteoblasts proliferation [11]. A further study has associated this negative effect on osteoblast proliferation with the presence of polyunsaturated fatty acids (FA) in the media [12]. In agreement with this in vitro data, studies looking at age-related changes in bone marrow fat in vivo have reported that increasing levels of FA oxidation by bone marrow adipocytes may inhibit osteoblast differentiation [13]. However, the mechanisms and the potential reversibility of this lipotoxic effect have not been assessed.
In this study, we have looked at the potential mechanisms that explain the lipotoxic effect of adipocyte infiltration on bone formation within the bone marrow. Using a system of co-cultures [11] we have exposed human osteoblasts to either human differentiating pre-adipocytes or their conditioned media. We have identified the predominant FA released by the adipocytes in our model and documented their effect on osteoblast differentiation, function and survival. Additionally, this lipotoxic effect was reversed by cerulenin, an inhibitor of FA synthase. Finally, we found that adipocytes affect not only osteoblast proliferation but also their function and survival through the inhibition of Runx2-nuclear binding and the activation of caspases. In conclusion, this evidence could provide a new understanding of the interaction between fat and bone within the marrow microenvironment and the potential regulation and prevention of lipotoxicity on bone metabolism.
Materials and methods
Normal human osteoblasts (NHOst)
NHOst as well as media were purchased from Lonza (Walkersville, MD, USA). Cells were obtained from healthy 24-year-old male donors (n= 3). Cells at passages three and four from time of marrow harvest were used in these experiments. NHOst were plated at a density of 5 × 105 cells in 100 cm2 dishes in growth media at 37°C in a humidified atmosphere of 5% CO2. Growth media were composed of osteoblast basal medium media (C-3208, Lonza) containing 10% of foetal bovine serum (FBS), 0.1% ascorbic acid and 0.1% gentamicin. The cells were allowed to grow for 2 days or until confluence at which point they were differentiated (day 0). Cells were differentiated into mature osteoblasts using a combination of osteoblast basal medium, the Growth Medium kit (CC-4193, Lonza) and finally the differentiation kit, which included 0.5 ml hydrocortisone (200 μM) and 5 ml β-glycerophosphate (1 M).
Normal human pre-adipocytes
Human Pre-adipocytes were obtained at Pennington Biomedical Research Center (Baton Rouge, LA, USA) from liposuction aspirates as previously described [14, 15]. Briefly, aspirates from subcutaneous adipose tissue sites were obtained from males and females (n= 6) undergoing elective procedures in plastic surgical offices of the Baton Rouge region. Ethical approval in accordance with the Human Tissue Act 65, 1983, was obtained from the Ethics Committee at Pennington Biomedical Research Center. The mean age and BMI (±S.D.) of the individuals were 38 ± 14 years and 30.4 ± 7.1, respectively. Tissues were washed three to four times with phosphate buffered saline (PBS) and suspended in an equal volume of PBS/0.5 mM calcium chloride supplemented with 1% bovine serum and 0.1% collagenase type I pre-warmed to 37°C. The tissue was placed in a shaking water bath at 37°C with continuous agitation for 60 min. and centrifuged for 5 min. at 300 ×g at room temperature. The supernatant was removed, and the pelleted stromal vascular fraction was resuspended in stromal medium (Dulbecco’s modified Eagle’s medium/F-12, from Invitrogen Grand Island NY, USA, 10% foetal bovine serum [FBS], 1% antibiotic/antimycotic) and plated at a density of 0.156 ml of tissue digest/cm2 of surface area in T225 flasks using stromal medium for expansion and culture. This initial passage of the primary cell culture is referred to as ‘passage 0’. Following the first 48 hrs of incubation at 37°C at 5% CO2, the cultures were washed with PBS and maintained in stromal medium until they achieved 80–90% confluence. Cells were harvested by trypsin digestion, suspended in 10% dimethylsulfoxide/10%DMEM/F12 Ham’s/80% bovine serum albumin (BSA) at 0.5 × 106 cells/ml, and cryopreserved in liquid nitrogen until required for experimental use.
Normal human pre-adipocytes differentiation
The cryopreserved cells were thawed and plated in stromal medium until they reached confluence. At this time, differentiation was induced using the adipogenic differentiation media composed of Dulbecco’s modified Eagle’s medium/F-12 (pH 8.4) with 3% FBS, 33 μM biotin, 17 μM pantothenate, 1 μM bovine insulin, 1 μM dexamethasone, 0.25 mM isobutylmethylxanthine, 100 units of penicillin, 100 μg of streptomycin and 0.25 μg of Fungizone. After 3 days, Adipogenic differentiation medium was changed to adipocyte maintenance medium, which was identical to the induction medium except for the removal of isobutylmethylxanthine [15].
Oil red O staining and its quantification
To demonstrate adipogenesis, oil red O staining was used as previously described [16]. Differentiated adipocytes were considered those polygonal in shape, with eccentrically located nuclei, considerable cytoplasm and lipid droplets scattered throughout. Mean adipocyte number was determined in 10 fields of cells for each condition. For oil red O quantification, cultures were fixed in Baker’s formal calcium, washed in 60% isopropanol and stained with double-filtered oil red O solution to show for lipid accumulation. Oil red O was extracted from cells using 100% ethanol and measured at 540 nm, using ELx800 Universal Microplate Reader (Bio-Tek Instruments Inc., Winooski, VT, USA). Oil red O concentration was determined against known standards of oil red O (0.02 mg/ml to 10.00 mg/ml).
Co-cultures of adipocytes and osteoblasts
Differentiating pre-adipocytes, were cultured in apical compartments of cell culture inserts (Pore size: 0.45 μm, growth area: 4.2 cm2, Falcon-BD Bioscience, Franklin Lakes, NJ, USA) with NHOst grown in the basal compartment of 6-well plates (growth area: 9.6 cm2). These inserts prevent cell-to-cell contact and allow unidirectional flow of media from the apical to basal compartments while contact between both media is avoided [11]. Pre-adipocytes were seeded on inserts (50,000/cm2) in the absence of underlying NHOst for 48 hrs to reach confluence. Inserts containing pre-adipocytes were transferred to plates with confluent NHOst monolayers (40,000/cm2) in the basal compartment. Both compartments received fresh culture medium and the co-culture was maintained for 3 weeks. Three types of co-culture were performed as previously described [11]: (1) differentiating pre-adipocytes were plated in the apical side of the insert with osteoblasts plated on the basal compartment (AD/OB), with unidirectional media flow going from the adipocyte to the osteoblast side; (2) pre-adipocytes were plated on the basal compartment and osteoblasts plated on apical side (OB/AD), with unidirectional media flow going from the osteoblast to the adipocyte side or (3) adipocytes differentiating media alone were placed in the apical side with NHOst plated in the basal compartment (-/OB). During all culture manipulations, contact between the medium in the bottom and the membrane was carefully avoided. Culture media from the both sides of the membrane were changed every 3 days. Supernatants were collected and placed at –20°C for further experiments and future analysis. After 21 days of co-culture, cells were stained for alkaline phosphatase (ALP) and alizarin red or trypsinized for protein extraction.
Cerulenin treatment
Human differentiating pre-adipocytes were plated in co-culture as previously described [14, 15]. Treatment with either cerulenin (2.5, 5 and 10 nM) or vehicle alone was started at day one of co-cultures. To analyse the effect of cerulenin on the co-cultures, six different conditions were analysed: AD/OB treated and untreated, OB/AD treated and untreated and -/OB treated and untreated. Cells were treated for 21 days with media being replaced every 3 days. Additionally, to assess the potential direct effect of cerulenin on NHOst, cells were plated in 6-well plates and treated for 21 days with either cerulenin (2.5, 5 and 10 nM) or vehicle alone. Media were replaced every 3 days.
Determination of cell proliferation
To determine the effect of cerulenin on adipocyte proliferation, differentiating pre-adipocytes were plated in 96-well plates at a density of 1×104 cells per well. After an initial 24-hr period in culture (time 0), cells were treated with either Cerulenin (10 nM) or vehicle alone. Cell proliferation was evaluated at timed intervals (24, 48 and 72 hrs) by the MTS® assay, according to the manufacturer’s (Promega, Madison, WI, USA) instructions. Proliferation was calculated in percentage of change as compared with time 0.
Determination of osteoblast function
As previously reported [17], osteoblasts placed in osteogenic media express ALP and fully mineralize at week 3 of differentiation. To identify changes in osteoblast function induced by the presence of adipocytes in co-culture ALP staining was performed at week 3 using 0.25% naphthol AS-MX ALP solution with fast blue RR salt (Sigma, St. Louis, MO, USA). After washing with PBS, cells were incubated with naphthol solution mixture for 1 hr at room temperature. The resulting purple, insoluble, granular dye deposit indicated sites of ALP activity. Additionally, calcium deposition was also quantified using 1% alizarin red S (Lab Chem Inc., Pittsburgh, PA, USA). Briefly, after alizarin red staining, matrix mineralization was quantified by extracting the alizarin red staining with 100 mM cetypyridinium chloride (Sigma) at room temperature for 3 hrs. The absorbance of the extracted alizarin red S staining was measured at 570 nm. Data represented as units of alizarin red S per milligram of protein in each culture after correction for cell number. Six wells were analysed per experimental condition. Experiments were performed in triplicate.
Determination of cell viability
To test whether adipocyte-secreted factors have any effect on cell survival, NHOst were seeded in 96-well plates. Upon reaching confluence, the cells were treated with 100 μl/well of supernatant obtained from the osteoblast side of the membrane under each of the six different co-culture plus treatment conditions. At timed intervals (24, 48, 72 and 96 hrs), MTS-formazan cell viability assays (Promega) were performed and corrected for cell number as previously described [18]. Briefly, a stock solution of MTS was dissolved in PBS at a concentration of 5 mg/ml and was added in a 1:10 ratio (MTS/DMEM) to each well, incubated at 37°C for 2 hrs and the optical density determined at a wavelength of 490 nm on a microplate reader model 3550 (Biorad, Hercules, CA, USA). In preliminary experiments the absorbance was found to be directly proportional to the number of cells over a wide range (2 × 102 to 5 × 104 cells/well). The percent survival was defined as ([experimental absorbance – blank absorbance]/control absorbance – blank absorbance]) × 100, where the control absorbance is the optical density obtained for 1 × 104 cells/well (number of cells plated at the start of the experiment), and blank absorbance is the optical density determined in wells containing medium and MTS alone.
Western blot analysis
Cells were plated in co-culture as previously described. At day 21, osteoblasts and adipocytes were lysed in ice-cold buffer (20 mM Tris, pH 7.9, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM EGTA, 0.5 mM DTT, 400 mM NaCl and 0.5 ml glycerol containing protease inhibitor tablets [Roche Diagnostics Canada, Laval, QC, Canada]), freeze-thawed three times in a dry ice-ethanol bath and centrifuged at 11,500 rpm for 15 min. to remove insoluble material. Protein concentrations were determined with a protein assay kit (Biorad). Samples were then aliquoted and stored at –80°C. Protein lysates (10 μg per lane) were separated on 15% SDS-polyacrylamide gels and blotted on to nitrocellulose membrane. For osteoblasts extracts, blots were blocked with 5% (w/v) skim milk in 1% Tween 20 in PBS and incubated overnight at 4°C with primary antibodies against Runx2 (PC287L, Calbiochem, San Diego, CA, USA), osterix (OSX) (sc-22538, Santa Cruz Biotechnology, Santa Cruz, CA, USA), two essential transcription factors for osteoblast differentiation and function [17] and osteocalcin (OCN) (sc-80902, Santa Cruz Biotechnology), an osteoblast specific protein downstream of Runx2 activation [17]. For adipocytes extracts, blots were blocked with 5% (w/v) skim milk in 1% Tween 20 in PBS and incubated overnight at 4°C with primary antibodies against peroxisome proliferator activator-γ2 (PPAR-γ2) (sc-22020, Santa Cruz Biotechnology). After three washes with the Tween solution, the membrane was incubated with rabbit anti-goat antibody conjugated with horseradish peroxidase. The secondary antibody was detected with chemiluminescence reagent (Perkin-Elmer Life Sciences, Inc., Boston, MA, USA) and exposed to X-ray film (Eastman Kodak Bio-max, Rochester, NY, USA).
Apoptosis detection
Two techniques were used to quantify apoptotic changes namely TUNEL and caspase 3/7 assays. For quantification of DNA cleavage TUNEL reaction was performed with the Apoptag Fluorescein Direct In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA) as previously described [19]. Briefly, 6 × 105 NHOst cells were seeded in 2-well glass chamber slides (Nalge Nunc, Rochester, NY, USA) and left in culture in osteoblast growth medium for 48 hrs at which point they were treated with supernatants from the osteoblast side of all co-culture systems. After 72 hrs of treatment, cells were fixed in 4% paraformaldehyde for 10 min., washed in 10 mM Tris–HCl, pH 8.0 and pre-incubated for 10 min. at room temperature in the reaction buffer for terminal deoxynucleotidyl transferase reaction (200 mM potassium cacodylate, 0.22 mg/ml BSA and 25-mM Tris–HCl, pH 6.6). The pre-incubation buffer was then removed, and a reaction mixture containing 500 U/ml terminal deoxynucleotidyl transferase, 25 mM CoCl2 and 40 μM biotinylated dUTP was added for 60 min. at 37°C. The reaction was terminated by the addition of 300 mM NaCl and 30 mM sodium citrate for 25 min. at room temperature and for 60 min. at room temperature in the dark. Propidium iodide (Sigma) was added to cell suspensions at a concentration of 5 μg/ml. Slides were mounted and observed through fluorescence microscopy. The proportion of apoptotic cells was quantified in 10 fields per well by three different observers. This experiment was repeated three times.
Furthermore, occurrence and mechanism of apoptosis were assessed by analysis of caspase-3/7 activity. NHOst (6 × 105) were seeded in 6-well plates and left in culture in osteoblast growth medium for 48 hrs at which point they were treated with supernatants from the osteoblast side of all co-culture systems. At timed intervals (24–72 hrs), osteoblasts were lysed in ice-cold buffer [20 mM Tris, pH 7.9, 1 mM EDTA, 1 mM EGTA, 0.5 mM DTT, 400 mM NaCl and 0.5 ml glycerol containing protease inhibitor tablets (Roche Diagnostics Canada)], freeze-thawed three times in a dry ice-ethanol bath and centrifuged at 11,500 rpm for 15 min. to remove insoluble material. Protein concentrations were determined as previously described. Caspase-3/7 activity was measured by using Caspase Glo-3/7 assay systems (Promega). Samples (100 μl) were gently mixed with Caspase-Glo substrate (100 μl) and the luminescence of each sample was measured by using Luciferase assay system (Promega).
Gas chromatography/mass spectrometry (GC/MS)
Supernatants (2.5 ml) obtained from both sides of the cerulenin-treated and untreated co-culture conditions were collected at week 3 of adipocyte differentiation. Samples were analysed for FA by GC/MS at Metabolic Solutions, Inc. (Nashua, NH, USA). To be considered as relevant as a potential lipotoxic factor in our model, any FA found by GC/MS of the analysed media should fulfil the following criteria: be significantly detected only when adipocytes are present in the co-culture system, have the capacity of being detected in the media obtained from the osteoblast side of the co-cultures, and finally, be significantly affected by the presence of cerulenin. In all experiments, supernatants obtained from normal adipocytes were analysed for FA by GC/MS as controls.
Treatment of osteoblasts with FA
After identification of the predominant FA in our model, NHOst were plated and treated with either stearate or palmitate at normal physiological concentrations employed in other published cell models [20, 21]. A FA known to stimulate osteoblast function (linolate) was used as control at a dose previously used in other study [22, 23]. Briefly, NHOst were plated at a density of 4 × 105 cells in 6-well plates containing osteoblast growth media at 37°C in a humidified atmosphere of 5% CO2. Sodium salt of the FA was dissolved at 37°C in PBS containing 350 mg/ml (5 mM) FA-free BSA to obtain a 10 mM FA stock solution. The molar ratio of FA to BSA is 2:1. The FA concentration in the medium was verified with NEFA kit (Wako Chemical, Richmond, VA, USA) as previously described [20]. Control cells were also treated with BSA. After 80% confluence media were replaced with osteoblast growth media alone or containing stearate (0.5–1 mM), palmitate (100–250 μM), or linolate (25–50 μM). At week 3 of treatment, cells were fixed and ALP and alizarin red staining and quantification were performed as previously described. Finally, to identify potential induction of adipocyte-like phenotype in osteoblast by FA, cells were treated for 3 weeks with FA and the stained with oil red O as previously described.
Runx2 activity measurement
Active Runx2 binding to DNA was determined using the ELISA-based Runx2 activation TransAM™ kit (Active Motif, Rixensart, Belgium) as previously described [24]. The Trans-AM Runx2 Kit contains a 96-well plate on which an oligonucleotide containing a Runx2 consensus-binding site (5′-AACCACA-3′) has been immobilized. The active form of Runx2 contained in nuclear extract specifically binds to this oligonucleotide. The primary antibody used in the Trans-AM Runx2 Kit recognizes an accessible epitope on Runx2 protein upon DNA binding. Addition of a secondary horseradish peroxidase-conjugated antibody provides a sensitive colorimetric readout easily quantified by spectophotometry (450 nm). To quantify active Runx2 binding, 15–20 mg of nuclear extract obtained from NHOst, cultured in osteoblast growth medium for 48 hrs and treated with supernatants from the osteoblast side of all co-culture systems, was measured using the Trans-AM Runx2 Kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA, USA).
Statistical analysis
All data are expressed as mean ± S.D. of three replicate determinations. Unless otherwise stated, all experiments were repeated three times. Statistical analysis was performed by two-way ANOVA for time course analysis and Student’s t-test for comparison between groups. A probability value of P < 0.05 was considered statistically significant.
Results
Effect of inhibition of FA synthase on adipocytes differentiation in co-cultures
The initial studies evaluated the effect of the FA synthase inhibitor cerulenin on NHOst adipogenesis. As shown in Fig. 1A (upper panels), neither the presence of osteoblasts nor the addition of cerulenin to the adipogenic medium affected the pre-adipocytes’ capacity to differentiate and produce fat droplets. Quantification of oil red O and protein expression levels of PPAR-γ showed no differences between cerulenin treated versus untreated differentiating adipocytes (data not shown). In addition, treatment with cerulenin did not show any effect on pre-adipocytes proliferation (Fig. 1B).
Fig 1.
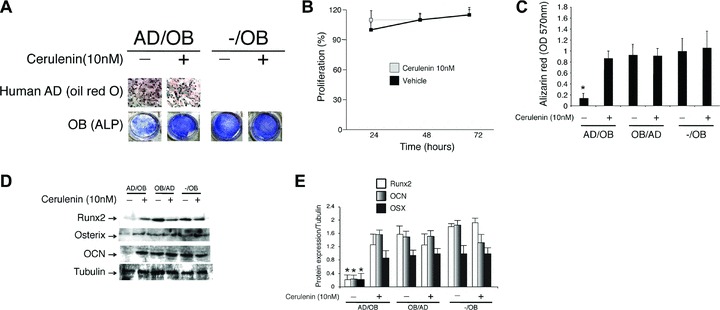
(A–C) Mature human osteoblasts were plated in co-culture with differentiating human adipocytes (AD) treated with either cerulenin (10 nM) or vehicle alone. After 3 weeks in co-culture, adipocytes were stained with oil red O (A, upper panels) to identify fat droplets and osteoblasts were stained with alkaline phosphatase (ALP) (A, lower panels) and alizarin red (C) to identify function and mineralization respectively. Addition of cerulenin to the adipocytes media did not affect adipocytes’ proliferation (B) and capacity to produce fat droplets (A, upper panels). In contrast, both osteoblast function and mineralization were significantly affected by the presence of adipocytes on top of the membrane. This inhibitory effect was prevented by addition of cerulenin (10 nM) to the media (P < 0.001). (D, E) Expression levels of Runx2, osterix (OSX) and osteocalcin (OCN) quantified by Western blot analysis were significantly reduced by the presence of adipocytes (*P < 0.001) and recovered by addition of cerulenin to the media. The data are representative of three different experiments.
Inhibition of FA synthase prevents the inhibitory effect of adipocytes on osteoblast function
NHOst were plated in co-culture with human pre-adipocytes induced to undergo adipogenesis in the absence or presence of the FA synthase inhibitor cerulenin or vehicle alone. After 3 weeks in co-culture, osteoblasts were stained with ALP and alizarin red to identify function and mineralization respectively. As shown in Fig. 1A and C, both osteoblast function and mineralization were significantly inhibited by the presence of adipocytes in the co-culture. This inhibitory effect was prevented by addition of cerulenin to the media (P < 0.001). Whereas addition of 2.5 nM of cerulenin showed no effect on osteoblast function (data not shown), both 5 and 10 nM cerulenin showed a similar inhibitory effect on adipocyte toxicity. Furthermore, expression of Runx2 and OCN was significantly reduced by the presence of adipocytes in the apical side of the co-cultures (Fig. 1D and E) (P < 0.001). Finally, this reduction in Runx2, OSX and OCN expression was prevented by addition of cerulenin (10 nM) to the media in the AD/OB condition (P < 0.001).
Inhibition of FA synthase prevents the inhibitory effect of adipocytes on osteoblast proliferation
Supernatants were collected from the osteoblast side of the membrane in the six different conditions (AD/OB, OB/AD, -/OB with and without cerulenin). NHOst were cultured for 24 hrs in 96-well plates. At 24 hrs, media were replaced with the respective conditioned media and cell proliferation was measured at timed intervals (24–96 hrs) as previously described [18]. Cell proliferation was significantly decreased in osteoblasts cultured with the supernatant from the AD/OB condition in the absence of cerulenin after 48, 72 and 96 hrs in culture (P < 0.001) (Fig. 2A). In contrast, osteoblasts exposed to supernatants from all other conditions either treated or untreated with cerulenin (5 and 10 nM) showed a progressive increase in cell proliferation at all timed intervals (Fig. 2A).
Fig 2.
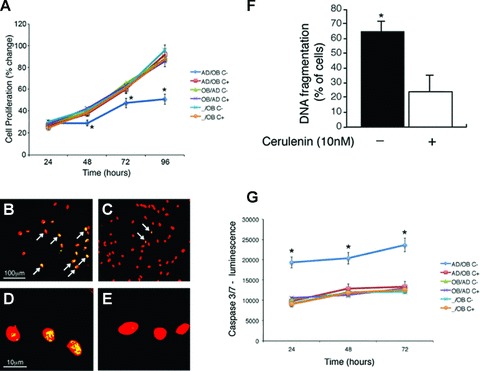
(A) Cerulenin prevents the inhibitory effect of adipocytes on osteoblasts survival– Supernatants were collected from the osteoblasts side of the membrane in the six different conditions (AD/OB, OB/AD, -/OB with and without cerulenin). Human osteoblasts were cultured for 24 hrs in 96-well plates. At 24 hrs, media were replaced with the supernatants and cell proliferation was measured at timed intervals (24–96 hrs). Cell proliferation was significantly decreased in osteoblasts cultured with the supernatant from the AD/OB untreated cells after 48, 72 and 96 hrs in culture (*P < 0.001). In contrast, osteoblasts exposed to supernatants from all other conditions either treated or untreated with cerulenin showed a progressive increase in cell proliferation at all timed intervals. (B–F) Inhibition of FA synthase prevents osteoblast apoptosis induced by adipocyte-secreted factors – Human osteoblasts were cultured in 2-well slides for 24 hrs. After 24 hrs, media were replaced with media obtained from normal confluent human adipocytes treated with either cerulenin (10 nM) or vehicle alone. After 72 hrs in culture, media were removed and cells showing apoptotic cells were identified (B–E) and quantified (F) using TUNEL assay. The TUNEL assay was able to detect a higher percentage of cells with DNA fragmentation (arrows) in the NHOst treated with supernatants obtained from untreated (B, D and F) as compared with cerulenin-treated (C, E and F) adipocytes (F) (*P < 0.01). (B and C: scale bar 100 μm), (D and E: 10 μm). (G) Caspase-3 and -7 activity was assayed using the Caspase-Glo luminescence assay and data represent the mean values (S.D.) of triplicate cultures. NHOst treated with supernatants obtained from untreated adipocytes (AD/OB-) showed higher caspase 3/7 activity as compared with cerulenin-treated adipocytes (*P < 0.001).
Inhibition of FA synthase prevents osteoblast apoptosis induced by adipocyte-secreted factors
NHOst were cultured in 2-well slides for 24 hrs. After 24 hrs, media were replaced with conditioned media obtained from normal confluent differentiated human adipocytes treated with either cerulenin (10 nM) or vehicle alone. After 72 hrs in culture, media were removed and the number of apoptotic cells was quantified using TUNEL assay. As shown in Fig. 2B–F, osteoblasts cultured in media obtained from untreated adipocytes showed significantly higher percentage of DNA fragmentation (Fig. 2B, D and F) than osteoblasts grown in media obtained from cerulenin-treated cells (Fig. 2C, E and F) (P < 0.01). Finally, no difference in the percentage of DNA fragmentation (± 8%) was found in NHOst maintained in routine culture medium and treated with either cerulenin or vehicle alone in the absence of adipocyte media.
Furthermore, we determined caspase 3/7 activity as these are important indicators of the caspase-activated apoptosis. As shown in Fig. 2G, osteoblasts grown in media obtained from the osteoblast side of the untreated AD/OB condition showed higher concentrations of caspase 3/7 activity than all the other conditions (P < 0.001). This effect was reverted by addition of cerulenin to the media (P < 0.001).
Identification of changes in FA after treatment with cerulenin
Supernatants were collected from the both sides of the membrane in the six different conditions (AD/OB, OB/AD, -/OB with and without cerulenin). GC/MS analysis of all supernatants identified the presence of seven FA (Fig. 3). However, palmitate and stearate represented 98% of all FA identified (P < 0.001) in the supernatants. In addition, supernatants obtained from the basal chamber of the AD/OB condition showed significantly higher levels of palmitate and stearate as compared with the osteoblastic side of all the other conditions (Fig. 3, P < 0.001). Furthermore, the levels of these two FA were significantly reduced to basal levels after treatment with cerulenin in the AD/OB condition (Fig. 3, P < 0.001).
Fig 3.
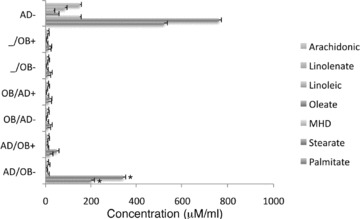
Fatty acid (FA) composition of supernatants collected from the osteoblast side in co-culture with human adipocytes treated with either cerulenin or vehicle alone and analysed by GC/MS showed the presence of seven FA. In addition, a significantly higher amount of two FA, palmitate and stearate, was found in the supernatants obtained from the AD/OB condition treated with vehicle alone (*P < 0.01; **P < 0.001 for untreated AD/OB versus all other conditions). The levels of these two FA were significantly reduced after treatment with cerulenin (δP < 0.001). MHD: Methylhexadecanoic acid.
Stearate and palmitate affect osteoblasts function and survival
As shown in Fig. 4, osteoblasts treated with stearate and palmitate, the predominant FA found in the supernatants, showed a significant and dose-dependent reduction in function (ALP) (Fig. 4A) and mineralization (alizarin red) (Fig. 4A and B) as compared with both untreated and linolate-treated cells (P < 0.01). Lower concentrations of palmitate and stearate showed minimal toxicity (data not shown). Additionally, linolate-treated osteoblast showed higher function and mineralization than untreated osteoblasts (Fig. 4A and B, P < 0.01). Furthermore, the inhibitory effect on mineralization was potentiated by combination of both palmitate and stearate FA in the media (Fig. 4B). FA did not induce adipocyte-like features in osteoblasts as per oil red O staining of either FA or vehicle-treated osteoblasts for 3 weeks (data not shown). Finally, higher levels of caspase 3/7 activity were found after addition of either palmitate or stearate into the media (Fig. 4C, P < 0.01).
Fig 4.
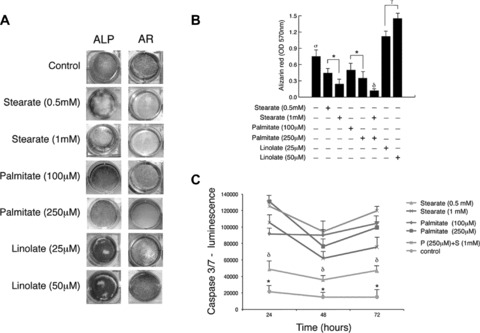
Stearate and palmitate affect osteoblasts function and mineralization – osteoblasts treated with either stearate or palmitate showed a significant reduction in alkaline phosphatase (ALP) (A, left panels) and alizarin red (A, right panels and B) as compared with both untreated and linolate -treated cells. Additionally, linolate-treated osteoblasts showed higher function and mineralization than untreated osteoblasts. Finally, combination of both FA was found to potentiate their negative effect on mineralization (B, δP < 0.001 versus all other conditions). *P < 0.001 for dose-dependent effect; **P < 0.01 untreated cells versus FA treated cells; γP < 0.01 for linolate dose-dependent effect; σP < 0.01 for control versus all other conditions. (C) Caspase-3/7 activity was assayed using the Caspase-Glo luminescence assay and data represent the mean values (S.D.) of triplicate cultures. NHOst treated with either stearate or palmitate showed higher caspase 3/7 activity as compared with untreated cells (*P < 0.001). δP < 0.01 Stearate (0.5 mM) versus all other conditions.
Runx2 transcriptional activity in osteoblasts is affected by the presence of adipocyte-secreted factors
We tested the effect of all lipotoxic conditions on Runx2 nuclear complex binding. As shown in Fig. 5, ELISA analysis of Runx2 complex binding showed a significant reduction in DNA binding activity in nuclear extracts obtained from osteoblasts exposed to supernatants from the basal chamber in the AD/OB condition as well as osteoblasts treated with either stearate and palmitate as compared with control (-/OB) (P < 0.001). The additional presence of cerulenin under the AD/OB condition reverted this inhibitory effect (P < 0.001). Finally, treatment of NHOst with cerulenin did not affect Runx2 nuclear binding as compared with untreated cells.
Fig 5.
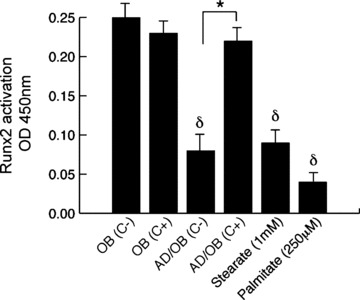
Effect of adipocyte-secreted factors on Runx2 nuclear-binding activity in human osteoblasts: Runx2 DNA-binding activity was determined using ELISA-based Runx2 activation kit and quantified by colorimetry. Osteoblasts exposed to supernatants obtained from adipocytes as well as treated with stearate and palmitate showed a significantly lower activity of the Runx2 nuclear complex in the nuclei as compared with controls. This effect was reverted by addition of cerulenin into the adipogenic media. Finally, addition of cerulenin into osteogenic media did not show a direct effect on the complex in normal osteoblasts. Values are mean ± S.E.M. of six wells per group in three independent experiments; *P < 0.001 cerulenin treated versus matched untreated cells. δP < 0.01 for cells exposed to adipogenic factors versus control (-/OB).
Discussion
In this study, we have demonstrated that the toxic effect of adipocyte-derived factors on osteoblasts in vitro not only includes a reduction in proliferation and function but also increasing levels of apoptosis. Additionally, we have found that this lipotoxic effect could be reverted by inhibition of FA biosynthesis. Finally, we have identified stearate and palmitate as the predominant FA secreted by human adipocytes that can account for the toxic effect of adipocytes in our model.
In our experiments, we exposed NHOst to human pre-adipocytes obtained from human subcutaneous fat [14]. With this model, we attempted to simulate the cell–cell interaction within the aging bone marrow milieu where predominantly toxic pre-adipocytes [2] are in close interaction with mature osteoblasts. Based on previous evidence indicating that FA could be responsible of the lipotoxic effect of fat cells on osteoblasts in vitro[12], and considering that differentiating pre-adipocytes release higher levels of FA than mature adipocytes, we tested whether inhibition of FA biosynthesis by cerulenin could prevent this lipotoxic effect of differentiating pre-adipocytes. First, we looked at the direct effect of cerulenin on NHOst in vitro. No direct effect of cerulenin was found on either the markers of osteoblast function or survival. Subsequently we tested whether, as in previous reports [25], the action that we observed after addition of cerulenin into the media was simply a reduction of the adipogenic process due to the inhibition of adipogenesis. We found that adipogenesis, cell proliferation and production of fat droplets were not affected by the presence of cerulenin at low doses whereas it was able to inhibit the synthesis of FA by differentiating adipocytes.
Furthermore, we found that in agreement with previous studies [11], the presence of adipocytes in co-cultures with osteoblasts affects the capacity of the latter to proliferate and function. We extended these observations by assessing the potential mechanism and by attempting to reverse this lipotoxic effect. Our results indicate that the presence of FA synthase inhibitor, cerulenin, reverted this negative effect as suggested by a recovery in the levels of osteoblasts proliferation, expression of Runx2, OSX, OCN and ALP expression, and mineralization capacity.
The toxicity of lipids, or lipotoxicity, and specifically lipid-induced apoptosis or lipoapoptosis, is a potential mechanism of several diseases such as non-alcoholic steatohepatitis [26] and type II diabetes mellitus [27]. The common apoptotic pathways activated by the presence of ectopic fat include the activation of caspase 3/7 and ceramide [26, 28]. In the case of bone, osteoblast apoptosis plays an important role in the pathophysiology of age-related bone loss [29]. However, the mechanisms that trigger osteoblast apoptosis remain in part unclear. Therefore, we suggested that fat infiltration could play a role in this process, thus constituting a new type of lipoapoptosis. To test this hypothesis, we determined the apoptotic changes (DNA fragmentation) as well as the activation of caspase 3/7 in osteoblasts grown in adipocyte-conditioned medium. We found that osteoblasts exposed to adipocyte-conditioned medium showed a significantly higher prevalence of apoptotic changes and higher levels of caspase 3/7 activation. As in our previous experiments, cerulenin reversed these effects suggesting that the synthesis of FA could be the mechanism accounting for lipoapoptosis in bone.
Furthermore, we attempted to identify the FA that could play a pivotal role in this lipotoxic effect. We found that palmitate and stearate are not only the predominant FA present in the lipotoxic media but also that these two FA significantly affect osteoblast function and survival. Interestingly, these two FA have been shown not only to have a toxic effect on several cell types including hepatocytes [26], beta cells [27] and osteoblasts [30], but also in the particular case of palmitate, it has been reported as one of the most prevalent FA secreted into the bone marrow [31] as well as a potent inducer of apoptosis in osteoblasts [20]. In agreement with previous studies looking at lipotoxicity in other cell models [20, 26], both, palmitate and stearate showed a lipotoxic effect in our model through the induction of apoptotic changes and the activation of caspases 3/7. Nevertheless, this toxic effect is not associated with all FA since, in agreement with previous studies [22, 23], addition of linolate into the media showed a protective effect on osteoblast function and survival.
Recently, Cornish et al.[32] have reported that FA could have a stimulatory effect on thymidine incorporation by mature osteoblasts, therefore having a positive effect on osteoblast function. However, their conclusions are hard to compare with our results not only because their cell model is considerably different but also because higher concentrations of FA were less consistent in their effect. In the same study, the authors report that saturated FA inhibit osteoclastogenesis in bone marrow cultures. This finding is relevant to understand the potential effect of FA on bone turnover in general and bone resorption in particular. However, further studies looking at the effect of inhibition of FA synthase on osteoclastogenesis should be pursued.
The significance of our findings goes beyond the concept of lipotoxicity. In contrast to pancreas where fat infiltration affects the function of normal beta cells without involving changes in cell differentiation [3], bone adipocytes and osteoblasts share the same precursor, indicating that increasing bone marrow adipogenesis not only happens at expense of osteoblast differentiation but also could have a direct inhibitory effect on osteoblast differentiation through the inhibition of critical transcription factors for osteoblastogenesis [10]. To assess this potential mechanism, we looked at the effect that the presence of adipocyte-secreted factors may have on Runx2-nuclear binding. Our results indicate that the presence of either differentiating adipocytes or FA in the same milieu with mature osteoblasts affects Runx2 nuclear binding. This, in turn, affects the expression of osteogenic proteins downstream of the Runx2-activated complex, such as OSX and OCN, constituting an additional mechanism of age-related bone loss.
In summary, in this study we have successfully reversed the lipotoxic effect of adipocytes on osteoblasts through the inhibition of FA synthase. In addition, we have identified the predominant FA that could explain the toxic effect of fat on bone cells in our particular model. Although this toxic effect could also be exerted through either the induction of PPAR-γ-related pathways [33] or the activation of other pro-apoptotic factors in the osteoblasts, further studies looking at the potential activated pathways should be pursued. Likewise, the use of FA synthase inhibitors as a potential therapeutic approach for senile osteoporosis merits further investigation.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research, the Nepean Medical Research Foundation and the Pennington Biomedical Research Foundation. Dr. G.D. holds a Fellowship from the University of Sydney-Medical Research Foundation.
References
- 1.Gimble JM, Robinson CE, Wu X, et al. The function of adipocytes in the bone marrow stroma: an update. Bone. 1996;19:421–8. doi: 10.1016/s8756-3282(96)00258-x. [DOI] [PubMed] [Google Scholar]
- 2.Cartwright MJ, Tchkonia T, Kirkland JL. Aging in adipocytes: potential impact of inherent, depot-specific mechanisms. Exp Gerontol. 2007;42:463–71. doi: 10.1016/j.exger.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–23. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 4.Duque G. Bone and fat connection, new perspectives on understanding age-related bone loss. Curr Op Rheum. 2008;20:429–34. doi: 10.1097/BOR.0b013e3283025e9c. [DOI] [PubMed] [Google Scholar]
- 5.Gimble JM, Zvonic S, Floyd ZE, et al. Playing with bone and fat. J Cell Biochem. 2006;98:251–66. doi: 10.1002/jcb.20777. [DOI] [PubMed] [Google Scholar]
- 6.Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2:35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- 7.Beresford JN, Bennett JH, Devlin C, et al. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci. 1992;102:341–51. doi: 10.1242/jcs.102.2.341. [DOI] [PubMed] [Google Scholar]
- 8.Pei L, Tontonoz P. Fat’s loss is bone’s gain. J Clin Invest. 2004;6:805–6. doi: 10.1172/JCI21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev. 2006;5:144–64. doi: 10.1016/j.arr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Diascro DD, Vogel RL, Johnson TE, et al. High fatty acid content in rabbit serum is responsible for the differentiation of osteoblasts into adipocyte-like cells. J Bone and Miner Res. 1998;13:96–106. doi: 10.1359/jbmr.1998.13.1.96. [DOI] [PubMed] [Google Scholar]
- 11.Maurin AC, Chavassieux PM, Frappart L, et al. Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone. 2000;26:485–9. doi: 10.1016/S8756-3282(00)00252-0. [DOI] [PubMed] [Google Scholar]
- 12.Maurin AC, Chavassieux PM, Vericel E, et al. Role of polyunsaturated fatty acids in the inhibitory effect of human adipocytes on osteoblastic proliferation. Bone. 2002;31:260–6. doi: 10.1016/s8756-3282(02)00805-0. [DOI] [PubMed] [Google Scholar]
- 13.Lecka-Czernik B, Moerman EJ, Grant DF, et al. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143:2376–84. doi: 10.1210/endo.143.6.8834. [DOI] [PubMed] [Google Scholar]
- 14.DeLany JP, Floyd ZE, Zvonic S, et al. Proteomic analysis of primary cultures of human adipose-derived stem cells: modulation by adipogenesis. Mol Cell Proteomics. 2005;4:731–40. doi: 10.1074/mcp.M400198-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Dubois SG, Floyd EZ, Zvonic S, et al. Isolation of human adipose-derived stem cells from biopsies and liposuction specimens. Methods Mol Biol. 2008;449:69–79. doi: 10.1007/978-1-60327-169-1_5. [DOI] [PubMed] [Google Scholar]
- 16.Duque G, Rivas D. Alendronate has an anabolic effect on bone through the differentiation of mesenchymal stem cells. J Bone Miner Res. 2007;22:1603–11. doi: 10.1359/jbmr.070701. [DOI] [PubMed] [Google Scholar]
- 17.Karsenty G. Minireview: transcriptional control of osteoblast differentiation. Endocrinology. 2001;142:2731–33. doi: 10.1210/endo.142.7.8306. [DOI] [PubMed] [Google Scholar]
- 18.Duque G, Abdaimi K, Henderson JE, et al. Vitamin D inhibits Fas ligand-induced apoptosis in human osteoblasts by regulating components of both the mitochondrial and Fas-related pathways. Bone. 2004;1:35–64. doi: 10.1016/j.bone.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Duque G, El Abdaimi K, Macoritto M, et al. Estrogens (E2) regulate expression and response of 1,25-dihydroxyvitamin D3 receptors in bone cells: changes with aging and hormone deprivation. Biochem Biophys Res Commun. 2002;299:446–54. doi: 10.1016/s0006-291x(02)02657-8. [DOI] [PubMed] [Google Scholar]
- 20.Kim JE, Ahn MW, Baek SH, et al. AICAR, inhibits palmitate-induced apoptosis in osteoblast. Bone. 2008;43:394–404. doi: 10.1016/j.bone.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 21.Eitel K, Staiger H, Brendel MD, et al. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem Biophys Res Commun. 2002;299:853–6. doi: 10.1016/s0006-291x(02)02752-3. [DOI] [PubMed] [Google Scholar]
- 22.Platt I, Rao LG, El-Sohemy A. Isomer-specific effects of conjugated linoleic acid on mineralized bone nodule formation from human osteoblast-like cells. Exp Biol Med. 2007;232:246–52. [PubMed] [Google Scholar]
- 23.Platt ID, El-Sohemy A. J Nutr Biochem. Epub: 2008. Regulation of osteoblast and adipocyte differentiation from human mesenchymal stem cells by conjugated linoleic acid. In Press. [DOI] [PubMed] [Google Scholar]
- 24.Akter R, Rivas D, Geneau G, et al. Effect of Lamin A/C Knockdown on Osteoblast Differentiation and Function. J Bone Miner Res. 2009;24:283–93. doi: 10.1359/jbmr.081010. [DOI] [PubMed] [Google Scholar]
- 25.Schmid B, Rippmann JF, Tadayyon M, et al. Inhibition of fatty acid synthase prevents preadipocyte differentiation. Biochem Biophys Res Commun. 2005;328:1073–1082. doi: 10.1016/j.bbrc.2005.01.067. [DOI] [PubMed] [Google Scholar]
- 26.Malhi H, Bronk SF, Werneburg NW, et al. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 27.El-Assaad W, Buteau J, Peyot ML, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–63. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 28.Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. 2002;1585:202–12. doi: 10.1016/s1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- 29.Weinstein RS, Manolagas SC. Apoptosis and osteoporosis. Am J Med. 2000;108:153–64. doi: 10.1016/s0002-9343(99)00420-9. [DOI] [PubMed] [Google Scholar]
- 30.Salari P, Rezaie A, Larijani B, et al. A systematic review of the impact of n-3 fatty acids in bone health and osteoporosis. Med Sci Monit. 2008;3:37–44. [PubMed] [Google Scholar]
- 31.Deshimaru R, Ishitani K, Makita K, et al. Analysis of fatty acid composition in human bone marrow aspirates. Keio J Med. 2005;54:150–5. doi: 10.2302/kjm.54.150. [DOI] [PubMed] [Google Scholar]
- 32.Cornish J, Macgibbon A, Lin JM, et al. Modulation of osteoclastogenesis by fatty acids. Endocrinology. 2008;149:5688–95. doi: 10.1210/en.2008-0111. [DOI] [PubMed] [Google Scholar]
- 33.Lecka-Czernik B, Suva LJ. Resolving the two “bony” faces of PPAR-gamma. PPAR Res. 2006:1–9. doi: 10.1155/PPAR/2006/27489. Article 27489, Pages. [DOI] [PMC free article] [PubMed] [Google Scholar]