

Abstract
The dietary isothiocyanates (ITCs) exhibit strong chemopreventive activities for a variety of neoplasms including breast cancer. However, the molecular mechanisms underlying ITC function in breast cancer cells have not been well established. Here, we found that phenethyl isothiocyanate (PEITC) acted more potently than the ‘pure’ anti-oestrogen ICI 182,780 to inhibit the growth of oestrogen receptor (ER)+ breast cancer MCF7 and H3396 cells and ER– MDA-MB-231 and SK-BR-3 cells. PEITC reduced the steady state levels of ER-α and its novel variant, ER-α36 in a dose-and time-dependent manner and inhibited oestrogen-induced activation of the mitogen activated protein kinase/ERK 1/2 signaling pathway. However, ICI 182,780 that is potent in destabilization of ER-α protein, failed to down-regulate ER-α36. Our results thus demonstrated that PEITC functions as a more potent ER-α‘disruptor’ than the well-known ICI 182,780 to abrogate ER-mediated mitogenic oestrogen signaling in breast cancer cells, which provides a molecular explanation for the strong growth inhibitory activity of ITCs in breast cancer cells, and a rational for further exploration of ITCs as chemopreventive agents for human mammary carcinogenesis.
Keywords: PEITC; breast cancer; oestrogen receptor; ER-α36; MAPK; ICI 182,780
Introduction
Breast cancer ranks second in cancer-related deaths among women in the United States [1]. Endocrine therapies, such as tamoxifen and the aromatase inhibitors, are widely used for the treatment of hormone-sensitive advanced breast cancer and successfully lead to tumour regression or disease stabilization in approximately 70% of patients for a certain period of time [2]. Most patients gradually gain resistance to initial endocrine therapy, although many retain hormone sensitivity and respond to subsequent endocrine treatments such as Fulvestrant (Faslodex®, ICI 182,780) [2]. ICI 182,780 (Fig. 1A) is a ‘pure’ anti-oestrogen, which induces degradation of oestrogen receptor-α (ER-α) protein, decreases transactivation function of ER-α and leads to a reduction in expression levels of oestrogen-regulated genes potentially important for cell growth [3, 4]. However, only 30% of patients who had progressed following prior aromatase inhibitor treatment gained clinical benefit from Fulvestrant with the median time to re-progression about 3.4 to 5.5 months [5, 6]. The object response rate was about 20.7% to 37% in advanced breast cancer of post-menopausal women whose disease progressed on prior endocrine therapy [5, 6].
Fig 1.
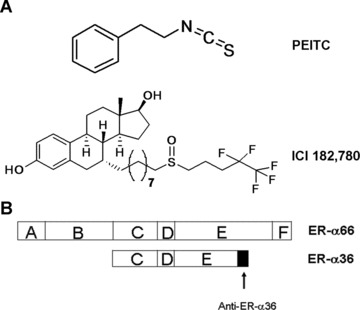
(A) The chemical structures of PEITC and ICI 182, 780. (B) The domain structures of ER-α66 and ER-α36, and the epitope of the anti-ER-α36 specific antibody.
Recently, we cloned a novel 36 kD variant of the ER-α, ER-α36 [7, 8]. The original 66 kD ER-α is referred to as ER-α66 hereafter. ER-α36 differs from ER-α66 by lacking both transcriptional activation domains (AF-1 and AF-2), but it retains the DNA-binding and dimerization domains, and partial ligand-binding domains [7, 8]. ER-α36 mediates membrane-initiated oestrogen and anti-oestrogen signaling by activation of the mitogen activated protein kinase (MAPK) signaling pathway [7, 8]. The finding that ER-α36 also mediates mitogenic anti-oestrogen signaling pathway may provide an alternative explanation for anti-oestrogen resistance observed in breast cancer patients undergoing anti-oestrogen therapy [9].
Epidemiological and pharmacological evidence demonstrate that dietary isothiocyanates (ITCs) have substantial chemopreventive activity against various types of cancers including breast cancer [10–14]. ITCs exist as conjugates in the genus Brassica of cruciferous vegetables and the genus Raphanus[15, 16]. ITCs also potently inhibit the malignant growth of cancer cells by inducing cell cycle arrest and apoptosis with little or no toxicity towards normal cells [17–19]. Phenethyl isothiocyanate (PEITC) (Fig. 1A) is one of the well-studied ITCs for their growth inhibitory functions in different cancer cells. PEITC was potent in inducing apoptosis and activating caspases in human bladder cancer cells and breast cancer cells [20, 21]. However, the molecular mechanisms by which dietary ITCs act as growth inhibitors and/or apoptosis inducers in breast cancer cells have not been elucidated.
In the present study, we have compared the effects of PEITC and ICI 182,780 treatment on proliferation and steady state levels of ER-α66 and -α36 in breast cancer MCF7, H3396, MDA-MB-231 and SK-BR-3 cells.
Materials and methods
Reagents
PEITC was purchased from Sigma-Aldrich (St. Louis, MO, USA) and ICI 182,780 was from Tocris Bioscience (Ellisville, MO, USA). The ER-α polyclonal antibody (RB-9016) was obtained from Lab Vision Corporation (Fremont, CA, USA). The ER-α36 antibody was generated as described before [7, 8]. The β-actin antibody, the goat antimouse IgG-HRP, the goat anti-rabbit IgG-HRP and the donkey anti-goat IgG-HRP antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). MG-132 was purchased from Calbiochem (San Diego, CA, USA). The phospho-p44/42 MAPK and p44/42 MAPK antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). The ECL Western blotting detection reagents were from GEHealthcare (Piscataway, NJ, USA). The ‘TRIzol’ RNA purification kit was obtained from Invitrogen (Carlsbad, CA, USA) and the ProtoScript II RT-PCR kit was purchased from New England BioLabs (Ipswich, MA, USA).
Cell culture
ER+ breast cancer cell line MCF7, ER– breast cancer cell line MDA-MB-231 and SK-BR-3, and human embryonic kidney (HEK) 293 cells were purchased from ATCC (Manassas, VA, USA). ER+ breast cancer H3396 cells were kindly provided by Dr. Leia Smith at Seattle Genetics, Inc. MCF7 and H3396 were maintained in improved minimal essential medium (IMEM) from Invitrogen with 10% heat-inactivated foetal bovine serum (FBS), 1% non-essential amino-acids, 1% HEPES buffer, 1% antibiotic-antimycotic from Invitrogen and 2 μg/ml bovine insulin (Sigma, St. Louis, MO, USA) at 37°C and 5% CO2 in a humidified incubator. MDA-MB-231, SK-BR-3 and HEK293 were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS and 1% antibiotic-antimycotic from Invitrogen at 37°C and 5% CO2 in a humidified incubator.
Cell growth assays
MCF7 and H3396 cells were seeded at 5 × 104 cells/dish in 35 mm culture dishes in IMEM supplemented with 10% FBS, while MDA-MB-231 and SK-BR-3 cells were seeded at 1 × 105 cells/dish in 35 mm culture dishes in DMEM supplemented with 2.5% charcoal dextran-stripped FBS. After treated with vehicle (DMSO) and different concentrations of PEITC or ICI 182,780 for 6 days, cells were trypsinized and counted with a haemocytometer. Three dishes were used for each concentration point and the experiments were repeated three times.
Western blot analysis
Cells were washed with cold phosphate buffered saline twice and lysed with the RIPA buffer containing 1% proteinase inhibitor cocktail solution and 1% phosphatase inhibitor cocktail solution (Sigma). The cell lysates were boiled for 5 min. in SDS gel-loading buffer and separated on a 10% SDS-PAGE gel. After electrophoresis, the proteins were transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were probed with appropriate primary antibodies and visualized with the corresponding secondary antibodies and the ECL kit (Thermo Scientific, Rockford, IL, USA). The same membranes were stripped and re-probed with an antibody against β-actin to ensure equal loading.
To study the effect of PEITC on the phosphorylation of the MAPK/ERK1/2, MCF7 cells, HEK293/V and HEK293/36 established as described before [7] were maintained in IMEM or DMEM supplemented with 2.5% dextran-charcoal stripped FBS for 48 hrs before being changed to serum-free medium for another 24 hrs. MCF7 cells were treated with vehicle (DMSO) or 5 μM of PEITC for 12 hrs before addition of vehicle (ethanol) or 1 nM 17β-estradiol (E2) for 30 min. HEK293/V and HEK293/36 cells were treated with vehicle, different concentrations of PEITC or EGF. Cells were lysed for Western blot analysis. The membranes were probed with anti-phospho-ERK antibody and visualized with the goat antimouse IgG-HRP secondary antibody and the ECL kit (Thermo Scientific). The same membranes were stripped and re-probed with an anti-p44/42 ERK antibody to ensure equal loading.
RNA purification and RT-PCR
MCF7 and H3396 cells maintained in IMEM medium containing 10% FBS were treated with different concentrations of PEITC or ICI 182,780 for 12 hrs. Total RNA was prepared with the ‘TRIzol’ RNA purification reagent. One microgram of total RNA was reverse transcribed using the ProtoScript II RT-PCR kit with random primers at 42°C for 1 hr. Semi-quantitative RT-PCR of ER-α66, ER-α36 and β-actin were performed with gene specific primers synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA). The following are the primer sequences for ER-α66, ER-α36 and β-actin.
ER-α66: forward primer: 5′-CACTCAACAGCGTGTCTCCGA-3′; reverse primer: 5′- CCAATCTTTCTCTGCCACCCTG-3′. ER-α36: forward primer: 5′-CAAGTGGTTTCCTCGTGTCTAAAG-3′; reverse primer: 5′-TGTTGAGTGTTGGTTGCCAGG-3′; β-actin: forward primer: 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′; reverse primer: 5′-CTAGAA GCATTTGCGGTGGACGATGGAGGG-3′.
The procedure of PCR for ER-α66 and β-actin was carried out as described before [22]. The PCR procedure for ER-α36 was started with a denaturing at 95°C for 3 min., then 94°C for 30 sec., 60°C for 30 sec. and 68°C for 1 min. (35 cycles) and at last elongate at 72°C for 7 min. All PCR products were analysed by electrophoresis in a 1.5% agarose gel and visualized by ethidium bromide staining under UV illumination.
Statistical analysis
Data were summarized as the mean ± standard error (S.E.) using GraphPad InStat software program. Tukey–Kramer multiple comparisons test was also used, and the significance was accepted for P-values less than 0.05.
Results
Both PEITC and ICI 182,780 repress the concentration of ER-α66
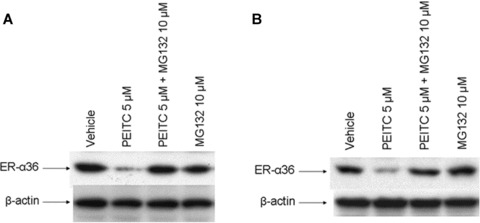
MCF7 and H3396 cells express high levels of ER-α66 and strongly respond to mitogenic oestrogen stimulation. In this study, we decided to examine the effects of PEITC on the steady state level of ER-α66 and the growth of ER+ breast cancer H3396 cells that highly express ER-α66 and HER2, a member of the epidermal growth factor receptor (EGFR) family. Cells were treated with vehicle or 5 μM of PEITC for 12 hrs and the steady state levels of ER-α66 were assessed with Western blot analysis using an anti-ER-α66 antibody. We found that the levels of ER-α66 expression were dramatically down-regulated in PEITC-treated H3396 cells (Fig. 2A), consistent with our previous report that PEITC down-regulated ER-α66 expression in ER+ breast cancer MCF7 and T47D cells [22]. As a control, the well-known ER ‘disruptor’ ICI 182, 780 was also reduced the steady state levels of ER-α66 protein (Fig. 2A), which was effectively restored by MG132, a proteasome inhibitor (Fig. 2A), consistent with the previous reports that ICI 182, 780 induces degradation of ER-α66 protein through the proteasome system [23, 24]. However, MG132 failed to recover the steady state levels of ER-α66 protein down-regulated by PEITC (Fig. 2A), suggesting that PEITC and ICI 182, 780 may use different mechanisms to down-regulate ER-α66 expression.
Fig 2.

(A) PEITC reduces the steady state levels of ER-α66 protein in H3396 cells. H3396 cells were treated with vehicle (DMSO), 5 μM PEITC or 5 μM ICI 182, 780, together with MG132 10 μM or MG132 10 μM alone for 12 hrs and lysed for Western blot analysis with anti-ER-α66 and anti-β-actin antibodies. (B) Effects of PEITC on the mRNA levels of ER-α66 in H3396 cells. H3396 cells were treated with vehicle, 2.5, 5 or 10 μM of PEITC for 12 hrs, total RNA were extracted and 1 μg of total RNA were used for RT-PCR using specific primers for ER-α66 and β-actin as described in methods. RT-PCR products were separated by 1.5% agarose gels and stained with ethidium bromide. Experiments were repeated three times, and the representative results are shown.
We then examined the steady state levels of ER-α66 mRNA in H3396 cells treated with different concentrations of PEITC for 12 hrs by RT-PCR analysis. We found that levels of ER-α66 mRNA were dramatically reduced after the treatment of PEITC starting at 2.5 μM (Fig. 2B), indicating that PEITC down-regulates ER-α66 expression at transcriptional level.
Both PEITC and ICI 182, 780 inhibit the growth of ER+ breast cancer cells
We then compared the effects of PEITC or ICI 182, 780 treatments on the growth of MCF7 and H3396 cells. MCF7 and H3396 cells were treated with different concentrations of PEITC or ICI 182, 780 for 6 days and the numbers of survived cells were then determined. PEITC or ICI 182, 780 treatments inhibited the growth of both cell lines in a dose-dependent manner but PEITC acted more potently than ICI 182, 780 (Fig. 3A, B). IC50 of PEITC in MCF7 cells was 1.6 ± 0.1 μM, and that in H3396 cells was 2.3 ± 0.2 μM. The decrease of cell number in PEITC treated cells was preceded by morphological changes that are characteristic of apoptosis, including membrane blebbing, shrunken cytoplasm, nuclear condensation and loss of adhesion, while the morphological changes induced by ICI 182, 780 treatment was not obvious (data not shown).
Fig 3.
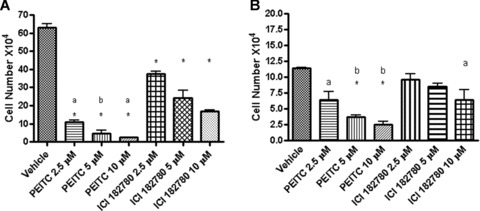
PEITC and ICI 182, 780 inhibit the proliferation of ER+ breast cancer MCF7 (A) and H3396 (B) cells. MCF7 and H3396 cells in normal medium were treated with vehicle, 2.5, 5 or 10 μM of PEITC or ICI 182, 780 for 6 days and then counted with a haemocytometer. (A) * As compared to vehicle group P < 0.001; a, P < 0.01, PEITC 2.5 μM versus ICI 182, 780 2.5 or 5 μM, PEITC 10 μM versus ICI 182, 780 10 μM; b, P < 0.05, PEITC 5 μM versus ICI 182780 5 or 10 μM. (B)* As compared to vehicle group P < 0.001; a, P < 0.01, Vehicle versus PEITC 2.5 μM or ICI 182, 780 10 μM; b, P < 0.05, PEITC 5 μM versus ICI 182, 780 5 μM, PEITC 10 μM versus ICI 182, 780 10 μM. Experiments were repeated three times, and the means ± S.E. are shown.
PEITC but not ICI 182, 780 down-regulates the steady state levels of ER-α36 protein in breast cancer cells
To probe the molecular mechanisms by which PEITC acted more potently than the ‘pure’ anti-oestrogen ICI 182, 780 in inhibiting growth and inducing apoptosis in ER+ breast cancer cells, we studied the effects of PEITC and ICI 182, 780 on the steady state levels of ER-α36, a novel variant of ER-α66 that mediates the membrane-initiated oestrogen signaling by activation of the MAPK/ERK pathway [7, 8]. MCF7 and H3396 cells were treated with different concentrations of PEITC or ICI 182, 780 for 12 hrs (Fig. 5A, B), or treated with 5 μM of PEITC or ICI 182, 780 for different time-points (Fig. 4C, D). In both ER+ breast cancer cell lines, Western blot analysis revealed that PEITC strongly reduced the steady state levels of ER-α36 protein in a dose-and time-dependent manner, but ICI 182, 780 failed to affect ER-α36 expression; ICI 182, 780 even increased the steady state levels of ER-α36 protein (Fig. 4C, D).
Fig 5.
The proteasome inhibitor MG132 restored levels of ER-α36 protein reduced by PEITC in MCF7 and H3396 cells. MCF7 (A) and H3396 (B) cells were incubated for 12 hrs in the vehicle (DMSO), 5 μM PEITC with or without MG 132 (10 μM), MG132 10 μM alone and lysed for Western blot analysis with anti-ER-α36 and anti-β-actin antibodies. Experiments were repeated three times, and the representative results are shown.
Fig 4.
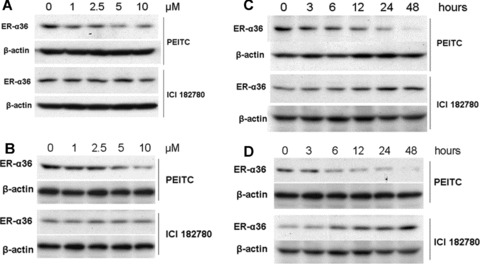
PEITC reduces the steady state levels of ER-α36 protein. MCF7 (A) and H3396 (B) cells were treated with vehicle (DMSO), 1, 2.5, 5 and 10 μM of PEITC or ICI 182780 for 12 hrs and lysed for Western blot analysis with anti-ER-α36 and anti-β-actin antibodies. MCF7 (C) and H3396 (D) cells were treated with 5 μM of PEITC or ICI 182, 780 for indicated time-points, and lysed for Western blot analysis with anti-ER-α36 and anti-β-actin antibodies. Experiments were repeated three times, and the representative results are shown.
We then probed the mechanisms underlying the down-regulation of ER-α36 expression by PEITC. MCF7 and H3396 cells were treated with vehicle, PEITC alone or together with the proteasome inhibitor, MG-132 for 12 hrs. Western blot analysis revealed that proteasome inhibitor MG132 treatment was able to recover the steady state levels of ER-α36 protein down-regulated by PEITC in both cell lines (Fig. 5A, B), suggesting that PEITC down-regulates the levels of ER-α36 protein through the proteasome-dependent proteolysis pathway.
We also examined the levels of ER-α36 mRNA in MCF7 and H3396 cells treated with different concentrations of PEITC or ICI 182, 780 for 12 hrs by RT-PCR analysis. We found that mRNA levels of ER-α36 in these breast cancer cells were unchanged after the treatment of PEITC or ICI 182, 780 (Fig. 6A–D), further indicating that the steady state levels of ER-α36 protein is down-regulated by PEITC via the proteasome proteolysis pathway and that ICI 182,780 was unable to influence ER-α36 expression at transcription level.
Fig 6.
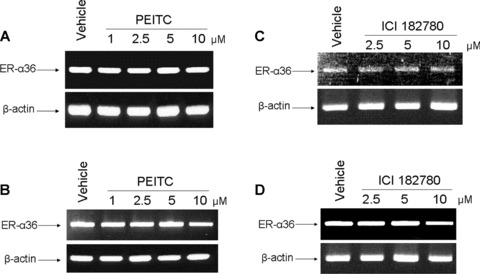
PEITC and ICI 182, 780 failed to influence the levels of ER-α36 mRNA. MCF7 and H3396 cells were incubated for 12 hrs in vehicle (DMSO) or 1, 2.5, 5 and 10 μM of PEITC or ICI 182, 780. Total RNA were extracted and 1 μg of total RNA were used for semi-quantitative RT-PCR using specific primers for ER-α36 and β-actin as described in methods. RT-PCR products were separated by 1.5% agarose gels and stained with ethidium bromide. (A) MCF7 cells treated by PEITC, (B) H3396 cells treated by PEITC, (C) MCF7 cells treated by ICI 182, 780, (D) H3396 cells treated by ICI 182, 780. Experiments were repeated three times, and the representative results were shown in the figure.
PEITC reduces the steady state levels of ER-α36 protein and inhibits the cell growth in ER– breast cancer cells
Recently, we found that ER-α36 is also expressed in specimens from ER– breast cancer patients and in established ER– breast cancer cells [7, 25]. Therefore, we decided to examine whether PEITC also inhibits the growth of ER– breast cancer cells. ER– MDA-MB-231 and SK-BR-3 cells were treated with vehicle, different concentrations of PEITC or ICI 182, 780 for 6 days and the survived cells were counted. In both ER– breast cancer cell lines, PEITC inhibited the cell growth more potently than ICI 182,780 (Fig. 7A) and down-regulated the steady state levels of ER-α36 protein while ICI 182,780 had no effect (Fig. 7B). IC50 in MDA-MB-231 cells was 2.6 ± 0.8 μM, and IC50 in SK-BR-3 cells was 1.0 ± 0.4 μM. These results further indicated that PEITC is a more potent growth inhibitory reagent for breast cancer cells compared to ICI 182,780.
Fig 7.
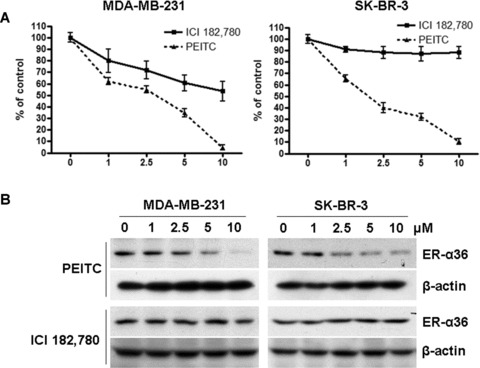
(A) PEITC inhibits the proliferation of ER– breast cancer MDA-MB-231 and SK-BR-3 cells. MDA-MB-231 and SK-BR-3 cells in oestrogen-free medium were treated with vehicle, 1, 2.5, 5 or 10 μM of PEITC or ICI 182, 780 for 6 days and then counted with a haemocytometer. Experiments were repeated three times, and the means ± S.E. are shown. (B) PEITC reduces the steady state levels of ER-α36 protein in MDA-MB-231 and SK-BR-3 cells. MDA-MB-231 and SK-BR-3 cells were treated with vehicle (DMSO), 1, 2.5, 5 and 10 μM of PEITC or ICI 182780 for 12 hrs and lysed for Western blot analysis with anti-ER-α36 and anti-β-actin antibodies. Experiments were repeated three times, and the representative results are shown.
PEITC induces ERK phosphorylation in ER-α36 expressing cells
To further probe the molecular mechanisms by which PEITC down-regulates ER-α36, we used stable cell lines from HEK293 cells transfected with the empty expression vector (HEK293/V) and the expression vector for ER-α36 (HEK293/36) established before [7]. After treatment of different concentrations of PEITC, the levels of phosphorylated ERK1/2 in HEK293/36 cells were increased at very low concentration of PEITC (0.1 pM), which was diminished when higher concentration of PEITC (>100 nM) was applied. PEITC failed to induce ERK phosphorylation in HEK293/V control cells. EGF, however, was able to induce ERK phosphorylation in control HEK293/V cells, indicating there is no global defect in the MAPK/ERK 1/2 signaling in HEK293 cells (Fig. 8). Our result thus indicated that PEITC at low concentrations is able to induce the activation of the MAPK/ERK 1/2 pathway while PEITC inhibits the MAPK/ERK 1/2 signaling at high concentrations. Our results also suggested that the PEITC activity is presumably through ER-α36.
Fig 8.
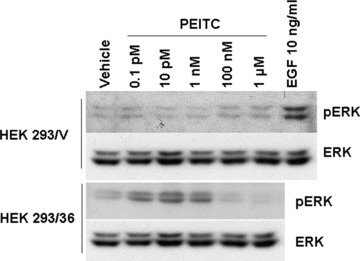
PEITC at low concentration induces ERK phosphorylation in ER-α36 expressing cells. HEK293/V and HEK293/36 cells were cultured in serum-free medium for 24 hrs and treated with vehicle (DMSO) or 0.1 pM, 10 pM, 1 nM, 100 nM or 1 μM of PEITC for 30 min. HEK293/V cells were also treated by EGF 10 ng/ml as positive control. Cells were then harvested and lysed for Western blot analysis with anti-phospho-p44/42 ERK and anti-p44/42 ERK antibodies. Experiments were repeated three times, and the representative result is shown.
PEITC inhibits non-genomic oestrogen signaling in MCF7 cells
Recently, we demonstrated that ER-α36 mediates non-genomic oestrogen signaling by activation of the MAPK/ERK signaling pathway [7]. We decided to examine the effects of PEITC on oestrogen-induced phosphorylation of the MAPK/ERK 1/2. ER+ breast cancer MCF7 cells maintained in steroid reduced medium were treated with PEITC 5 μM for 12 hrs before stimulation with oestrogen. Western blot analysis with anti-phopho-ERK antibody revealed that PEITC at 5 μM inhibited oestrogen-stimulated phosphorylation of the MAPK/ERK1/2 in MCF7 cells, suggesting that PEITC abrogates non-genomic oestrogen signaling presumably through the down-regulation of steady state levels of ER-α36 protein (Fig. 9).
Fig 9.

PEITC at high concentration inhibits oestrogen-induced phosphorylation of the MAPK/ERK in MCF7 cells. MCF7 cells were cultured in serum-free medium for 24 hrs and treated with vehicle (DMSO) or 5 μM of PEITC for 12 hrs before addition of vehicle (ethanol) or 17β-estradiol (E2) 1 nM for 1 or 2 hrs. Cells were then harvested and lysed for Western blot analysis with anti-phospho-p44/42 ERK and anti-p44/42 ERK antibodies. Experiments were repeated three times, and the representative result is shown. The volume shown in the figure were mean ± S.E., n= 3, *P < 0.05.
Discussion
Recently, we identified and characterized a novel ER-α variant, ER-α36 that mediates membrane-initiated mitogenic oestrogen signaling through activation of the MAPK/ERK pathway [7, 8]. ER-α36 also mediates activation of the MAPK/ERK signaling induced by anti-oestrogens, such as tamoxifen and ICI 182, 780 [7]. In this report, we found that the ‘pure’ anti-oestrogen ICI 182, 780 effectively down-regulated the levels of ER-α66 protein but increased the steady state levels of ER-α36 protein, while the dietary ITC, PEITC potently down-regulated the steady state levels of both ER-α66 and ER-α36. In growth inhibition assays, PEITC acted more potently than the ‘pure’ anti-oestrogen ICI 182, 780 to inhibit the proliferation of breast cancer cells. Our results thus demonstrated that the dietary ITC, PEITC functioned as a potent ER ‘disruptor’ to inhibit both genomic oestrogen signaling mediated by ER-α66 and the non-genomic oestrogen signaling mediated by ER-α36. On the other hand, ICI 182, 780, acted only to inhibit the genomic oestrogen signaling but failed to inhibit the non-genomic oestrogen signaling mediated by ER-α36 [7, 26].
Transient co-transfection of ER-α36 and ER-α66 demonstrated that ER-α66 suppresses ER-α36 promoter activity in an oestrogen-independent manner [27]. Therefore, with the releasing of the suppression from ER-α66 by ICI 182,780, ER-α36 expression may be increased in breast cancer cells. Our results thus provide a molecular explanation for the low response rate of ICI 182, 780 in breast cancer patients.
The exact mechanism underlying the failure of ICI 182, 780 to down-regulate ER-α36 was unknown. It has been reported that alterations in amino acids of the helix 12 at the ligand-binding domain of ER-α66 result in stabilization of ER-α66 protein in the presence of ICI 182, 780 [28–30], indicating that the helix 12 domain is involved in protein degradation induced by ICI 182, 780. ER-α36 has a truncated ligand-binding domain that lacks the last 4 helices (helices 9–12) of ER-α66 (Fig. 1B). The lack of the helix 12 in ER-α36 protein, an important domain for ICI 182, 780 to induce ER-α66 protein degradation by proteasome proteolysis, provides an explanation for the failure of ICI 182, 780 to induce degradation of ER-α36 protein.
The proteasome inhibitor, MG132 efficiently restored the levels of ER-α36 protein in PEITC treated cells, but failed to recover the down-regulated levels of ER-α66 protein mediated by PEITC [22], suggesting PEITC uses a mechanism different from ICI 182, 780 to negatively regulate levels of ER-α66 protein. Further experiments indicated that PEITC may down-regulate ER-α66 at transcriptional level and ER-α36 at post-transcriptional level. PEITC was reported to repress androgen receptor (AR) expression in prostate cancer cells by reducing levels of Sp1 protein, an important transcription factor for optimum AR transcription [31]. Since there are several binding sites for the Sp1 family of transcription factors in the minimal promoter region of ER-α66 that are essential for ER-α66 expression, it is thus possible that PEITC may down-regulates ER-α66 expression with mechanisms similar to its effect on AR expression in prostate cancer cells [32].
ER-α36 transcripts are generated from the ER-α66 genomic DNA through alternative promoter usage and alternative splicing [7, 8]. The transcription of ER-α36 is initiated from a previously unidentified promoter in the first intron of the ER-α66 genomic DNA [7, 8]. Thus, ER-α66 and ER-α36 are subjected to different transcription regulation through different promoter, which may explain why PEITC down-regulated ER-α66 expression at transcription level but was unable to change the levels of ER-α36 transcripts.
The finding that PEITC at low concentration (<100 nM) activated the MAPK/ERK 1/2 in ER-α36 expressing cells suggested that PEITC may interact with ER-α36 and induce membrane-initiated signaling mediated by ER-α36. PEITC may contain a ‘core’ structure that fits the ligand-binding ‘pocket’ of ER-α36 exerting an oestrogen-like effect. At high concentration (>100 nM), however, PEITC failed to induce the phosphorylation of the MAPK/ERK 1/2. Thus, PEITC acts with biphasic effect; at low concentration, PEITC exhibits oestrogenic effect while at high concentration it may function as an anti-oestrogen through down-regulation of ER-α expression. The exact mechanism underlying PEITC’s effect on the steady state levels of ER-α36 protein is unknown. Ligands such as 17β-estradiol induce ER-α66 degradation by proteasome [33]. Since PEITC functions as a ligand for ER-α36, it may use a similar mechanism to induce ER-α36 degradation through the ubiquitin/proteasome system.
The achievable concentration of PEITC from a rich vegetable diet may reach as high as 9.2–42.1 μM in human plasma [34, 35]. Therefore, the PEITC concentration required to produce significant inhibition of cell growth and down-regulation of ER-α expression in human breast cancer cells is well achievable in vivo. Our study thus supports the idea that the ITCs from cruciferous vegetables are important dietary factors for breast cancer chemoprevention, and might even provide a ‘core’ chemical structure for designing novel endocrine agents for breast cancer treatment.
Acknowledgments
This work was supported by a NIH grant DK070016 (Z.Y. Wang) and Nebraska Tobacco Settlement Biomedical Research Program Awards (LB-595 and LB692) to Z.Y. Wang.
References
- 1.American Cancer Society. Cancer facts & figures 2008. Atlanta, Georgia: American Cancer Society; 2008. [Google Scholar]
- 2.Cheung KL, Owers R, Robertson JF. Endocrine response after prior treatment with fulvestrant in postmenopausal women with advanced breast cancer: experience from a single centre. Endocr Relat Cancer. 2006;13:251–5. doi: 10.1677/erc.1.01108. [DOI] [PubMed] [Google Scholar]
- 3.Wakeling AE, Dukes M, Bowler J. A potent specific pure antioestrogen with clinical potential. Cancer Res. 1991;51:3867–73. [PubMed] [Google Scholar]
- 4.Robertson JFR, Nicholson RI, Anderson E, et al. The antitumor effects of single dose, long acting Faslodex™ (ICI 182780) compared with tamoxifen in post-menopausal primary breast cancer patients treated before surgery. Breast Cancer Res Treat. 2000;59:99. [Google Scholar]
- 5.Perey L, Paridaens R, Hawle H, et al. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00) Ann Oncol. 2007;18:64–9. doi: 10.1093/annonc/mdl341. [DOI] [PubMed] [Google Scholar]
- 6.Howell A, Robertson JF, Quaresma AlbanoJ, et al. Fulvestrant, formerly ICI 182780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. 2002;20:3396–403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Zhang X, Shen P, et al. A variant of estrogen receptor-α, hER-α36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. 2006;103:9063–68. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Zhang X, Shen P, et al. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336:1023–7. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 9.Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 10.Verhoeven DT, Goldbohm RA, van Poppel G, et al. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidemiology Biomarkers Prev. 1996;5:733–48. [PubMed] [Google Scholar]
- 11.Xiao D, Vogel V, Singh SV. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–45. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 12.Tang L, Zhang Y. Mitochondria are the primary target in isothiocyanate-induced apoptosis in human bladder cancer cells. Mol Cancer Ther. 2005;4:1250–9. doi: 10.1158/1535-7163.MCT-05-0041. [DOI] [PubMed] [Google Scholar]
- 13.Fowke JH, Chung FL, Jin F, et al. Urinary isothiocyanate levels, Brassica, and human breast cancer. Cancer Res. 2003;63:3980–6. [PubMed] [Google Scholar]
- 14.Ambrosone CB, McCann SE, Freudenheim JL, et al. Breast Cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J Nutr. 2004;134:1134–8. doi: 10.1093/jn/134.5.1134. [DOI] [PubMed] [Google Scholar]
- 15.Fenwick GR, Heaney RK, Mullin WJ. Glucosinolates and their breakdown products in food and food plants. Crit Rev Food Sci Nutr. 1983;18:123–201. doi: 10.1080/10408398209527361. [DOI] [PubMed] [Google Scholar]
- 16.Getahun SM, Chung FL. Conversion of glucosinolates to isothiocyanates in humans after ingestion of cooked watercress. Cancer Epidemiol Biomark Prev. 1999;8:447–51. [PubMed] [Google Scholar]
- 17.Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–7. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 18.Hu R, Kim BR, Chen C, et al. The role of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cell. Carcinogenesis. 2003;24:1361–7. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava SK, Singh SV. Cell cycle arrest and apoptosis-induced by benzyl isothiocyanate are associated with inhibition of nuclear factor kappa B activation in human pancreatic cancer cells. Carcinogenesis. 2004;25:1701–9. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 20.Tang L, Zhang Y. Dietary isothiocyanates inhibit the growth of human bladder carcinoma cells. J Nutr. 2004;134:2004–10. doi: 10.1093/jn/134.8.2004. [DOI] [PubMed] [Google Scholar]
- 21.Tseng E, Scott-Ramsay EA, Morris ME. Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and Mammary epithelial MCF-12A cell lines. Exp Biol Med. 2004;229:835–42. doi: 10.1177/153537020422900817. [DOI] [PubMed] [Google Scholar]
- 22.Kang L, Ding L, Wang ZY. Isothiocyanates repress estrogen receptor alpha expression in breast cancer cells. Oncol Rep. 2009;21:185–92. [PMC free article] [PubMed] [Google Scholar]
- 23.Preisler-Mashek MT, Solodin N, Stark BL, et al. Ligand-specific regulation of proteasome-mediated proteolysis of estrogen receptor-alpha. Am J Physiol Endocrinol Metab. 2002;282:E891–8. doi: 10.1152/ajpendo.00353.2001. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson RI, Gee JM, Manning DL, et al. Responses to pure antiestrogens (ICI 164384, ICI 182780) in estrogen-sensitive and -resistant experimental and clinical breast cancer. Ann NY Acad Sci. 1995;761:148–63. doi: 10.1111/j.1749-6632.1995.tb31376.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee LM, Cao J, Deng H, et al. ER-alpha36, a novel variant of ER-alpha, is expressed in ER-positive and -negative human breast carcinomas. Anticancer Res. 2008;28:479–83. [PMC free article] [PubMed] [Google Scholar]
- 26.Nethrapalli IS, Tinnikov AA, Krishnan V, et al. Estrogen activates mitogen-activated protein kinase in native, nontransfected CHO-K1, COS-7, and RAT2 fibroblast cell lines. Endocrinology. 2005;146:56–63. doi: 10.1210/en.2004-1106. [DOI] [PubMed] [Google Scholar]
- 27.Zou Y, Ding L, Coleman M, et al. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett. 2009;583:1368–74. doi: 10.1016/j.febslet.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahfoudi A, Roulet E, Dauvois S, et al. Specific mutations in the estrogen receptor change the properties of antiestrogens to full agonists. Proc Nat Acad Sci USA. 1995;92:4206–10. doi: 10.1073/pnas.92.10.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montano MM, Ekena K, Krueger KD, et al. Human estrogen receptor ligand activity inversion mutants: receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens. Mol Endocrinol. 1996;10:230–42. doi: 10.1210/mend.10.3.8833652. [DOI] [PubMed] [Google Scholar]
- 30.Pearce ST, Liu H, Jordan VC. Modulation of estrogen receptor alpha function and stability by tamoxifen and a critical amino acid (Asp-538) in helix 12. J Biol Chem. 2003;278:7630–8. doi: 10.1074/jbc.M211129200. [DOI] [PubMed] [Google Scholar]
- 31.Wang LG, Liu XM, Chiao JW. Repression of androgen receptor in prostate cancer cells by phenethyl isothiocyanate. Carcinogenesis. 2006;27:2124–32. doi: 10.1093/carcin/bgl075. [DOI] [PubMed] [Google Scholar]
- 32.deGraffenried LA, Hopp TA, Valente AJ, et al. Regulation of the estrogen receptor alpha minimal promoter by Sp1, USF-1 and ERalpha. Breast Cancer Res Treat. 2004;85:111–20. doi: 10.1023/B:BREA.0000025398.93829.78. [DOI] [PubMed] [Google Scholar]
- 33.Wijayaratne AL, McDonnell D P. The human estrogen receptor-α is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. 2001;276:35684–92. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 34.Ji Y, Morris ME. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal Biochem. 2003;323:39–47. doi: 10.1016/j.ab.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Liebes L, Conaway CC, Hochster H, et al. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal Biochem. 2001;291:279–89. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]