

Abstract
To explore the molecular mechanisms by which glioblastomas are resistant to tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), we examined TRAIL signalling pathways in the tumours. TRAIL has four membrane-anchored receptors, death receptor 4/5 (DR4/5) and decoy receptor 1/2 (DcR1/2). Of these receptors, only DR5 was expressed consistently in glioblastoma cell lines and tumour tissues, ruling out the role of DcR1/2 in TRAIL resistance. Upon TRAIL binding, DR5 was homotrimerized and recruited Fas-associated death domain (FADD) and caspase-8 for the assembly of death-inducing signalling complex (DISC) in the lipid rafts of the plasma membrane. In the DISC, caspase-8 was cleaved and initiated apoptosis by cleaving downstream caspases in TRAIL-sensitive glioblastoma cells. In TRAIL-resistant cells, however, DR5-mediated DISC was modified by receptor-interacting protein (RIP), cellular FADD-like interleukin-1β-converting enzyme inhibitory protein (c-FLIP) and phosphoprotein enriched in diabetes or in astrocyte-15 (PED/PEA-15). This DISC modification occurred in the non-raft fractions of the plasma membrane and resulted in the inhibition of caspase-8 cleavage and activation of nuclear factor-κB (NF-κB). Treatment of resistant cells with parthenolide, an inhibitor of inhibitor of κB (I-κB), eliminated TRAIL-induced NF-κB activity but not TRAIL resistance. In contrast, however, targeting of RIP, c-FLIP or PED/PEA-15 with small interfering RNA (siRNA) led to the redistribution of the DISC from non-rafts to lipid rafts and eliminated the inhibition of caspase-8 cleavage and thereby TRAIL resistance. Taken together, this study indicates that the DISC modification by RIP, c-FLIP and PED/PEA-15 is the most upstream event in TRAIL resistance in glioblastomas.
Keywords: glioblastoma, apoptosis, caspase-8, DR5, TRAIL
Introduction
Glioblastoma is the most common and malignant brain tumour and current treatment modalities include surgical resection, adjuvant radiotherapy and temozolomide chemotherapy [1]. The goal of cancer therapies is either to inhibit cell growth or to activate cell death pathways [2]. Several cell death pathways have been identified, including apoptosis, autophagy, mitotic catastrophe and necrosis. Glioblastoma is diffusely invasive and the invasive tumour cells become resistant to the treatments because of the activation of cell growth and/or inhibition of cell death pathways [3, 4]. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) has recently emerged as a cancer therapeutic agent because of its capability of inducing apoptosis in glioblastoma cells [5, 6]. Further studies, however, have shown that the majority of glioblastomas are resistant to TRAIL-induced apoptosis [7]. Here, we report the molecular mechanisms that control TRAIL resistance in the tumours.
TRAIL is expressed in natural killer cells and lymphocytes [8] as a type II membrane protein, with a short intracellular N-terminal tail and a long extracellular C-terminal [9]. The C-terminal has the receptor-binding site and can generate a soluble biologically active recombinant human TRAIL (rhTRAIL) [10]. Although clinical development of rhTRAIL as a therapeutic agent was stalled when polyhistidine-tagged rhTRAIL was reported to be hepatotoxic [11], the subsequent studies have shown that non-tagged rhTRAIL is not toxic to human hepatocytes in vitro[12] and in vivo[13]. Recently, a phase I clinical trial has confirmed that the non-tagged rhTRAIL is well tolerated in patients [14]. The clinical trial, however, has further shown the resistance of the majority of human cancers to TRAIL.
TRAIL has four membrane-anchored receptors, two death receptors, DR4/5, and two decoy receptors, DcR1/2 [9]. The death receptors are type I membrane proteins, each with two extracellular cysteine-rich domains for TRAIL binding and a cytoplasmic death domain (DD) that recruits Fas-associated death domain (FADD) and caspase-8 for the formation of a death-inducing signalling complex (DISC) [15] in the lipid rafts of the plasma membrane [16]. The homotrimeric formation of death receptors [17] is required for the receptor-mediated assembly of the DISC [18]. Unlike death receptors, decoy receptors have the cysteine-rich domains but lack the functional cytoplasmic DD, so they inhibit TRAIL-induced apoptosis [19, 20] by interrupting the homotrimeric formation of death receptors [17, 21].
Caspase-8 cleavage in the DISC is the upstream event in TRAIL-induced apoptosis. Caspase-8 is synthesized as a zymogen and exists in two isoforms (p55 and p53), each consisting of two death effector domains (DEDs) and a protease domain of p18 and p10 subunits [22]. Through the DED, caspase-8 zymogens are recruited by FADD to the DISC and cleaved through two-step autoproteolytic processes for the release of p18 and p10 subunits [23, 24] that initiate apoptotic cascade by cleavage of caspase-3 and Bcl-2 inhibitory BH3-domain protein (Bid) and subsequent activation of mitochondrial pathway [9]. The DISC can be modified by intracellular adaptors, leading to the inhibition of caspase-8 and activation of cell survival pathways [25]. Through the DD, DR4 and DR5 can recruit a DD adaptor, tumour necrosis factor receptor 1 (TNFR1)-associated death domain (TRADD), which in turn recruits receptor-interacting protein (RIP) and inhibitor of κB (I-κB) kinases (IKK) to the DISC, leading to IKK activation, I-κB phosphorylation and nuclear factor-κB (NF-κB) activation [26]. Through the DED, FADD recruits intracellular DED adaptors, cellular FADD-like interleukin-1β-converting enzyme inhibitory protein (c-FLIP) [27] and phosphoprotein enriched in diabetes or in astrocyte-15 (PED/PEA-15) [28] and thus couples the DISC to NF-κB and extracellular signal-regulated kinases 1/2 (ERK1/2) pathway [29, 30].
These DISC models, however, are mainly based on the studies in which protein–protein interactions were shown upon the overexpression of proteins and thus remain to be established in native cancer cells. In the examination of glioblastoma cells, we show here that DR5 is the only functional receptor that, upon TRAIL ligation, forms homotrimers and mediates the formation of the DISC. DR5-mediated DISC can be classified in two types: the DISC formed in lipid rafts by DR5, FADD and caspase-8 for TRAIL-induced apoptosis; and the DISC assembled in non-rafts of plasma membrane by DR5, FADD, caspase-8, RIP, c-FLIP and PED/PEA-15, leading to the inhibition of caspase-8 cleavage and activation of NF-κB pathway. The inhibition of RIP, c-FLIP and PED/PEA-15 leads to the DISC redistribution from non-rafts to lipid rafts and elimination of the caspase-8 inhibition and resistance of glioblastoma cells to TRAIL-induced apoptosis.
Materials and methods
Antibodies and reagents
Flag-tagged rhTRAIL was purchased from Alexis (San Diego, CA, USA), protein G-sepharose was from Invitrogen (Carlsbad, CA, USA) and rhTRAIL was from PeproTech, Inc. (Rocky Hill, NJ, USA). The antibodies used in the study included those against Flag M2 (Sigma-Aldrich, Saint Louis, MO, USA), DcR1 (Calbiochem, San Diego, CA, USA), DcR2 (Imgenex, San Diego, CA, USA), DR4 (Ab1-DR4, Diaclone, Besancon, France; Ab2-DR4, Chemicon, Danvers, MA, USA; Ab3, Imgenex), DR5 (pROsCI., Inc., Poway, CA, USA), FADD, caspase-8 (Medical & Biological Laboratories, Nagoya, Japan), c-FLIP (NF6 clone from Alexis), PED/PEA-15 (a kind gift from Dr. Francesco Beguinot, Naples, Italy), DNA fragmentation factor (DFF45) and caspase-3 (StressGen, Ann Harbor, MI, USA), acetyl-CoA carboxylase and phosphorylated and unphosphorylated I-κB (Cell Signaling Technology, Beverly, MA, USA), Src kinase Fyn (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and caveolin-1 (BD Biosciences, San Jose, California, USA). The antibodies also included horseradish peroxidase (HRP)-conjugated goat-antimouse IgG2a and IgG2b (Southern Biotech, Birmingham, AL, USA) and HRP-conjugated goat-anti-rabbit antibody and goat-antimouse IgG1 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Phycoerythrin (PE)-conjugated anti-human DR4, DR5, DcR1 and DcR2 and mouse IgG1 were from eBioscience (San Diego, CA, USA) and PE-conjugated IgG1 was from BD PharMingen (San Diego, CA, USA). Carbobenzyloxy-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl ketone (z-IETD-fmk) was from R & D Systems (Minneapolis, MN, USA). Parthenolide was purchased from Sigma (St. Louis, MO, USA), dual-luciferase reporter assay system was from Promega (Madison, WI, USA), cholesterol assay kit was from Wako Pure Chemicals (Richmond, VA, USA) and protease inhibitor mixture, Triton X-100 and Tween-20 were from Sigma-Aldrich. Chemiluminescence was from Pierce (Rockford, IL, USA), NF-κB oligonucleotides (5′-AGTTGAGGGGACTTTCCCAGGC-3′) were from Promega and pNF-κB-Luc reporter plasmid and Renilla luciferase control vector (pRL-TK) were from Stratagene (La Jolla, CA, USA). Annexin V Apoptosis Detection Kit I was purchased from BD Biosciences, ViraPower lentiviral Expression System was from Invitrogen, biotinylated valine-alanine-aspartate-fluoromethyl keton (bVAD-fmk) was from ICN pharmaceuticals (Costa Mesa, CA, USA) and streptavidin-agarose was from Novagen (Gibbstown, NJ, USA).
Glioblastoma cell lines and tumour tissues
Human glioblastoma cell lines D247MG, LN18, LN71, LN443 and T98G (kind gifts from N. De Tribolet, Lausanne, Switzerland) and U343MG, U87MG, U138MG and U343MG (American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM supplemented with 10% FBS in a humidified 5% CO2 and 37°C incubator. Samples of glioblastomas and normal brain tissues were kindly provided by the London (Ontario) Brain Tumor Tissue Bank (London Health Sciences Center, London, Ontario, Canada) [31]. Total protein was extracted from the tissues by homogenization in 1% Triton X-100 lysis buffer and subjected to Western blotting.
Detection of cell death and apoptosis
Cell death was measured by crystal violet cell viability assay and calculated based on the formula: 1 – (optical density of cells treated/optical density at 550 nm of cells untreated) × 100 [15]. For apoptotic cell death, Annexin V Apoptosis Detection Kit I was used according to the manufacture’s protocol (BD Biosciences). In brief, annexin V was conjugated to PE for the detection of early stage of apoptotic cells in conjunction with a vital dye 7-amino-actinomycin (7-AAD) that measures the membrane integrity. The apoptotic cell death was further examined by Western blot detection of cleavage of caspase-8, caspase-3 and DFF45.
Flow cytometry
Annexin V-PE assay was carried out by flow cytometry, as described above. The cell surface expression of TRAIL receptors was examined by flow cytometry. In brief, 0.1 μg/ml of PE-conjugated anti-human DR4, DR5, DcR1 and DcR2 (mouse IgG1) or mouse IgG1, a negative control, was added to 106 cells in 200 μl of immunofluorescence (IF) buffer (PBS containing 2% FBS and 0.02% sodium azide). After 1 hr of incubation in the dark at 4°C, 10,000 cells were analysed using a Becton and Dickinson FACScan™ (Mountain View, CA, USA). The results were processed using Cell Quest™ software (Becton and Dickinson).
Reducing and non-reducing SDS and Western blots
To detect ligand-induced high-molecular receptor complex, the cells were treated with TRAIL, lysed in 1% Triton X-100 lysis buffer and run on an SDS-PAGE gel under non-reducing conditions. For Western blots, protein samples were subjected to SDS-PAGE electrophoresis and transferred to nitrocellulose membranes; the membranes were incubated overnight at 4°C with the primary antibodies and at room temperature for 1 hr with HRP-conjugated secondary antibodies and developed by chemiluminescence.
DcR1 and DcR2 construct and transfection
Both pLenti-DcR1 (a lentiviral vector harbouring DcR1, which was constructed using the pLenti6/V5 Directional TOPO Cloning kit from Invitrogen) and pT-easy-DcR2 were cut with SpeI and ApaI. The released fragment containing DcR2 cDNA was cloned into the digested pLenti6/V5 vector and the resultant constructs were named pLenti6-DcR1 and pLenti6-DcR2 [20]. Lentiviruses were produced using ViraPower Lentiviral Expression System. The supernatants containing lentiviral particles were filtered with MILLEX-HV Syringe Driven Filter Unit (Millipore, Billerica, MA, USA) and followed by a concentration using Amicon Ultra Centrifugal Filter Devices (Millipore). The viral titres were determined by following the manufacturer’s manual. For transient expression, the cells were infected and then selected in the presence of 50 μg/ml blasticidin, 24 hrs after infection. After 8 days, the cells were subjected to the given experiments.
In vivo trapping of active caspases
This assay was carried out based on a previous report [32]. LN443 and U343MG cells were incubated with 50μM bVAD-fmk for 2 hrs at 37°C and then treated for 90 or 180 min. with 300 ng/ml of TRAIL. The cells were lysed in 500 μl lysis buffer (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 10% glycerol and 2 mM EDTA) and centrifuged at 15,000 ×g for 10 min. The supernatants were added with 30 μl of streptavidin-agarose. After overnight rotation at 4°C, the agarose beads were extensively washed with a lysis buffer containing 1 M NaCl. The proteins were eluted from the beads by the addition of 60 μl of SDS sample buffer and by incubation at 95°C for 5 min. and subjected to Western blotting, in which acetyl-CoA carboxylase was used as a control for precipitation and loading.
Immunoprecipitation of the DISC
TRAIL-induced DISC was immunoprecipitated and subjected to Western blot analysis, according to a previously reported protocol [15]. In brief, the cells were treated with mixed Flag-TRAIL and anti-Flag antibody and Flag-TRAIL-induced DISC was immunoprecipitated with protein G-sepharose. For the unstimulated control, the cells were first lysed, the cell lysates were then incubated with mixed Flag-TRAIL/anti-Flag M2 and the unstimulated DR5 complexes were immunoprecipitated with protein G-sepharose. The samples were subjected to Western blots. The supernatants, with the depletion of the complex, were subjected to Western blot analysis of caspase-8 cleavage.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from the cells according to the method described in a previous report [33]. The annealed NF-κB oligonucleotides (5′-AGTTGAGGGGACTTTCCCAGGC-3′) were end-labelled with 50 μCi [γ-32P]ATP by T4 polynucleotide kinase (Promega) at 37°C for 1 hr in a 20 μl reaction solution containing 2 μl of annealed probe and 4 μl of 5× forward reaction buffer. The unincorporated radionucleotides were removed by Microspin G-25 columns (GE healthcare (Piscataway, NJ, USA)). The binding reaction was carried out for 20 min. on ice in a 20 μl solution containing 20 mM HEPES (pH 7.5), 50 mM NaCl, 1.5 mM MgCl2, 1 mM DTT, 1 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 1 μg poly (dI:dC), 5 μg nuclear protein extract and 50 fmol radiolabelled probe. Protein–DNA complexes were separated on pre-electrophoresed 6% non-denaturing polyacrylamide gel. After electrophoresis, the gel was dried at 80°C for 1.5 hrs and protein–DNA interaction was visualized by autoradiography.
NF-κB-responsive luciferase reporter assay
The cells were co-transfected with 0.8 μg NF-κB-inducible reporter plasmid (pNF-κB-Luc) and 0.2 μg Renilla luciferase control vector (pRL-TK) for 5 hrs in OptiMEM (Invitrogen, Carlsbad, CA, USA) and recovered in full medium for 24 hrs. After harvesting, cell extracts were assayed for luciferase activity using a dual-luciferase reporter assay system. Light emission was quantified in a microplate luminescence reader (LUMIstar Galaxy; BMG Lab Technologies, Cary, NC, USA). Transfection was done in triplicate and the results were calculated as the activity of firefly luciferase relative to that of the pRL-TK.
DR4, DR5, RIP, PED/PEA-15 and c-FLIP siRNA
Double-stranded small interfering RNA (siRNA) duplexes specific to nucleotides 535–555 of the c-FLIP gene (Gene Bank accession number U97074) or 188–208 of the PED/PEA-15 gene (Gene Bank accession number Y13736) were synthesized through Qiagen service (Valencia, CA, USA). Four siRNA oligonucleotides specific to the RIP nucleotides 244–264, 573–593, 622–642 and 837–857 were designed and chemically synthesized by Qiagen–Xeragon. Of the four siRNA duplexes, the siRNA specific to RIP nucleotides 837–857 was selected for specific knockdown of RIP in glioblastoma cells. DR4 and DR5 siRNA were purchased from Qiagen, each with mixed two target sequences (TAGCTCAGCTGCAACCATCAA and CAGGCAATCGACATAATATAT for DR4; ACCAGGTGTGATTCAGGTGAA and CCGACTTCACTTGATACTATA for DR5). The synthetic siRNA and scramble siRNA (Qiagen) were transfected using HiPerfect transfection reagent (Qiagen), according to the manufacturer’s protocol. The transfected cells were allowed to grow for 72 hrs and then experimentally treated, as outlined in ‘Results’.
Lipid raft and non-raft fractionation and cholesterol analysis
Lipid raft and non-raft fractions were separated by discontinuous sucrose density gradients of Triton-X 100 cell lysates [34]. In brief, subconfluent cells from ten 15-cm culture dishes (1 × 108 cells) were transfected or untransfected with siRNA specific to RIP, PED/PEA-15 or c-FLIP for 72 hrs and treated or untreated with TRAIL for 15 min. The cells were lysed on ice in 2 ml of MNX buffer (1% Triton X-100 in 25 mM MES and 150 mM NaCl [pH 6.5]). The homogenates were mixed with 2 ml of 90% sucrose made with MNX buffer and placed on the bottom of a centrifuge tube. The samples were then overlaid with 4 ml of 35% sucrose and 4 ml of 5% sucrose and centrifuged at 175,000 ×g in a SW32Ti rotor with OptimaTM L-80 XP centrifuge (Beckman, Brea, CA, USA) for 16 hrs at 4°C. Twelve fractions of 1 ml were collected from the top to the bottom of the gradient and 2–11 factions were analysed by Western blot. To identify lipid raft fractions, the fractions were examined for cholesterol content with the cholesterol assay kit and Western blots with lipid raft markers such as Fyn and caveolin-1.
Results
DcR1 and DcR2 are not expressed in human glioblastomas
To explore the molecular basis in TRAIL resistance, we first analysed the expression of TRAIL receptors in human glioblatoma cell lines and tumour tissues. A panel of nine glioblastoma cell lines were examined for their sensitivity to TRAIL by the cell viability assay (Fig. 1A) and annexin V-PE apoptotic assay (Fig. 1B). TRAIL-sensitive (U343MG, LN-18, LN-71 and T98G) and -resistant lines (D247MG, U87MG, LN443, U138MG and U118MG) were examined by Western blotting for the expression of DR4/5, DcR1/2, RIP and c-FLIP. Ten human glioblastoma tumours were included in the study, together with five normal human brain tissues as controls. Western blot analysis showed a strong and consistent expression of DR5 protein in all the cell lines and tumour tissues (Fig. 1C); the results are consistent with earlier studies of glioblastoma cell lines [6].
Fig 1.
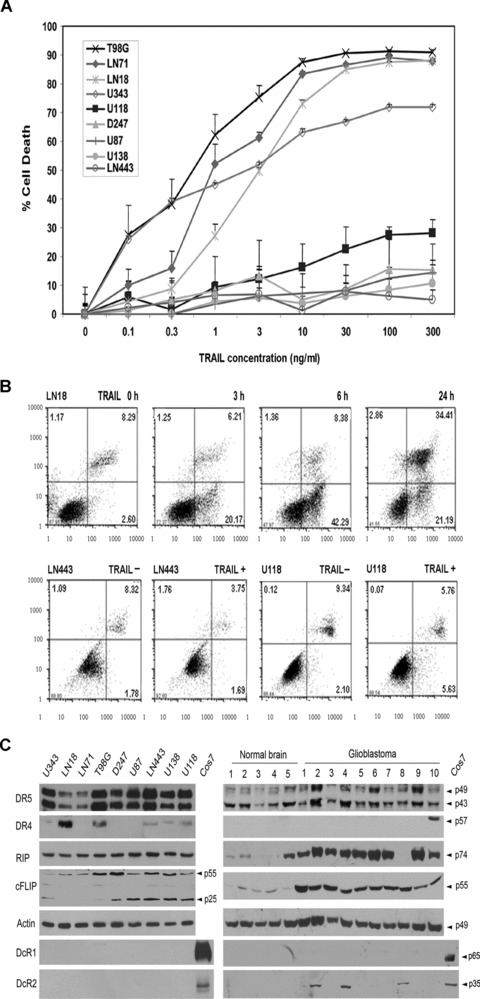
Expression of TRAIL signalling proteins in correlation with TRAIL sensitivity. (A) Cell death analysis of nine glioblastoma cell lines treated with serial dilutions of TRAIL (starting from 300 ng/ml) by crystal violet cell viability assay (mean ± S.E.M., n= 8). (B) Annexin V/7ADD analysis of TRAIL-induced apoptosis in sensitive LN18 and resistant LN443 and U118MG cell lines treated with 10 ng/ml TRAIL for the times indicated above the panels. The percent annexin V-positive cells are indicated in the right-bottom quadrant and the percent annexin V and 7ADD cells are indicated in the right-top quadrant. (C) The expression of DR5, DR4, RIP, c-FLIP, DcR1 and DcR2 in nine cell lines, Cos7 cells transfected with DcR1 and DcR2 cDNA (left panel) and normal human brain and glioblastoma tumour tissues (right panel), as determined by Western blotting, with the proteins indicated on the left side and molecular weights on the right side of the panel, with actin as a loading control.
In contrast to DR5, however, earlier studies of DR4 have generated inconsistent results in cell lines, mainly because of the use of different DR4 antibodies. To clarify this issue, we transfected LN18 cell line with DR4 siRNA and Western blotting showed that only one (Diaclone) of the three antibodies could detect the DR4 knockdown (data not shown) and thus suggested the specificity of the Diaclone antibody. Using this antibody, a strong DR4 band was seen in two cell lines and a weak band in three of the nine cell lines (Fig. 1C). DR4 was detected only in 1 of the 10 tumours but not in any of the normal brain tissues (Fig. 1C). The cell lines were then analysed by flow cytometry; DR5 was consistently expressed on the cell surface of all the cell lines, whereas DR4 was detected weakly in LN18 and T98G cell lines (data not shown), keeping in line with the Western blot findings (Fig. 1C).
To examine the expression of DcR1 and DcR2, we first transfected human DcR1 and DcR2 cDNA in Cos-7 cells and used the transfected Cos-7 cell lysates as specific positive control. Western blots detected the ectopically expressed DcR1 and DcR2 in Cos-7 cells but not the endogenously expressed DcR1 and DcR2 in any of the nine cell lines (Fig. 1C). DcR1 was not detected in any of the normal brain and tumour tissues, whereas DcR2 was seen in 3 of 10 tumours (Fig. 1C). Flow cytometry detected neither DcR1 nor DcR2 in any of the cell lines (data not shown). The failure to detect DcR1 and DcR2 in glioblatoma cell lines and tumour tissues has therefore ruled out the role of the decoy receptors in TRAIL resistance.
DD and DED adaptors, RIP, c-FLIP and PED/PEA-15 may play a role in TRAIL resistance. PED has been reported to be highly expressed in human glioblastomas [35]. In this study, we further examined RIP and c-FLIP expression. Relatively consistent levels of RIP were detected in all the cell lines (Fig. 1C). RIP was detected in 9 of 10 glioblastoma tumours and 1 of 5 normal brain tissue; the results suggest that RIP is highly expressed in glioblastomas. There are two isoforms of c-FLIP, the long form (c-FLIPL) and the short form (c-FLIPS) [27], and both were detected at higher levels in the resistant cell lines than in the sensitive cell lines, whereas c-FLIPL, but not c-FLIPS, was detected highly in all the glioblatoma tumours as compared with normal brain tissues. These studies suggest the potential role of RIP and c-FLIP in TRAIL resistance.
DR5 is the TRAIL functional receptor in glioblastomas
To further determine whether DR4 or DR5 or both are the functional receptors, we examined the interaction of TRAIL with each of the receptors. TRAIL-sensitive (U343MG and LN18) and -resistant cell lines (LN443 and U138MG) were treated with mixed Flag-tagged TRAIL and Flag M2 antibody and immunoprecipitated with protein G-sepharose. This approach allowed us to examine the interaction between TRAIL and its receptors on the cell surface. To examine the total receptors, the cells were lysed, treated with mixed Flag-TRAIL and Flag antibody and immunoprecipitated with protein G-sepharose. Western blots detected DR5 in all the immunoprecipitants (Fig. 2A). Although DR4 protein was detected in LN18 and LN443 cells (Fig. 1C), Western blot analysis of the immunoprecipitants from these two cell lines detected no DR4 (Fig. 2B); the result suggests that DR4 fails to interact with TRAIL either on cell surface or intracellularly.
Fig 2.
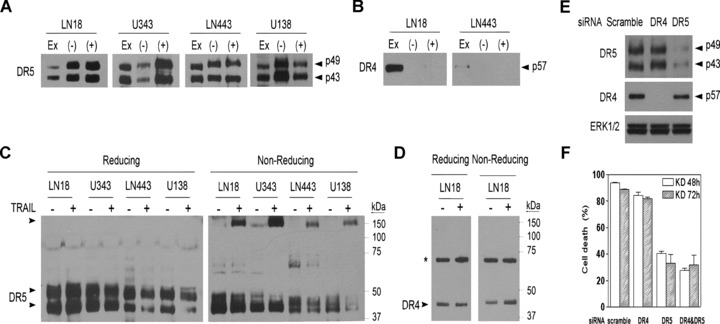
DR5 is the TRAIL functional receptor. Glioblastoma cell lines, as indicated above the panel, were immunoprecipitated by Flag-TRAIL/M2 antibody before (+) and after (–) the cells were lysed and subjected to Western blotting with DR5 (A) and DR4 antibody (B). Cell extract (EX) was included as a control of the endogenous proteins. Glioblastoma cell lines were treated (+) or untreated (–) with 300 ng/ml TRAIL for 30 min. and examined by Western blots under reducing and non-reducing conditions for the presence of the homotrimeric complex of DR5 (C) and DR4 (D). * indicates a non-specific band. (E) TRAIL-sensitive LN18 cells were transfected with scramble, DR4 and DR5 siRNA for 72 hrs and examined by Western blot for the expression of DR4 and DR5, with ERK1/2 used as a loading control. (F). LN18 cells were transfected with scramble, DR4 and DR5 siRNA or double-transfected with DR4 and DR5 siRNA cells, treated with 300 ng/ml TRAIL and examined for cell death by the cell viability assay (mean ± S.E.M., n= 4).
Next, we analysed the homotrimeric formation of the death receptors. The cell lines were treated with TRAIL; Western blots under non-reducing conditions detected the homotrimeric DR5 complex in each of the cell lines after TRAIL treatment (Fig. 2C). DR4 protein was detected in LN18 cells in the reducing and non-reducing gel, but the non-reducing gel failed to show DR4 homotrimer (Fig. 2D). Finally, we transfected LN18 cells with DR4 and DR5 siRNA to knock down the receptors (Fig. 2E) and showed that the knockdown of DR5 but not DR4 significantly inhibited TRAIL-induced cell death (Fig. 2F). These results clearly demonstrate that DR5, but not DR4, is the functional TRAIL receptor.
Caspase-8 cleavage is inhibited in TRAIL resistance
Caspase-8 is the initiator caspase in TRAIL-induced apoptosis. To determine whether TRAIL resistance results from the inhibition of caspase-8, we trapped the caspase by pre-treating cells with a cell-permeable biotinylated pan-caspase inhibitor, bVAD-fmk, which can bind covalently and irreversibly to the active cysteine site of caspases [36]. If present in a cell when apoptosis is induced, bVAD-fmk can bind only to the initiator caspase(s), halt apoptotic cascade and be used with immobilized streptavidin to isolate initiator caspase(s) in vivo in cells [32]. In this study, we first examined whether bVAD-fmk inhibits the cleavage of caspase-8 and caspase-3, a caspase-8 downstream effector caspase. TRAIL-sensitive U343MG cells and TRAIL-resistant LN443 cells were untreated or treated with bVAD-fmk for 2 hrs and apoptosis was induced with TRAIL stimulation. The enzymatically active cleavage subunits of caspase-8 (p43/41 and p18) and caspase-3 (p17) were detected in the U343MG cells treated only with TRAIL and the presence of bVAD-fmk inhibited the cleavage of both the caspases (Fig. 3A). In contrast, the cleavage of caspase-8 and caspase-3 was inhibited in LN443 cells regardless of the presence or absence of bVAD-fmk (Fig. 3A).
Fig 3.
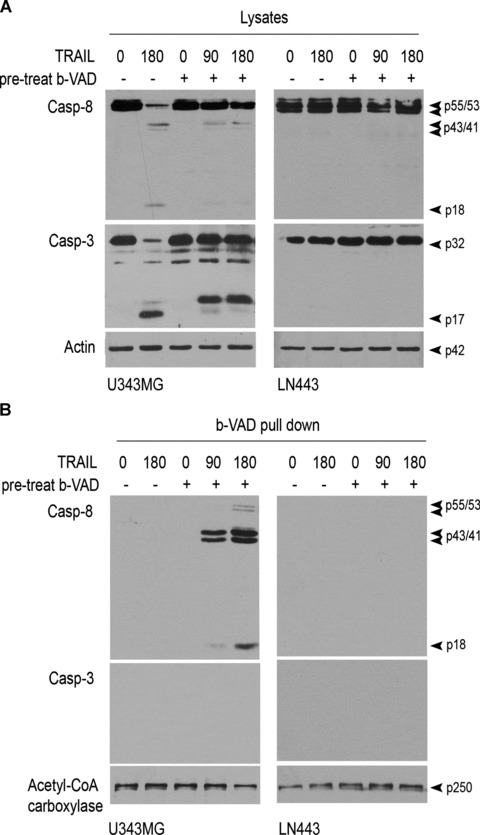
bVAD-fmk trapping of activated initiator caspase-8. (A) TRAIL-sensitive U343MG cells and TRAIL-resistant LN443 cells were pre-treated with bVAD-fmk for 2 hrs and stimulated with 300 ng/ml TRAIL for the times indicated above the panel. The cells were lysed and subjected to Western blotting for cleavage of caspase-8 (Casp-8) and caspase-3 (Casp-3). (B) U343MG and LN443 cells were treated with bVAD-fmk and then TRAIL- and bVAD-fmk-trapped activated caspases were pulled down by streptavidin and examined by Western blotting, with acetyl-CoA carboxylase as a control for precipitation and loading.
Next, we treated U343MG and LN443 cells first with bVAD-fmk for 2 hrs and then with TRAIL. The cells were lysed and the cell lysates were subjected to streptavidin pull-down. Western blotting of the streptavidin pull-down revealed the enzymatically active cleavage products of caspase-8 (p43/41 and p18) but not of caspase-3 in TRAIL-sensitive U343MG cells (Fig. 3B); the result indicates that caspase-8 is the initiator protease of downstream caspases in TRAIL-induced apoptosis. In contrast, however, no cleavage product of either caspase-8 or caspase-3 was seen in LN443 cells (Fig. 3B), which indicates that the initiator caspase-8-initiated caspase cascade is inhibited in TRAIL resistance.
Caspase-8 is recruited but not cleaved in TRAIL resistance
To investigate the molecular mechanisms involved in the inhibition of caspase-8 cleavage, we compared the DISC components of TRAIL-sensitive U343MG cells with those of TRAIL-resistant LN443 cells. For the DISC analysis, the cells were treated with mixed Flag-tagged TRAIL and Flag antibody for 15 min. and immunoprecipitated with protein G-sepharose [15]. For the unstimulated control, the cells were lysed first, treated with mixed Flag-TRAIL/M2 antibody and subjected to immunoprecipitation. The immunoprecipitants were examined on Western blots and DR5, FADD and caspase-8 were detected in the DISC in both U343MG and LN443 cells (Fig. 4A). Western blots detected the caspase-8 zymogens (p55/53), the first-step cleavage products (p43/41) in the DISC of the sensitive U343MG cells (Fig. 4A). In contrast, the caspase-8 zymogens but not the cleavage products were detected in the DISC of LN443 cells; the result indicates that the zymogens are recruited to the DISC but fail to be cleaved in the DISC in the resistant cells. Next, we examined the DISC-depleted supernatants and found the caspase-8 p18 subunit in the supernatants from the sensitive U343MG but not the resistant LN443 cells (Fig. 4B), which indicates the release of the caspase-8 protease subunits from the DISC to the cytosol, in which they cleave downstream caspase-3 in TRAIL-induced apoptosis.
Fig 4.
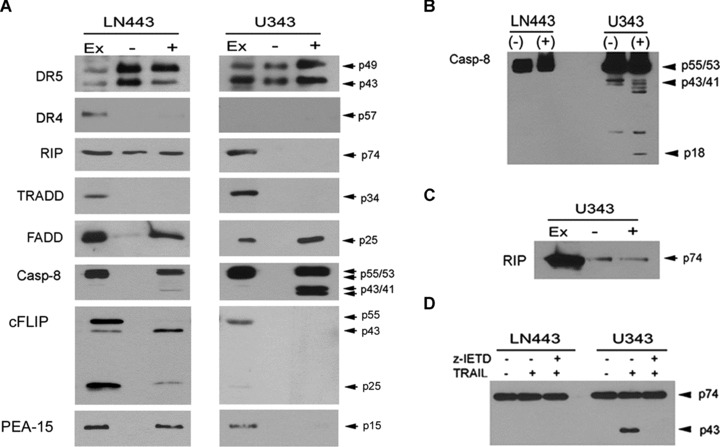
The DISC modification leads to the inhibition of caspase-8 cleavage. (A) TRAIL-induced DISC was immunoprecipitated by treating LN443 (left panel) and U343MG (right panel) cells with Flag-TRAIL/M2 antibody for 15 min. (+). For the unstimulated control, the cells were lysed first and then incubated with Flag-TRAIL/M2 antibody for 15 min. (–). Cell lysates (EX) were included as controls. The samples were examined by Western blotting for the presence of the proteins indicated on the left panel, with molecular weights on the right panel. (B) The supernatants collected from LN443 and U343MG cells after the DISC depleted, as described above in (A), were examined by Western blots for caspase-8 cleavage products, with the molecular weights indicated on the right side of the panel. (C) Western blot of the DISC analysis with RIP antibody (right third blot in (A)) was overexposed to show RIP in the unstimulated control (–) and the DISC (+) of U343MG cells. (D) LN443 and U343MG cells were treated with 300 ng/ml TRAIL in the absence or presence of caspase-8 inhibitor z-IETD for 6 hrs and subjected to Western blotting for the expression of RIP (p74) and its cleavage product (p43).
The DISC analysis further detected RIP, c-FLIP (both c-FLIPL and c-FLIPS) and PED/PEA-15 in the DISC in the resistant LN443 but not the sensitive U343MG cells (Fig. 4A). TRADD was not detected in the DISC in any of the cell lines, regardless of the earlier report that RIP is recruited to the DISC through TRADD in the transfectant [26]. Interestingly, RIP was expressed at consistent levels in sensitive and resistant cell lines (Fig. 1C) but only detected in the DISC of the resistant line (Fig. 4A). To examine this further, we first overexposed the Western blots and showed weak bands of RIP in the DISC in the sensitive U343MG cells (Fig. 4C), which suggests that RIP might be associated with DR5 but is cleaved off the DISC, perhaps by the activated caspase-8 in the DISC (Fig. 4A). To test this, we treated U343MG and LN443 cells with TRAIL and showed that TRAIL treatment resulted in the RIP cleavage from the full length (74 kD) to the cleavage (43 kD) product in U343MG but not LN443 cells and the presence of caspase-8 inhibitor z-IETD-fmk eliminated the RIP cleavage in the sensitive U343MG cells (Fig. 4D).
NF-κB activity has no effect on TRAIL resistance
The detection of RIP in the DISC in resistant cells suggests the possibility that RIP may mediate TRAIL-induced NF-κB signalling in the cells. To test this, U343MG and LN443 cells were treated with TRAIL and examined first by EMSA for NF-κB DNA-binding activity. Both cell lines showed a low constitutive NF-κB activity (Fig. 5A), consistent with an earlier report [37]. TRAIL treatment increased slightly the NF-κB activity in the resistant LN443 but not the sensitive U343MG cells (Fig. 5A). To further explore the role of NF-κB, a panel of two TRAIL-sensitive (LN71 and LN18) and four TRAIL-resistant cell lines were examined for NF-κB transcriptional activity by an NF-κB-responsive luciferase reporter assay. The results showed that TRAIL treatment increased the NF-κB activity in all the resistant but not the sensitive lines (Fig. 5B). To determine the role of DR5 in TRAIL-induced NF-κB activity, LN443 cells were transfected with DR5 siRNA and the luciferase reporter assays showed that transfection of DR5 siRNA eliminated TRAIL-induced NF-κB activity (Fig. 5C); the results indicate that DR5 is required for TRAIL-induced NF-κB activity.
Fig 5.
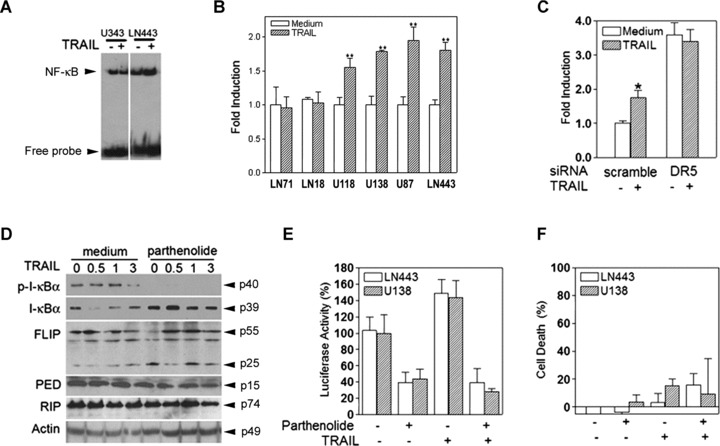
TRAIL-induced NF-κB activity does not contribute to TRAIL resistance. (A) U343MG and LN443 cells were untreated (–) or treated (+) with 300 ng/ml TRAIL for 30 min. and examined for NF-κB DNA-binding activity by EMSA. (B) TRAIL-sensitive (LN71 and LN18) and TRAIL-resistant cell lines (U118MG, U138MG, U87MG and LN443) were treated with 300 ng/ml TRAIL for 6 hrs and examined by an NF-κB-responsive luciferase reporter assay. (C) LN443 cell were transfected with either scramble or DR5 siRNA for 72 hrs, treated with 300 ng/ml TRAIL for 6 hrs and examined by the luciferase reporter assay. (D) LN443 cells were treated or untreated with 10 μM parthenolide for 16 hrs and followed with 300 ng/ml TRAIL treatment for the hours as indicated before being subjected to Western blot analysis of the expression of phosphorylated I-κB (p-I-κB), total I-κB, c-FLIP, PED/PEA-15 and RIP, with actin as a loading control. TRAIL-resistant LN443 and U138MG cells were treated with 10 μM parthenolide for 16 hrs and then with 300 ng/ml TRAIL for 3 hrs for the luciferase reporter assay (E) and for 24 hrs for the cell death assay (F).
To further examine the role of TRAIL-induced NF-κB activity in TRAIL resistance, TRAIL-resistant LN443 cells were treated with parthenolide, an inhibitor of the I-κB phosphorylation [38].
Parthenolide treatment eliminated the TRAIL-induced phosphorylation of I-κB in LN443 (Fig. 5D) but did not affect the protein expression of c-FLIP, PED/PEA-15 and RIP in the cells (Fig. 5D). We then treated TRAIL-resistant LN443 and U138MG cells first with parthenolide and then with TRAIL for the luciferase reporter and cell viability assays; the results showed that the parthenolide treatment eliminated TRAIL-induced NF-κB activity (Fig. 5E); however, the inhibition of NF-κB activity by parthenolide had no effect on the cell resistance to TRAIL (Fig. 5F). Taken together, these studies establish the role of DR5 in TRAIL-induced activation of NF-κB; however, TRAIL-induced NF-κB activity does not contribute to TRAIL resistance in glioblastoma cells.
RIP, c-FLIP and PED/PEA-15 contribute to TRAIL resistance
To further examine the role of the DD and DED adaptors in TRAIL resistance, we targeted each of the genes with siRNA in the resistant glioblastoma cells. Transfection of siRNA specific to RIP nucleotides 837–857, c-FLIP nucleotides 535–555 and PED/PEA-15 nucleotides 188–208 markedly reduced the expression of RIP, c-FLIP (c-FLIPL and c-FLIPS) and PED/PEA-15, respectively, in D247MG and U118MG cells (Fig. 6A). The transfected cells were treated with TRAIL and Western blot revealed the cleavage products of caspase-8, caspase-3 and DFF45 in the cells transfected with RIP, c-FLIP or PED/PEA-15 siRNA but not with scramble siRNA (Fig. 6B). The cell viability assay further showed a significant cell death in the TRAIL-treated cells transfected with siRNA specific to RIP, c-FLIP and PED/PEA-15 as compared with scramble siRNA (Fig. 6C).
Fig 6.
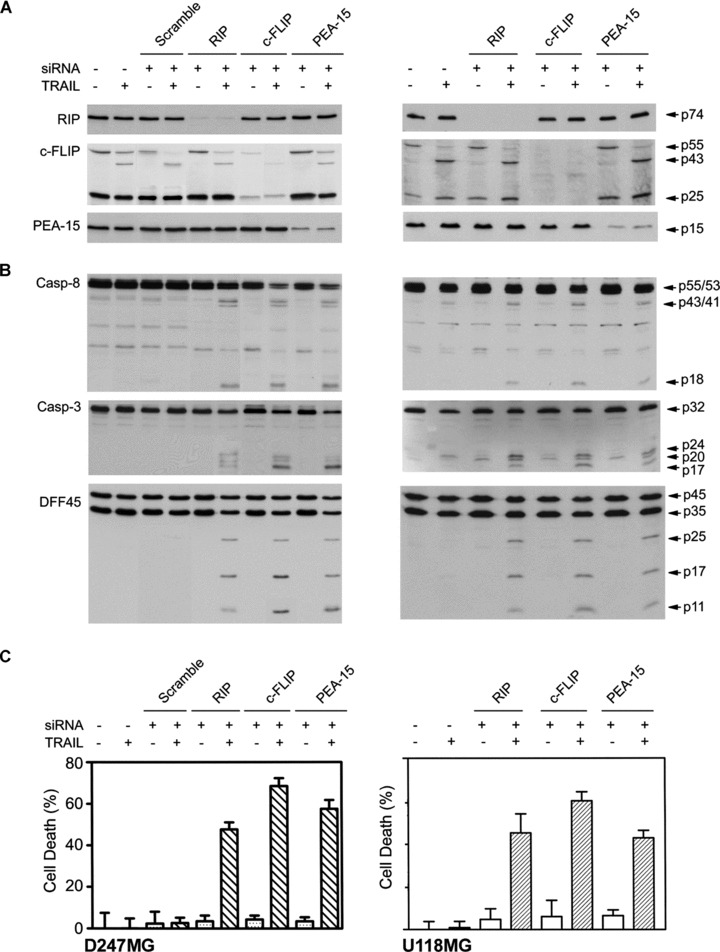
Selective knockdown of RIP, c-FLIP and PED/PEA-15 sensitizes the resistant cells to TRAIL. (A) D247MG and U118MG cells were transfected with RIP, c-FLIP, PED/PEA-15 siRNA and scramble siRNA for 72 hrs. The expression of RIP, c-FLIP and PED/PEA-15 was examined by Western blots. (B) The cleavage of caspases and DFF45 was examined by Western blotting in the transfected cells following treatment with 300 ng/ml TRAIL for 3 hrs. Proteins are indicated on the left, and the molecular weights of the proteins and cleavage products are on the right side of the panels. (C) Cell death was measured by the cell viability assay in the transfected cells after treatment with 300 ng/ml TRAIL for 24 hrs (mean ± S.E.M., n = 8).
Transfection of c-FLIP siRNA did not alter the expression of caspase-8 (Fig. 6B), a homologue of c-FLIP [27]; the results indicate the specific knockdown of c-FLIP by the siRNA. The knockdown of RIP had no effect on the expression of c-FLIP, ruling out the possibility that RIP-mediated NF-κB activity may up-regulate c-FLIP in TRAIL resistance, as reported in TNF pathway [39]. The finding that the knockdown of RIP, c-FLIP or PED/PEA-15 eliminates the inhibition of caspase-8 cleavage indicates that these adaptors are required for maintaining TRAIL resistance.
RIP, c-FLIP and PED/PEA-15 localize the DISC in non-rafts in TRAIL resistance
DR5-mediated DISC is a membrane-associated complex and we have therefore examined the role of lipid rafts and non-rafts of the plasma membrane in the DISC assembly and TRAIL-induced apoptosis. To this end, lipid raft and non-raft fractions of the plasma membrane were generated from TRAIL-sensitive U343MG cells by a discontinuous sucrose density gradient. Twelve fractions of 1 ml were collected from the top to the bottom of the gradient; fractions 2–11 were analysed by Western blot and fractions 4 and 5 were identified as lipid rafts using an antibody to lipid raft markers caveolin-1 and Src kinase Fyn (Fig. 7A). Lipid rafts are rich in cholesterol and the examination of cholesterol showed a higher content of cholesterol in fractions 4 and 5 of lipid rafts (Fig. 7B). Western blots further detected DR5 but not caspase-8 and FADD in lipid raft fractions (Fig. 7C); however, treatment with TRAIL led to the redistribution of caspase-8 and FADD to lipid rafts (Fig. 7D). These results suggest that TRAIL-induced DISC assembly occurs in lipid rafts for TRAIL-induced apoptosis in glioblastoma cells.
Fig 7.
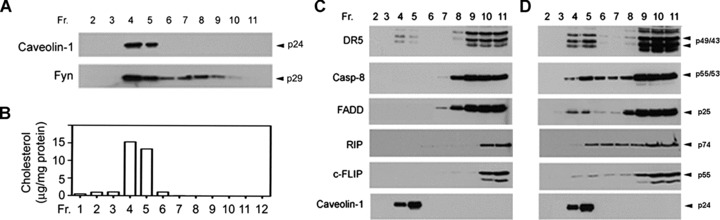
TRAIL treatment leads to the DISC redistribution to lipid rafts. (A) TRAIL-sensitive U343MG cells were subjected to discontinuous sucrose density gradients for separation of lipid raft and non-raft fractions. Lipid raft fractions 4 and 5 were identified by Western blots using lipid raft markers caveolin-1 and Fyn. (B) The concentration of cholesterol was measured in each of the fractions and a higher cholesterol content was observed in fractions 4 and 5. (C) The lipid raft and non-raft fractions from U343MG cells were subjected to Western blot analysis of the expression of DR5, caspase-8 (Casp-8), FADD, RIP and c-FLIPL, with caveolin-1 included as a lipid raft marker. (D) U343MG cells were treated with 300 ng/ml TRAIL for 15 min. and lipid raft and non-raft fractions were generated and examined by Western blot for the expression of the proteins indicated on the left side of the panel.
Next, we examined the role of lipid rafts in TRAIL resistance. TRAIL-resistant LN443 cells were subjected to discontinuous sucrose density gradients for the generation of lipid raft and non-raft fractions. Western blotting with lipid raft markers (Fig. 8A) and cholesterol examination (Fig. 8B) identified fractions 4 and 5 as lipid rafts. Western blots detected DR5 in lipid rafts and non-rafts, whereas FADD, caspase-8, RIP and c-FLIP were seen only in non-raft fractions in the cells with or without TRAIL treatment (Fig. 8C). These results suggest that the DISC distribution in non-rafts results in TRAIL resistance. The detection of RIP, PED/PEA-15 and c-FLIP in the DISC in LN443 cells (Fig. 4A) suggests the possibility that these proteins may play a role in the DISC distribution in the non-rafts. To test this, LN443 cells were transfected with siRNA specific to RIP, c-FLIP and PED/PEA-15, respectively. The transfectants were treated with TRAIL and subjected to the discontinuous sucrose density gradients for the generation of lipid raft and non-raft fractions. TRAIL treatment led to the redistribution of caspase-8 from non-rafts to lipid rafts (Fig. 8D). These results suggest that RIP, c-FLIP and PED are required for the localization of the DISC in non-rafts, thus maintaining TRAIL resistance. Targeting of each of the adaptors results in the switch of the DISC assembly from non-rafts to lipid rafts for TRAIL-induced apoptosis.
Fig 8.
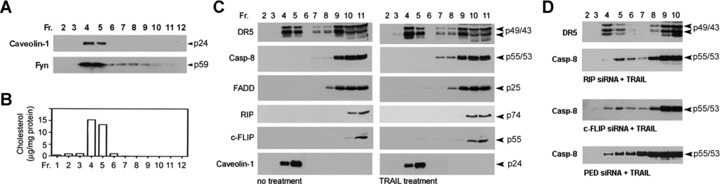
RIP, c-FLIP and PED/PEA-15 localize the DISC in non-rafts in TRAIL resistance. (A) The lipid raft and non-raft fractions from TRAIL-resistant LN443 cells were subjected to Western blotting of lipid raft markers caveolin-1 and Fyn. (B) Chemical analysis of cholesterol content in the lipid raft and non-raft fractions of LN443 cells. (C) LN443 cells were untreated (left panel) or treated with 300 ng/ml TRAIL for 15 min. (right panel) and lipid raft and non-raft fractions were separated and examined on Western blots for DR5, caspase-8 (Casp-8), FADD, RIP, c-FLIP and caveolin-1. (D) LN443 cells were transfected with RIP, c-FLIP and PED/PEA-15 siRNA, respectively, and then treated with 300 ng/ml TRAIL and subjected to discontinuous sucrose density gradients. In total, 2–10 fractions were examined by Western blot analysis of DR5 and caspase-8.
Discussion
Our earlier work has characterized human glioblastoma cell lines into TRAIL-sensitive and -resistant phenotypes [5]. TRAIL-induced apoptosis has been well documented in the sensitive cells, in which TRAIL binds on the cell surface death receptors for the recruitment of FADD and caspase-8 [15], the autoproteolytic cleavage of caspase-8 and the initiation of apoptosis by cleavage of downstream caspase-3 and Bid [6]. These studies have provided the basis for clinical development of rhTRAIL as a glioblastoma therapeutic agent. These studies have also shown the resistance of the majority of glioblastoma cells to TRAIL treatment [5–7]. In the investigation of the molecular mechanisms, we have shown that TRAIL resistance occurs in the DISC in TRAIL-resistant glioblastoma cells.
TRAIL signalling begins by binding of its receptors on the cell surface [9]. DR4 and DR5 have been shown to be able to transduce intracellular signals; however, several fundamental questions remain. Why does TRAIL have two death receptors? What are the biological functions of each of the death receptors? How does each of the death receptors contribute to TRAIL signalling? While these questions remain largely unclear, studies have suggested several possibilities. First, each cancer type may use one of the receptors: human Jurkat [40] and fibrosarcoma HT1080 [41] express DR5, whereas chronic lymphocytic leukaemia undergoes apoptosis through DR4 [42]. Second, each of the receptors may induce different intracellular signals. DR5 may contribute to TRAIL-induced apoptosis [43], whereas DR4 may mediate TRAIL-induced NF-κB signals [44]. In this study, we have shown that DR5 but not DR4 is the functional receptor that is abundantly expressed on the cell surface (Fig. 1), forms homotrimer (Fig. 2) and mediates the DISC assembly (Fig. 4) for TRAIL-induced apoptotic (Fig. 3) and NF-κB signals (Fig. 5) in human glioblastoma cells. This study suggests that targeting of DR5 with rhTRAIL and DR5 agonistic antibody should be considered in order to activate TRAIL pathways in glioblastomas.
In contrast to death receptors, DcR1/2 have been considered as TRAIL-resistant receptors, as established by overexpression studies [19–21]. The transcripts of DcR1/2 were detected in some human cancer cells, thus raising the concern on the usefulness of rhTRAIL as a therapeutic agent [14]. To explore the role of endogenous DcR1/2, we have shown that DcR1/2 proteins are not expressed in glioblastoma cell lines and tumour tissues (Fig. 1). This study rules out the potential role of endogenous DcR1/2 in TRAIL resistance in glioblastomas.
TRAIL resistance may result from the DISC modifications by intracellular DD and DED adaptors [25]. Studies of transfectants have shown that DR5 interacts with TRADD and through TRADD it recruits RIP to the DISC [26] for the NF-κB activation [45]. Through the DED–DED interaction, FADD recruits c-FLIP and PED/PEA-15 to the DISC, where caspase-8 cleavage is inhibited [15]. While these molecular models have been well documented in transfectants, our studies have established several unique features of the DISC modifications in TRAIL-resistant glioblastoma cells (Fig. 4). First, TRADD is not present in the DISC of either the sensitive or the resistant cells. Second, RIP is not recruited by TRADD to the DISC. Instead, RIP is associated with DR5 prior to TRAIL treatment and remains in the subsequently formed DISC in TRAIL-resistant cells. In the sensitive cells, third, RIP is cleaved off the DISC by caspase-8, perhaps because of the absence of c-FLIP and PED/PEA-15 in the DISC. Finally, we have confirmed the role of RIP, PED/PEA-15 and c-FLIP in TRAIL resistance in glioblastoma cells by specifically targeting each of the adaptor proteins (Fig. 6).
RIP can recruit IKK complex to the DISC for the phosphorylation and degradation of I-κB and activation of NF-κB [26, 46]. NF-κB signalling may induce the expression of c-FLIP, thereby inhibiting TRAIL-induced apoptosis [39]. Studies of human pancreatic cancer cells have suggested that TRAIL-induced NF-κB signalling promotes tumour migration and metastasis [47]. In this study, we have shown that the DISC is the bifurcation site in which RIP inhibits caspase-8 cleavage (Fig. 4) and couples to NF-κB pathway (Fig. 5). Our studies, however, have further clarified that it is the inhibition of caspase-8 in the DISC (Fig. 4) but not the NF-κB activity that inhibits TRAIL-induced apoptosis in glioblastoma cells (Fig. 5). This study suggests that targeting of NF-κB pathways is able to eliminate TRAIL resistance in glioblastoma cells.
DR5-mediated DISC is a membrane-associated complex and lipid rafts have been implicated in Fas- [48] and DR5-induced apoptosis [16]. In contrast, however, lipid rafts have been reported for TNFR1-initiated activation of NF-κB and ERK1/2 [49, 50]. In examining the role of lipid rafts, we have shown that lipid rafts are required for the DISC assembly in TRAIL-induced apoptosis in the sensitive glioblastoma cells (Fig. 7); however, the lipid raft model fails to explain the DISC assembly in TRAIL-induced NF-κB signalling in the resistant cells. In search of the molecular mechanisms, we have shown that the non-raft phase of the plasma membrane mediates TRAIL-induced DISC assembly in the resistant cells, thereby contributing to the TRAIL resistance (Fig. 8). The DD and DED adaptors, RIP, PED/PEA-15 and c-FLIP, are localized in non-rafts and targeting of each of these adaptors with siRNA results in the DISC redistribution to lipid rafts and TRAIL-induced apoptosis in the resistant cells (Fig. 8).
In conclusion, this study has established DR5 as the TRAIL functional receptor that can induce apoptotic and non-apoptotic signals in glioblastoma cells. The study has further identified two types of the DISC: the DISC formed in lipid rafts by DR5, FADD and caspase-8 in TRAIL-sensitive glioblastoma cells; and the DISC assembled in non-rafts of the plasma membrane by DR5, FADD, caspase-8, RIP, c-FLIP and PED/PEA-15 in TRAIL-resistant glioblastoma cells. The RIP-, c-FLIP- and PED/PEA-15-mediated modification of the DISC leads to the inhibition of caspase-8 cleavage and NF-κB activation. These intracellular adaptors are required for TRAIL-induced NF-κB activity; however, it is the inhibition of caspase-8 cleavage by these adaptors that is responsible for TRAIL resistance. Targeting of these intracellular adaptors but not of the DISC downstream NF-κB pathways can therefore eliminate TRAIL resistance in human glioblastomas.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health/National Cancer Institute (CA129687) and the Southeastern Brain Tumor Foundation. Chunhai Hao is the Georgia Cancer Coalition Distinguished Scholar.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12:1395–403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- 3.Giese A, Bjerkvig R, Berens ME, et al. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–36. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 4.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–22. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 5.Hao C, Beguinot F, Condorelli G, et al. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–70. [PubMed] [Google Scholar]
- 6.Song JH, Song DK, Pyrzynska B, et al. TRAIL triggers apoptosis in malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 2003;13:539–53. doi: 10.1111/j.1750-3639.2003.tb00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li YC, Tzeng CC, Song JH, et al. Genomic alterations in human malignant glioma cells associate with the cell resistance to the combination treatment with tumor necrosis factor-related apoptosis-inducing ligand and chemotherapy. Clin Cancer Res. 2006;12:2716–29. doi: 10.1158/1078-0432.CCR-05-1980. [DOI] [PubMed] [Google Scholar]
- 8.Smyth MJ, Takeda K, Hayakawa Y, et al. Nature’s TRAIL-on a path to cancer immunotherapy. Immunity. 2003;18:1–6. doi: 10.1016/s1074-7613(02)00502-2. [DOI] [PubMed] [Google Scholar]
- 9.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–48. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 10.Mariani SM, Krammer PH. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur J Immunol. 1998;28:973–82. doi: 10.1002/(SICI)1521-4141(199803)28:03<973::AID-IMMU973>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 11.Jo M, Kim TH, Seol DW, et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med. 2000;6:564–7. doi: 10.1038/75045. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence D, Shahrokh Z, Marsters S, et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 2001;7:383–5. doi: 10.1038/86397. [DOI] [PubMed] [Google Scholar]
- 13.Hao C, Song JH, Hsi B, et al. TRAIL inhibits tumor growth but is nontoxic to human hepatocytes in chimeric mice. Cancer Res. 2004;64:8502–6. doi: 10.1158/0008-5472.CAN-04-2599. [DOI] [PubMed] [Google Scholar]
- 14.Gajewski TF. On the TRAIL toward death receptor-based cancer therapeutics. J Clin Oncol. 2007;25:1305–7. doi: 10.1200/JCO.2006.09.9804. [DOI] [PubMed] [Google Scholar]
- 15.Xiao C, Yang BF, Asadi N, et al. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem. 2002;277:25020–5. doi: 10.1074/jbc.M202946200. [DOI] [PubMed] [Google Scholar]
- 16.Song JH, Tse MCL, Bellail A, et al. Lipid rafts and non-rafts mediate TRAIL-induced apoptotic and non-apoptotic signals in non-small cell lung carcinoma cells. Cancer Res. 2007;67:1–10. doi: 10.1158/0008-5472.CAN-06-3896. [DOI] [PubMed] [Google Scholar]
- 17.Clancy L, Mruk K, Archer K, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci U S A. 2005;102:18099–104. doi: 10.1073/pnas.0507329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner KW, Punnoose EA, Januario T, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070–7. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 19.Sheikh MS, Huang Y, Fernandez-Salas EA, et al. The antiapoptotic decoy receptor TRID/TRAIL-R3 is a p53-regulated DNA damage-inducible gene that is overexpressed in primary tumors of the gastrointestinal tract. Oncogene. 1999;18:4153–9. doi: 10.1038/sj.onc.1202763. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Yue P, Khur FR, et al. Decoy receptor 2 (DcR2) is a p53 target gene and regulates chemosensitivity. Cancer Res. 2005;65:9169–75. doi: 10.1158/0008-5472.CAN-05-0939. [DOI] [PubMed] [Google Scholar]
- 21.Merino D, Lalaoui N, Morizot A, et al. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–55. doi: 10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scaffidi C, Medema JP, Krammer PH, et al. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J Biol Chem. 1997;272:26953–8. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- 23.Medema JP, Scaffidi C, Kischkel FC, et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J. 1997;16:2794–804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell. 1998;1:319–25. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 25.Hao C, Song JH, Vilimanovich U, et al. Modulation of TRAIL signaling complex. Vitam Horm. 2004;67:81–99. doi: 10.1016/S0083-6729(04)67006-3. [DOI] [PubMed] [Google Scholar]
- 26.Chaudhary PM, Eby M, Jasmin A, et al. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–30. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 27.Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 28.Condorelli G, Vigliotta G, Iavarone C, et al. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. EMBO J. 1998;17:3858–66. doi: 10.1093/emboj/17.14.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol. 2004;24:2627–36. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krueger J, Chou FL, Glading A, et al. Phosphorylation of phosphoprotein enriched in astrocytes (PEA-15) regulates extracellular signal-regulated kinase-dependent transcription and cell proliferation. Mol Biol Cell. 2005;16:3552–61. doi: 10.1091/mbc.E04-11-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao C, Parney IF, Roa WH, et al. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol. 2002;103:171–8. doi: 10.1007/s004010100448. [DOI] [PubMed] [Google Scholar]
- 32.Tu S, McStay GP, Boucher LM, et al. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8:72–7. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- 33.Lassar AB, Davis RL, Wright WE, et al. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–15. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 34.Schuck S, Honsho M, Ekroos K, et al. Resistance of cell membranes to different detergents. Proc Natl Acad Sci U S A. 2003;100:5795–800. doi: 10.1073/pnas.0631579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckert A, Bock BC, Tagscherer KE, et al. The PEA-15/PED protein protects glioblastoma cells from glucose deprivation-induced apoptosis via the ERK/MAP kinase pathway. Oncogene. 2008;27:1155–66. doi: 10.1038/sj.onc.1210732. [DOI] [PubMed] [Google Scholar]
- 36.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 37.La Ferla-Bruhl K, Westhoff MA, Karl S, et al. NF-kappaB-independent sensitization of glioblastoma cells for TRAIL-induced apoptosis by proteasome inhibition. Oncogene. 2007;26:571–82. doi: 10.1038/sj.onc.1209841. [DOI] [PubMed] [Google Scholar]
- 38.Hehner SP, Heinrich M, Bork PM, et al. Sesquiterpene lactones specifically inhibit activation of NF-kappa B by preventing the degradation of I kappa B-alpha and I kappa B-beta. J Biol Chem. 1998;273:1288–97. doi: 10.1074/jbc.273.3.1288. [DOI] [PubMed] [Google Scholar]
- 39.Micheau O, Lens S, Gaide O, et al. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bodmer JL, Holler N, Reynard S, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–3. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 41.Varfolomeev E, Maecker H, Sharp D, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. 2005;280:40599–608. doi: 10.1074/jbc.M509560200. [DOI] [PubMed] [Google Scholar]
- 42.MacFarlane M, Inoue S, Kohlhaas SL, et al. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005;12:773–82. doi: 10.1038/sj.cdd.4401649. [DOI] [PubMed] [Google Scholar]
- 43.Kelley RF, Totpal K, Lindstrom SH, et al. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J Biol Chem. 2005;280:2205–12. doi: 10.1074/jbc.M410660200. [DOI] [PubMed] [Google Scholar]
- 44.Kurbanov BM, Fecker LF, Geilen CC, et al. Resistance of melanoma cells to TRAIL does not result from upregulation of antiapoptotic proteins by NF-kappaB but is related to downregulation of initiator caspases and DR4. Oncogene. 2007;26:3364–77. doi: 10.1038/sj.onc.1210134. [DOI] [PubMed] [Google Scholar]
- 45.Lin Y, Devin A, Cook A, et al. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of I-kappaB kinase and c-Jun N-terminal kinase. Mol Cell Biol. 2000;20:6638–45. doi: 10.1128/mcb.20.18.6638-6645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harper N, Farrow SN, Kaptein A, et al. Modulation of tumor necrosis factor apoptosis-inducing ligand-induced NF-kappa B activation by inhibition of apical caspases. J Biol Chem. 2001;276:34743–52. doi: 10.1074/jbc.M105693200. [DOI] [PubMed] [Google Scholar]
- 47.Trauzold A, Siegmund D, Schniewind B, et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene. 2006;25:7434–9. doi: 10.1038/sj.onc.1209719. [DOI] [PubMed] [Google Scholar]
- 48.Muppidi JR, Siegel RM. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat Immunol. 2004;5:182–9. doi: 10.1038/ni1024. [DOI] [PubMed] [Google Scholar]
- 49.Legler DF, Micheau O, Doucey MA, et al. Recruitment of TNF receptor 1 to lipid rafts is essential for TNF-alpha-mediated NF-kappaB activation. Immunity. 2003;18:655–64. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 50.Doan JE, Windmiller DA, Riches DW. Differential regulation of TNF-R1 signaling: lipid raft dependency of p42mapk/erk2 activation, but not NF-kappaB activation. J Immunol. 2004;172:7654–60. doi: 10.4049/jimmunol.172.12.7654. [DOI] [PubMed] [Google Scholar]