

Abstract
The formation of G-quadruplex structures within the NHE III1 region of the c-myc promoter and the ability of these structures to repress c-myc transcription has been well established. However, just how these extremely stable DNA secondary structures are transformed to activate c-myc transcription is still unknown. NM23-H2/NDP kinase B has been recognized as an activator of c-myc transcription via interactions with the NHE III1 region of the c-myc gene promoter. Through the use of RNAi, we confirmed the transcriptional regulatory role of NM23-H2. In addition, we show that NM23-H2 binds to both the single-stranded guanine- and cytosine-rich strands of the c-myc NHE III1, and to a lesser extent, to a random single-stranded DNA template. However, it does not bind to the NHE III1 in duplex form. Significantly, potassium ions and compounds that stabilize the G-quadruplex and i-motif structures have an inhibitory effect on NM23-H2 DNA binding activity. Mutation of Arg88 to Ala88 (R88A) reduces both DNA and nucleotide binding but had minimal effect on the NM23-H2 crystal structure. On the basis of these data and molecular modeling studies, we have proposed a stepwise trapping-out of the NHE III1 region in a single-stranded form, thus allowing single-stranded transcription factors to bind and activate c-myc transcription. Furthermore, this model provides a rationale for how the stabilization of the G-quadruplex or i-motif structures formed within the c-myc gene promoter region can inhibit NM23-H2 from activating c-myc gene expression.
Keywords: NM23-H2
INTRODUCTION
A number of nuclease hypersensitive elements (NHEs) have been identified that play important roles in the regulation of c-myc transcription (1). Notably, one of these, the NHE III1, has been the focus of considerable research over the past two decades (2-4) because of its major role in controlling 75–95% of the total c-myc transcription (Figure 1) (5, 6). In vitro characterization of the DNA within this region revealed that it is capable of engaging in a slow equilibrium between a typical Watson-Crick base-paired double helix and both single-stranded and atypical DNA secondary structures (7). Specifically, oligonucleotides representing the purine-rich and pyrimidine-rich strands have been shown to adopt G-quadruplex and i-motif structures, respectively (8-13). In addition to the DNA structural transitions within the NHE III1, many human protein factors that recognize this element either in vitro or in vivo have also been documented, including hnRNP A1, A2, and B1 (14), hnRNP K (14-16), NM23-H2/NDP kinase B (3, 17), CNBP (18), NSEP-1 (19), Sp1 (20, 21), Sp3 (21, 22), CTCF (23), MAZi (24), THZif-1 (25, 26), and c-MYB (27, 28). It is interesting to note that only a small number of these proteins exclusively recognize the duplex B-DNA conformation of the NHE III1, while the majority of these proteins bind sequence specifically to either the purine-rich or the pyrimidine-rich strand in unwound or non-B-DNA conformations. On the basis of these and several other observations, many models have been suggested to explain the details of c-myc transcription with respect to the NHE III1 region; however, none of these models have been confirmed.
Figure 1.
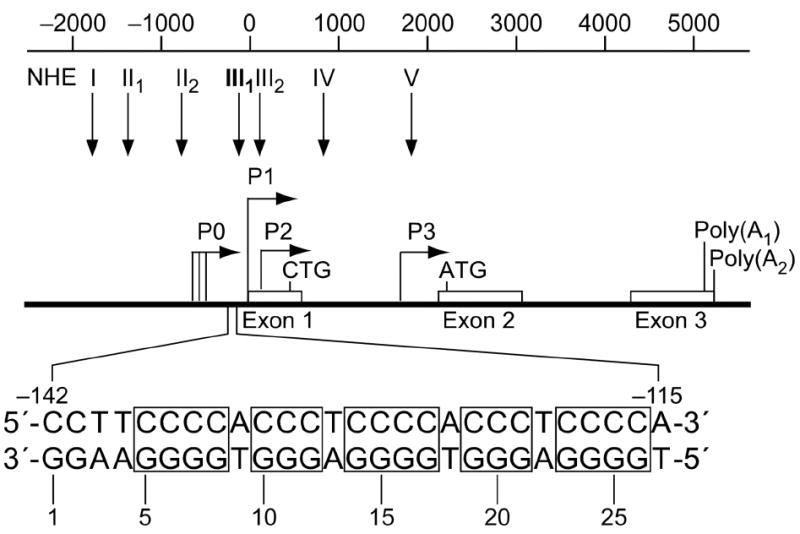
Promoter structure of the c-myc gene. Shown in inset is the 27-bp sequence of the NHE III1 (1). Polypurine and polypyrimidine tracts are shown in boxes.
NM23-H2/NDP kinase B (from the list above, hereafter referred to as NM23-H2) comes from a family of proteins that have been known for decades as housekeeping enzymes that catalyze the transfer of α-phosphate between nucleoside triphosphates and diphosphates (reviewed in ref. 29). The genes that encode nucleoside diphosphate (NDP) kinases have been sequenced and cloned from bacteria to humans and all have displayed significant sequence (approx. 44% from E. coli to human) and structural homology. However, despite their similarity in sequence and monomeric structure, variations occur in the tertiary structures of NDP kinases. Most bacterial enzymes are tetrameric, whereas all known eukaryotic enzymes have been shown to form hexamers (29, 30). In general, NDP kinases have been acknowledged as a large family of highly conserved proteins that participate in nucleotide metabolism, yet the actual biological significance of their enzymatic activity has remained elusive.
Over the past decade, extensive experimental evidence has suggested that the biological activities of NDP kinases extend beyond their originally described enzymatic means as phosphotransferases, functioning in several other unrelated regulatory roles including, but not limited to, development and differentiation, proliferation, metastasis, and apoptosis. In humans, eight different NDP kinases genes, designated nm23-H1 to nm23-H8, have been sequenced and characterized (reviewed in ref. 31). The most studied and most abundant isoforms in human cells are NM23-H1 and NM23-H2, which are encoded by the genes nm23-H1 and nm23-H2, respectively. The nm23-H1 gene was cloned by Steeg et al. on the basis of its potential as a metastasis suppressor gene (32), whereas NM23-H2 was isolated as a result of sequence homology to NM23-H1 (33). Since their discovery, a variety of metastasis model systems have demonstrated that expression levels of NM23-H1, and to a lesser extent, NM23-H2, inversely correlate with metastatic potential (34-38).
A potential explanation for some of the observed regulatory properties of NDP kinases was initially described when the purine-binding transcription factor, which activates transcription of the c-myc oncogene via the NHE III1 region, was unexpectedly identified as NM23-H2 (17). Subsequently, NM23-H2 was shown to possess equivalent DNA binding properties to those of the purine-binding transcription factor and the ability to stimulate transcription from the c-myc promoter both in vitro and in cell transfection assays (6, 17, 39). Both of these activities are independent from the originally discovered phosphotransferase activity of NM23-H2, since the DNA binding and transcriptional activation properties are retained in a catalytically inactive mutant of NM23-H2 (H118F) (40). In addition, NM23-H2 has been shown to localize within the nucleus and associated with chromatin, which is in agreement with its putative role in transcriptional regulation (41, 42). However, negative results from a study by Michelotti et al. suggest that NM23-H2 is not a conventional transcription factor because it lacks a characteristic transcriptional activation domain and is unable to stimulate c-myc transcription on its own (43).
Despite the many reports, controversy still remains in terms of the molecular mechanisms behind the activation of c-myc transcription by NM23-H2. Thus far, the DNA binding properties of NM23-H2 have been somewhat perplexing, given that disparity occurs depending on the DNA sequence, the enzyme source, and the assay employed. Indeed, in vitro and in vivo footprinting experiments have both demonstrated sequence-specific interactions between NM23-H2 and the c-myc NHE III1 (3, 39), which suggests a defined consensus binding sequence for NM23-H2. On the other hand, NM23-H2 has also been shown to bind to both single-stranded and duplex portions of the c-myc NHE III1 (44, 45), as well as to bind with high affinity to single-stranded DNA in a non-sequence-specific manner (46-48). In addition, the affinity of NM23-H2 has been shown to depend on the length of the DNA substrate in that longer c-myc NHE III1–containing DNA fragments bind better than shorter fragments (45). These results indicate that the DNA recognition properties of NM23-H2 may be related to the structural conformation of DNA rather than to a specific DNA sequence. In addition, to further complicate matters, in the course of DNA binding studies, it became evident that NM23-H2 apparently exhibits an intrinsic nuclease activity, wherein NM23-H2 reversibly cleaves duplex DNA substrates containing the c-myc NHE III1 and produces site-specific double-stranded breaks within the repeated polypurine/polypyrimidine sequence of the NHE III1 (49). This has been proposed to be an active site-directed reaction, in which a covalent protein–DNA complex is formed between Lys12, a residue located in the nucleotide-binding site of NM23-H2, and the DNA phosphodiester backbone (50). Moreover, in relation to c-myc transcription, it has been suggested that NM23-H2, through the breaking and rejoining of DNA strands, might be involved in the alteration or removal of unusual DNA conformations within the c-myc NHE III1 (45). At present, two closely related models of how genes are regulated by NM23-H2 have been presented. Both allow for DNA topological changes or the elimination of DNA secondary structures through protein–DNA interactions; however, the mechanisms governing these transformations differ, given that one involves DNA local unwinding, while the other entails DNA strand cleavage (45).
The current study focuses on evaluating the biochemical mechanisms in connection with the two aforementioned models of how NM23-H2 activates c-myc transcription. In addition to providing supplementary evidence for the ability of NM23-H2 to specifically activate c-myc gene expression through the use of RNAi technologies, we also demonstrate that the addition of a size-exclusion chromatography step or a heparin affinity chromatography step to the purification of recombinant NM23-H2 protein leads to separation of the nuclease activity from the NM23-H2-containing fractions, which seemingly eliminates the DNA strand cleavage as a potential mechanism for c-myc transcriptional activation by NM23-H2. However, the single-stranded DNA binding activity of NM23-H2 is preserved after either of these additional purification steps. Therefore, on the basis of our DNA binding data and molecular modeling studies, we propose a new model wherein the transcriptional activation of c-myc by NM23-H2 is a result of its ability to stabilize or trap-out the c-myc NHE III1 region in a single-stranded form, and subsequently allowing single-strand-specific transcription factors, such as hnRNP K and CNBP, to recognize their DNA binding sites more efficiently. In addition, this model provides a rationale for how ligand-induced stabilization of the DNA secondary structures formed within the NHE III1 region of the c-myc gene promoter inhibits NM23-H2 from activating c-myc gene expression.
EXPERIMENTAL PROCEDURES
RNA Interference
Commercially available NM23-H2 small interference RNA (siRNA) SMARTpool and siRNA SMARTpool nontargeting control were purchased from Dharmacon. Both siRNAs were transiently transfected into HeLa cells at a final concentration of 10 nM using siLentFect reagent (Bio-Rad) according to the manufacturer’s instructions. Cells were harvested 24 and 48 h post-transfection. Transcript and protein levels were determined by real-time RT-PCR and Western blot, respectively.
Quantitative Real-Time RT-PCR
After siRNA transfection, total RNA was extracted from cultured cells using the Nucleospin RNA extraction kit (Clontech) according to the manufacturer’s instructions. First-strand cDNA synthesis was then performed with 1 μg of total RNA using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative real-time PCR was performed in triplicate in 16 μL reaction volumes containing iQ SYBR-Green Supermix (Bio-Rad), 200 nM of each primer, and 1 μL cDNA using the iCycler MyiQ real-time PCR detection system (Bio-Rad). The following primers were used: NM23-H2 forward, 5′-GGGCTGAACGTGGTGAAGAC-3′; NM23-H2 reverse, 5′-TTCAGGCTTAAACCATAGGC-3′; NM23-H1 forward, 5′-GGGTCTTGTGGGAGAGATTATC-3′; NM23-H1 reverse, 5′-GACGGTCCTTCAGGTCAACG-3′; c-myc forward, 5′-GCTGCTTAGACGCTGGATT-3′; c-myc reverse, 5′-TCCTCCTCGTCGCAGTAGA-3′; β-actin forward, 5′-CTGGAACGGTGAAGGTGACA-3′; β-actin reverse, 5′-AAGGGACTTCTGTAACAACGCA-3′. For each set of primers, melting curve analysis yielded a single peak consistent with one PCR product.
Western Blot
After siRNA transfection, HeLa cells were lysed with 1% NP-40 lysis buffer. Equal amounts of protein (50 μg/lane) were resolved on a 4–15% sodium dodecyl sulfate (SDS)-polyacrylamide gel. The proteins were then transferred to nitrocellulose membranes and probed with human anti-NM23-H2 antibody (1:1000; Seikagaku). Signal detection was carried out using a secondary antibody conjugated to horseradish peroxidase (1:3000; Bio-Rad) and an enhanced chemiluminescence kit (Cell Signaling Technology).
Recombinant Protein and Purification
The wild-type and R88A mutant NM23-H2 expression vectors were both provided by Dr. Edith Postel’s laboratory (17). Briefly, expression was in E. coli strain BL21(DE3)pLysS followed by ammonium sulfate fractionation and DEAE and hydroxyapatite column chromatography steps. An additional size-exclusion chromatography step on a HiPrep 26/60 Sephacryl S-100 High Resolution column (GE Healthcare) was added that led to separation of nuclease activity from the NM23-H2-containing fraction. The wild-type protein displayed excellent NDP kinase activity in a coupled enzyme assay involving pyruvate kinase and lactate dehydrogenase, as previously described (44). The protein concentration was determined by absorbance at 280 nM using a calculated extinction coefficient of 1.3 mL cm−1 mg−1.
Preparation and End-Labeling of Oligonucleotides
Oligonucleotides were 5′-end-labeled using T4 polynucleotide kinase. Unincorporated radioactive nucleotides were removed using a Bio-Spin 6 chromatography column (Bio-Rad) after inactivation of the kinase by heating for 5 min at 95 °C. Labeled single-stranded oligonucleotides were purified using denaturing gel electrophoresis (16% PAGE). For construction of double-stranded oligonucleotides, labeled single-stranded oligonucleotides were annealed with their complementary strand and then purified using nondenaturing gel electrophoresis (16% PAGE).
Gel Mobility Shift Assay
Labeled DNA substrates were mixed with varying amounts of recombinant NM23-H2 (as specified in the figures) in reaction buffer (12 mM HEPES, 4 mM Tris·HCl [pH 7.4], 1 mM EDTA, 5% glycerol, 1 mM DTT, 0.5 μg BSA, 1.5 mM MgCl2) for ~20 min at 4 °C in a total volume of 20 μL. Reactions containing G-quadruplex-interactive agents or KCl were incubated for 1 h at 25 °C prior to the addition of NM23-H2. To separate the protein–DNA complexes from free DNA, the reactions were electrophoresed on 5% native polyacrylamide gels in the presence of 0.5 TBE at 4 °C for ~1 h at 150 V. All gel mobility shift assays were performed three times. Representative gels are shown in the figures.
DNA Strand Breakage Assay
Reactions were prepared as described above for gel mobility shift assays. After incubation, the reactions were terminated by adding an equal volume of alkaline stop buffer (95% formamide, 10 mM EDTA, 10 mM NaOH, 0.1% xylene cyanol, 0.1% bromphenol blue). The samples were subsequently heated to 95 °C for 5 min and analyzed on a 16% sequencing PAGE gel.
Surface Plasmon Resonance (SPR)
Biotinylated G-strand (5′- CGCTTATGGGGAGGGTGGGGAGGGTGGGGAAGGTGGGGAGGAGA-Biotin-3′) and C-strand (5′-Biotin-TCTCCTCCCCACCTTCCCCACCCTCCCCACCCTCCCCATAAGCG-3′) oligonucleotides were cartridge purified (Biosearch Technologies, Novato, CA). SPR experiments were performed using a BIAcore 2000 optical biosensor system (BIAcore, Inc.). Streptavidin chips were docked into the instrument and preconditioned with three consecutive one-minute injections of 1 M NaCl in 50 mM NaOH at a flow rate of 30 μL/min. Biotinylated G-strand and C-strand oligonucleotides at a concentration of 50 μg/mL were flowed through the streptavidin chip surface for 7 min at a flow rate 15 μL/min. Protein alone or in the presence of compounds (at the indicated concentrations) were mixed in running buffer (12 mM HEPES, 4 mM Tris·HCl [pH 7.4], 1 mM EDTA, 1.5 mM MgCl2). The buffer solution was filtered through a 0.22-μM filter and degassed thoroughly before use.
Heparin Affinity Column Chromatography
A column was packed with heparin (Sigma) and equilibrated with buffer B (25 mM Tris·HCl [pH 7.6], 50 mM NaCl, 0.5 mM MgCl2, 1 mM EDTA, 5 mM β-mercaptoethanol, 1 mM DTT, 10% glycerol). A sample of recombinant NM23-H2 in buffer B was applied to the column, which was then washed twice with 1 mL of buffer B. Elution of the protein was carried out using a linear gradient of 0–1.0 M NaCl in buffer B. Fractions (0.25 mL) were collected and analyzed by SDS/PAGE. DNA binding activity and DNA cleavage activity were analyzed using EMSA and the DNA strand breakage assay, respectively.
Isothermal Titration Calorimetry
Nucleotide binding constants were measured using a MicroCal VP isothermal titration calorimetry system following the manufacturer’s instructions. A solution containing either 17 μM NM23-H2 protein, 20 mM Tris·HCl buffer [pH 8.0], and 0.5 mM EDTA or 20 mM potassium phosphate buffer [pH 7.1], 75 mM KCl, 5 mM MgCl2 and 1 mM dithiothreitol was injected with 0.3 mM nucleotide (GDP or ADP) or 0.1 mM dinucleotide (d(AG), 7 μL per injection). All experiments were performed at 25 °C. Thermodynamic constants were extracted using the Origin software package provided by MicroCal.
Crystallography
The wild-type NM23 and the R88A mutant proteins were buffered with 20 mM Tris·HCl [pH 7.0] and concentrated by ultrafiltration to 17 and 10 mg/mL, respectively. Crystallization was by the hanging drop method, in which 2 μL of protein solution were combined with 2 μL of the precipitant solution and equilibrated through vapor diffusion over the precipitant solution. A total of nine crystal forms were obtained, three of which diffracted well and were further pursued. Crystals of the wild-type protein were obtained using a precipitant solution of 19% PEG 1500, 50 mM sodium citrate [pH 6.5], 20 mM MgCl2, and 5 mM DTT. The complex with the dinucleotide d(AG) (2′-deoxyadenylyl(3′,5′)-2′-deoxyguanosine) was obtained by soaking an NM23 crystal for 30 min in crystallization buffer supplemented to contain 10 mM d(AG) and 30% PEG 1500, followed by flash-freezing in liquid nitrogen. Co-crystallization attempts with d(AG) and longer oligonucleotides were unsuccessful. The GDP complex was obtained by mixing a protein solution containing 13.6 mg/mL NM23 and 14 mM GDP (sodium salt) in Tris·HCl buffer [pH 7], with precipitant solution containing 50 mM Tris·HCl [pH 7.5], 20% PEG 1500, 200 mM MgCl2, and 5 mM DTT, and crystallizing at 10 °C. Crystals of the R88A mutant were obtained from 20% PEG 1500, 100 mM sodium citrate [pH 5.6], 20 mM MgCl2, and 2 mM DTT. Crystals were flash frozen after briefly soaking in crystallization buffer containing 35% PEG 1500.
Diffraction data were measured at BioCars Sector 14, Advanced Photon Source, Argonne National Laboratory, and reduced with d*TREK (51). The structures were determined by difference Fourier (GDP complex) or molecular replacement using PDB entry 1NUE (52) as the starting model and the program MOLREP in the CCP4 program suite (53). Model building was with the program COOT (54) and model refinement with REFMAC as implemented in CCP4. All three structures contain a full hexamer in the asymmetric unit; non-crystallographic restraints were not applied. Final crystallographic parameters are listed in Table 1. The X-ray figures (shown in Figure 9) were prepared with PyMOL (DeLano Scientific LLC, Palo Alto, CA, USA).
Table 1.
Crystallographic Data.
| d(AG) Complex | GDP Complex | R88A Mutant | |
|---|---|---|---|
| Data Collection Statistics: | |||
| X-ray source | APS 14BM-C | APS 14ID-B | APS 14BM-C |
| Detector | Quantum 4 | MARCCD | Quantum 4 |
| Wavelength (Å) | 0.9000 | 1.1271 | 0.9000 |
| Resolution (Å) | 1.3 | 1.7 | 1.7 |
| Space group | P212121 | P212121 | P212121 |
| Cell | 52.5, 118.2, 128.9 | 69.8, 86.8, 159.1 | 71.8, 104.6, 118.1 |
| Z (hexamers) | 4 | 4 | 4 |
| Unique reflections | 195789 | 103224 | 93076 |
| Completeness (%)* | 99.3 (98.3) | 96.6 (81.4) | 99.8 (99.4) |
| Multiplicity* | 8.0 (6.8) | 6.5 (4.3) | 7.2 (6.9) |
| I/σ(I)* | 16.4 (2.3) | 25.4 (3.3) | 16.9 (3.4) |
| Rmerge* | 0.086 (0.42) | 0.062 (0.33) | 0.093 (0.43) |
| Refinement: | |||
| Rcryst | 0.18 | 0.20 | 0.17 |
| Rfree | 0.22 | 0.24 | 0.22 |
| r.m.s.d. bonds (Å) | 0.02 | 0.02 | 0.02 |
| r.m.s.d. angles (°) | 1.9 | 1.7 | 1.7 |
All data (outer shell)
Figure 9.
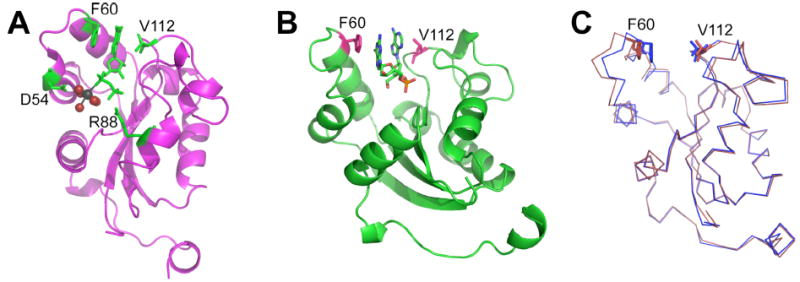
Ribbon diagrams indicating nucleotide binding and helix-turn-helix subdomain movements in individual subunits. (A) GDP binding to the wild-type protein, indicating Phe 60 and Val 112 interactions with the nucleotide base, Arg88 contact with the β phosphate, and Asp 54 hydrogen bonding to the magnesium-water complex, which are shown as spheres. (B) Approximate orientation for dinucleotide binding. (C) Superimposition of the GDP (blue) and dinucleotide (red) subunits, highlighting the ~3 Å shift in the helix-turn-helix subdomain positions.
Molecular Modeling
Modeling of NM23-H2 with the single-stranded DNA sequences corresponding to the guanine- and cytosine-rich strands of the c-myc NHE III1 was based upon the known crystal structure of NM23-H2 bound to GDP (1NUE) (52). The single-stranded DNA sequences were built using the Biopolymer module within the Insight II molecular modeling software. Electrostatic potential maps for NM23-H2 were calculated using the Delphi program and displayed using GRASP. The model for the complex between NM23-H2 and the single-stranded DNA was built using manual docking and the Discover_3 minimization program within Insight II. The complex was soaked in a TIP3P water layer of 10Å thickness and then subjected to molecular dynamics simulations with equilibration for 40 picoseconds and simulation for 100 picoseconds. Trajectories were collected every 0.1 picosecond. Low potential energy frames were minimized using 3000 steps of Discover_3 minimization. Minimized structures of the complex were used to analyze the interactions.
RESULTS
Knockdown of Endogenous NM23-H2 Expression by siRNA Results in Repression of c-myc Transcription
Since several studies have previously demonstrated that overexpression of NM23-H2 augments the levels of c-myc transcription, we decided to expand on these studies and examine whether depletion of endogenous NM23-H2 decreases such transcription reactions. To answer this question, we took advantage of RNA interference technologies, which have proven to be an effective tool for identifying transcription factor target genes (55). First, to verify the optimal silencing conditions, we transiently transfected increasing concentrations of an NM23-H2 siRNA pool into HeLa cells and determined the minimal amount needed to achieve maximal reduction of NM23-H2. At all concentrations, >80% knockdown of NM23-H2 expression was observed, whereas no effect was detected on the expression of NM23-H1, a homologue of NM23-H2 (data not shown). Therefore, the effects of siRNA-mediated knockdown of NM23-H2 on c-myc expression were examined in HeLa cells following 24 and 48 h of treatment. Within 24 h of transfection of the NM23-H2-siRNA, the mRNA (Figure 2A) and protein (Figure 2B) levels of NM23-H2 were reduced to nearly undetectable levels, which were maintained for the duration of the experiment. More interestingly, knockdown of NM23-H2 resulted in a reduction of c-myc mRNA expression levels of approximately 40% after 24 h (p < 0.05), which increased to almost 50% after 48 hours (p < 0.001) (Figure 2A). In addition, there was no effect on either NM23-H2 or c-myc expression in the untreated cells or the cells treated with a control non-targeting siRNA pool (Figure 2A). These results add to previous findings and strengthen the evidence for the involvement of NM23-H2 in the transcriptional activation of the c-myc gene.
Figure 2.
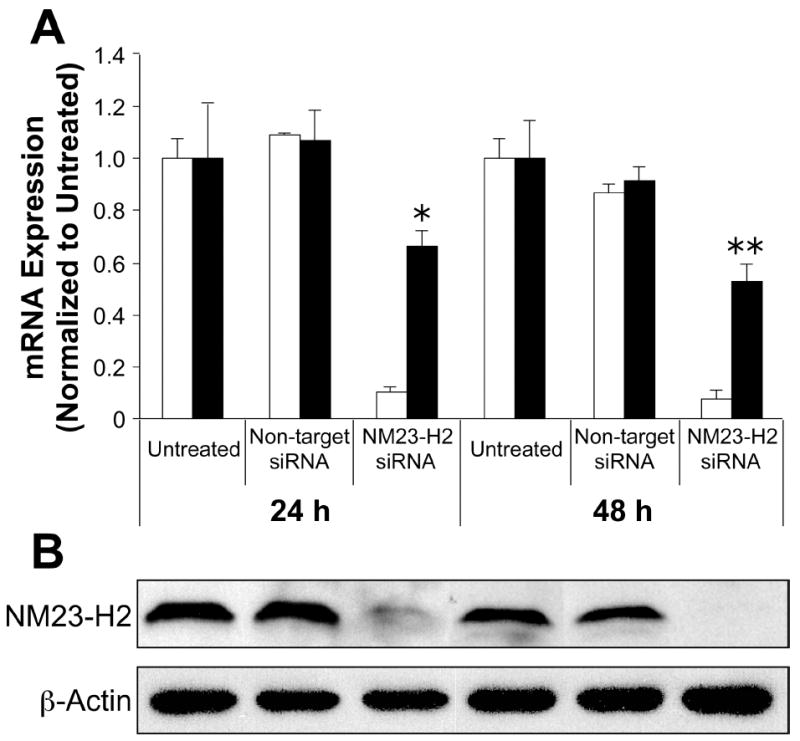
Effect of siRNA-mediated knockdown of NM23-H2 on c-myc transcription in HeLa cells. (A) The mRNA expression levels of NM23-H2 (white bars) and c-myc (black bars) measured by real-time PCR 24 h and 48 h after the different treatments shown (*, p < 0.05, ** p < 0.001). (B) Corresponding protein levels of NM23-H2 measured by Western blot analysis.
Relationship between NM23-H2 and c-myc Expression in Colon Tumor Cell Lines
It has been demonstrated that NM23-H2 mRNA levels are among the most abundant in colorectal cancers (56). Likewise, the c-myc gene is overexpressed in nearly 70% of colorectal cancers. Therefore, in a straightforward attempt to link NM23-H2 to the regulation of the c-myc gene, we analyzed the expression levels of both genes in five colorectal cancer cell lines (SW480, SW620, CaCo2, HT29, and HCT116). Total RNA from normal colon tissue was used as a control. As anticipated, there was a clear positive correlation (R2 = 0.856) between NM23-H2 and c-myc expression in the small number of cell lines analyzed (Figure 3). Such a correlation is in line with the involvement of NM23-H2 in the activation of c-myc transcription in colorectal cancer. However, despite several efforts, the molecular basis for this relationship has remained elusive.
Figure 3.
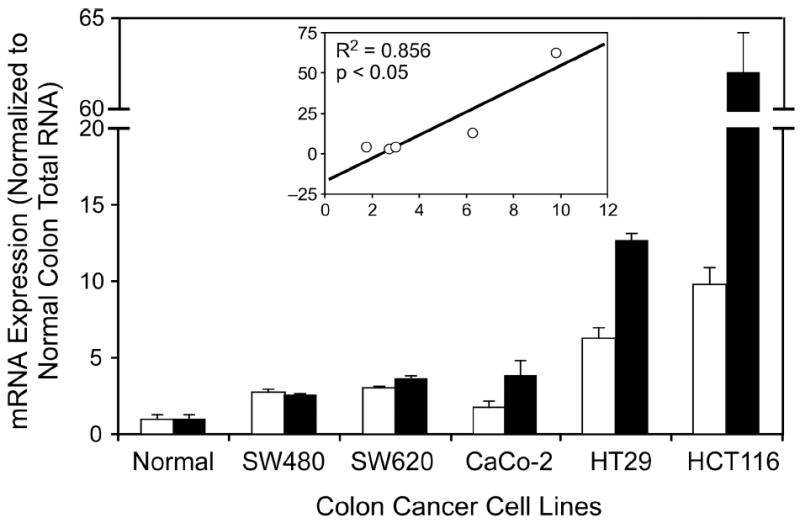
The mRNA expression levels of NM23-H2 (white bars) and c-myc (black bars) in human colon cancer cell lines versus normal colon measured by real-time PCR. Positive correlation between NM23-H2 and c-myc (R2 = 0.856) is shown in the inset (x-axis, NM23-H2; y-axis, c-myc). SW480 and SW620 are cell lines from the same patient (SW480 is from the primary colon tumor and SW620 is from a lymph node metastasis site).
Nuclease Activity Does Not Associate with NM23-H2 during Size-Exclusion Chromatography or Heparin Affinity Chromatography
In general, the ambiguity surrounding the possible DNA cleaving activity of NM23-H2 may perhaps be attributable to the differing sources and the purity of the recombinant protein. In this study, recombinant NM23-H2 was expressed and purified as previously described by Postel et al. (17). While we did observe DNA cleavage products in the presence of this recombinant NM23-H2 protein (Figure 4, right panel), we were unable to detect any gel retardation signal corresponding to a protein–DNA complex in native polyacrylamide gel analysis (data not shown). These results are inconsistent with previous studies, which have shown the presence of three separate species, a protein–DNA complex, free DNA, and cleaved DNA. When the recombinant NM23-H2 was subjected to further purification by size-exclusion chromatography, we reproducibly found that the protein lost its nuclease activity (Figure 4, left panel). The protein eluted at a volume consistent with a molecular mass of ~100 kDa, the expected size for the hexameric NM23-H2 protein, making it unlikely that the contaminating activity remained associated with the protein. Despite the fact that equal amounts of the protein were loaded on the left- and right-hand gels (Figure 4), we cannot ignore the possibility that the absence of the nuclease activity in the size-excluded preparation is a consequence of inactivation of the nuclease in this preparation relative to the conventionally purified NM23-H2.
Figure 4.
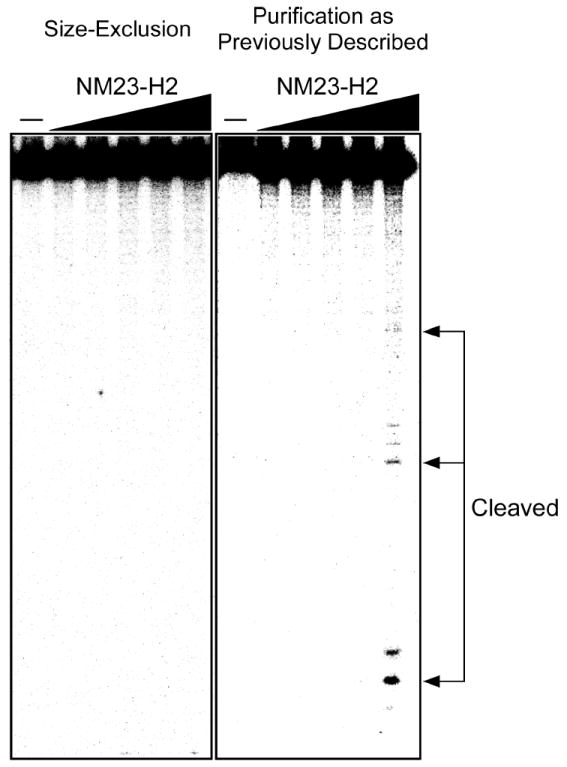
Effects of purification of the DNA cleavage activity of recombinant NM23-H2. Protein was serial diluted 2-fold from a maximum concentration of 1000 nM in both panels. Arrows indicate cleavage products.
In an attempt to further determine whether the apparent nuclease activity was associated with NM23-H2, the recombinant protein that displayed the DNA cleavage activity was applied to a heparin affinity column and eluted with a linear gradient of 0 to 1.0 M NaCl. As shown in Figure 5A, the major protein peak was eluted in fractions 8–12 at approximately 0.3 M NaCl. SDS/PAGE of the fractions across this peak revealed a single band corresponding to a polypeptide with a molecular mass of ~17 kDa (Figure 5B), which is in agreement with the molecular mass determined from the NM23-H2 amino acid sequence. The DNA binding activity and DNA cleavage activity of the eluted fractions were analyzed using a gel mobility shift assay and a DNA cleavage assay, respectively. Interestingly, the DNA binding activity co-eluted with the NM23-H2 protein peak, while the DNA cleavage activity was associated with the 1.0 M NaCl fractions (Figure 5A). These results suggest that the DNA cleavage activity might be a consequence of either a contaminating nuclease or an NM23-H2-interacting protein that becomes dissociated upon further purification. However, the identity of this nuclease remains elusive in view of the fact that proteomic analysis of the fractions containing the nuclease activity was unable to identify any known proteins with the ability to cause DNA strand breaks. Nevertheless, these observations clearly demonstrate that the nuclease activity detected was not associated with 17 kDa NM23-H2.
Figure 5.
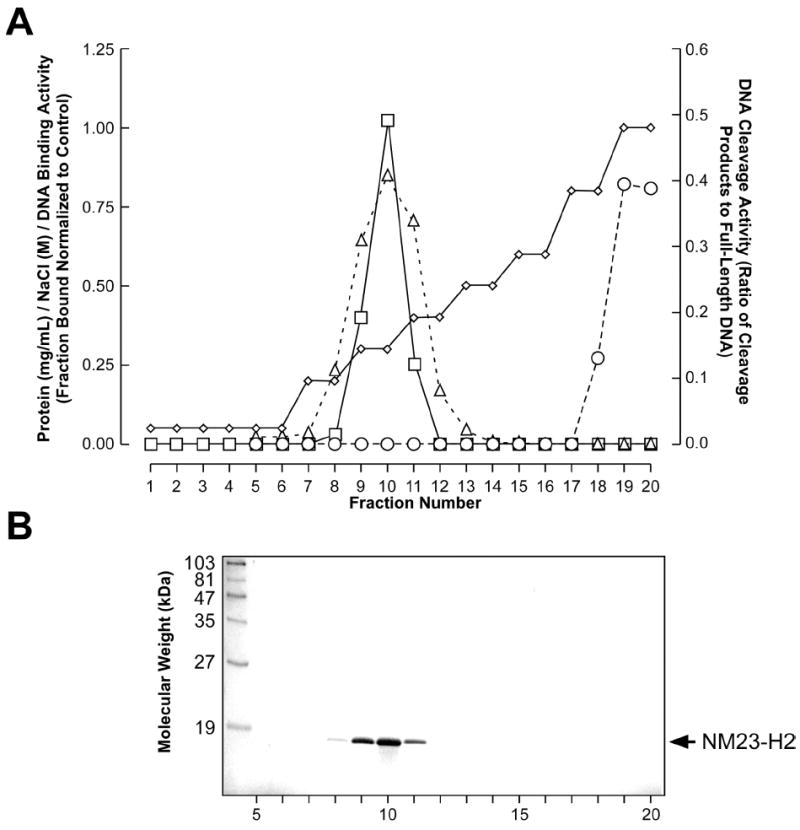
Heparin affinity column elution profile of recombinant NM23-H2. (A) Graphical representation of the NaCl concentration gradient (diamonds), protein concentration (squares), DNA binding activity (triangles), and DNA cleavage activity (circles) of each eluted fraction are shown. (B) 4–15% SDS/PAGE of eluted fractions. The molecular weight standards are indicated on the left side of the gel; an arrow shows the location of the NM23-H2 band on the right side.
DNA Binding Activity of NM23-H2
Having demonstrated that NM23-H2 does not display inherent nuclease activity, the next step was to examine its ability to interact with DNA. However, due to mixed reports pertaining to the DNA binding specificity of NM23-H2 for the c-myc NHE III1 region, we found it necessary to reevaluate some of the previously described studies. Initially, in order to assess directly the relative affinity of NM23-H2 for single-stranded versus duplex portions of the c-myc NHE III1 region, we carried out gel mobility shift assays under essentially the same conditions as Hildebrandt et al. (47), with the exception of the addition of a nonspecific DNA competitor and the use of slightly longer DNA substrates (47 base pairs). As shown in the representative gels in Figure 6A, protein–DNA complexes were formed between NM23-H2 and the single-stranded oligonucleotides corresponding to the purine-rich (Pu47) and the pyrimidine-rich (Py47) strands of the c-myc NHE III1 region, as evidenced by the occurrence of gel retardation bands; however, no such complex was observed with a duplex oligonucleotide of the same sequence (ds47). We also found that NM23-H2 appeared to favor the Pu47 sequence over the complementary Py47 sequence; however, the noticeable smear of radioactivity in the gel shift employing the Py47 sequence could represent dissociation of the bound complex during electrophoresis, which is indicative of the formation of an unstable complex with Py47. NM23-H2 also bound to a random 47-mer sequence but only at about a 10-fold higher concentration than to the Pu47.
Figure 6.
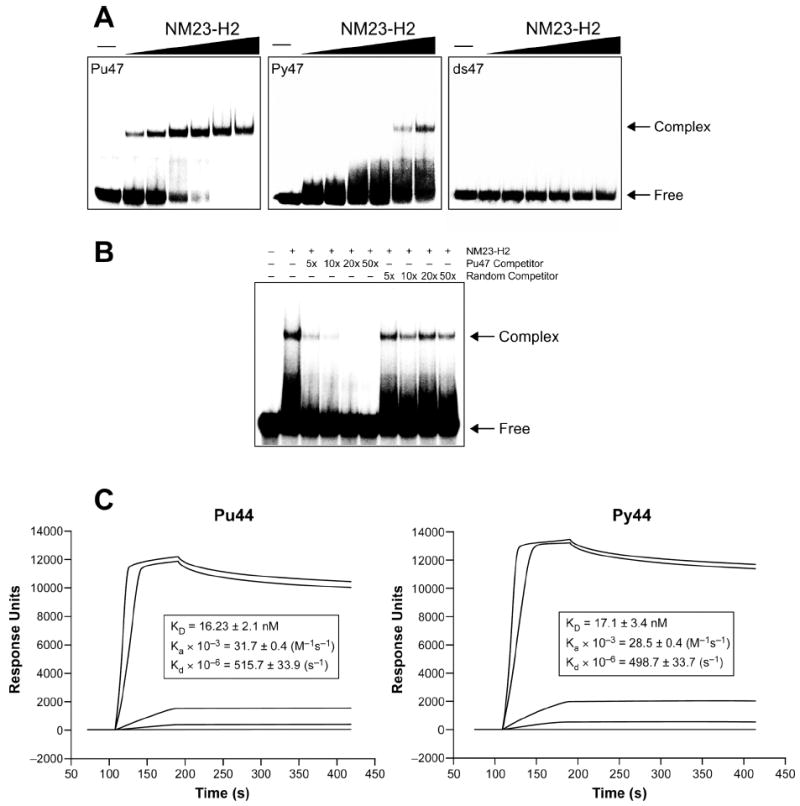
DNA binding specificity of NM23-H2 to oligonucleotides corresponding to the c-myc NHE III1 region. (A) Representative gel mobility shift assays of NM23-H2 (10, 25, 50, 100, 200, and 400 nM) binding to the single-stranded guanine-rich strand (Pu47), the single-stranded cytosine-rich strand (Py47), and the duplex (ds47). (B) Competition of NM23-H2 (150 nM) binding to the Pu47 sequence. Ranges of 5-fold to 50-fold excess of single-stranded oligonucleotides corresponding to the Pu47 sequence or a random sequence were used as competitors. Arrows in (A) and (B) indicate the protein–DNA complex and free DNA. (C) SPR sensorgrams for binding of increasing concentrations of NM23-H2 to the G-strand (left panel) and the C-strand (right panel) of the NHE III1 region of the c-myc promoter. The concentrations of the protein NM23-H2 range from 1 nM for the lowest curve to 1000 nM for the top curve.
To further examine the stability of the NM23-H2–Pu47 complex, a competition experiment was carried out. Briefly, protein–DNA complexes were equilibrated for approximately 30 min, and then incubated an additional 30 min with unlabeled competitor oligonucleotides corresponding to the Pu47 sequence or an unrelated sequence. Figure 6B shows the results of an experiment in which the complex between Pu47 and NM23-H2 was exposed to only a 5-fold excess of unlabeled Pu47 oligonucleotide. Disruption of the complex was found under these conditions but not by a random oligonucleotide sequence up to 50-fold excess. In addition to the instability of the Py47–NM23-H2 complex, these results also suggest that NM23-H2 might only weakly bind to the guanine-rich strand of the c-myc NHE III1 given that the preformed protein–DNA complex is easily disrupted by the presence of a minor amount of specific competitor. This observation is in agreement with the previously suggested role of NM23-H2 as an accessory protein, which temporarily binds to and prepares the DNA for interactions with other specific transcription factors.
To quantitatively determine the specificity of NM23-H2 to the purine- and pyrimidine-rich stands of the NHE III1 region of the c-Myc promoter, SPR was used. Two biotinylated 44-mers, which correspond to the guanine- and cytosine-rich strands of the NHE III1 region of the c-Myc promoter, were immobilized on a streptavidin-coated sensor chip. Increasing concentrations of purified NM23-H2 (1 nM to 1000 nM) were injected and the interaction of the protein with the DNA was measured. Binding between NM23-H2 and the guanine-rich strand (Figure 6C, left panel) and cytosine-rich strand (Figure 6C, right panel) was observed at concentrations as low as 50 nM, as illustrated by the increase in response that remained after the completion of the initial protein injection. The intensity of the observed response increased with increasing concentrations of protein in a dose-dependent manner. By fitting the sensorgrams using a 1:1 Langmuir model, the KD for the guanine- and cytosine-rich strands were calculated as 14.3 and 13.7 nM, respectively. These data strongly suggest that NM23-H2 binds equally well to the guanine- and cytosine-rich strands. The difference between the results of the gel mobility shift assay and the SPR results is most likely related to the instability of the NM23-H2–Py47 complex versus the NM23-H2–Pu47 complex (see Figure 6A, center panel). SPR allows for analysis of complex association and dissociation in “real time,” whereas gel shift assays are a non-equilibrium measurement that reflect the maintenance of an intact complex in a complicated environment. Thus, less stable complexes (i.e. the NM23-H2-Py47) may partially dissociate during gel loading or running, in which the affinity would be underestimated.
Stabilization of G-Quadruplex and i-Motif Structures Inhibits Binding of NM23-H2 to the c-myc NHE III1
Again using SPR, the effect of the G-quadruplex/i-motif-interactive agent TMPyP4 on NM23-H2 DNA binding was studied (11, 57). TMPyP2, a positional isomer of TMPyP4 unable to interact well with the G-quadruplex or i-motif, was used as a control (11, 57).
Before the protein was co-injected with the G-quadruplex-interactive agents, the compounds were injected alone to determine if the compounds are able to interact with the DNA. The interaction of TMPyP4 with both the guanine- and cytosine-rich 44-mers was observed weakly at 0.1 μM and strongly at 1 and 10 μM (KD: 0.49 μM [G-strand], KD: 0.45 μM [C-strand]) (unpublished results). Due to their similar KD values, no specificity of TMPyP4 between strands was concluded. Upon the co-injection of 300 nM NM23-H2 with increasing concentrations of TMPyP4, significant decreases in the Rmax values of both strands were detected beginning at 1 μM and continuing at 10 μM in a dose-dependent manner (Rmax decrease of 72.5% [G-strand] and 73.2% [C-strand] at 10 μM TMPyP4 compared to protein alone) (Figure 7, A and B). This decrease in Rmax values indicates an inhibition of NM23-H2 binding to both strands of the NHE III1 region of the c-Myc promoter in the presence of TMPyP4. This was expected, since TMPyP4 is known to bind to both G-quadruplexes and i-motifs (11, 57). TMPyP2 was used to determine if a compound of similar structure to TMPyP4 can non-specifically inhibit NM23-H2 binding to the DNA of interest. In the absence of protein, TMPyP2 did interact with both oligomers (KD: 15 μM [G-strand], KD: 4.22 μM [C-strand]); however, this interaction was less effective than that of TMPyP4 (Figure 7, A and B). When the ability of TMPyP2 to inhibit NM23-H2 binding was measured, only a small decrease in Rmax was observed (23.0% [G-strand], 32.8% [C-strand] at 10 μM TMPyP2 compared to protein alone), in comparison to the decrease due to TMPyP4 treatment (compare A and B in Figure 7). These data confirm that the stabilization of the G-quadruplex and i-motif by KCl and TMPyP4 prevents NM23-H2 binding to the NHE III1. We would expect to see a preferential effect of KCl on the binding of NM23-H2 to the purine-rich strand since KCl facilitates the formation of and stabilizes the G-quadruplex. Indeed, this is what we observe in Figure 7C. At a concentration of 20 mM KCl, there is a significant reduction of binding of the purine-rich strand to NM23-H2, which is more than twice the effect on the pyrimidine-rich strand. At higher concentrations (50–100 mM KCl), a more general non-specific effect of KCl is observed on both strands. Thus, conditions (TMPyP4 and KCl) that are known to stabilize the secondary DNA structures decrease the binding of NM23-H2 to these purine-rich and pyrimidine-rich strands.
Figure 7.
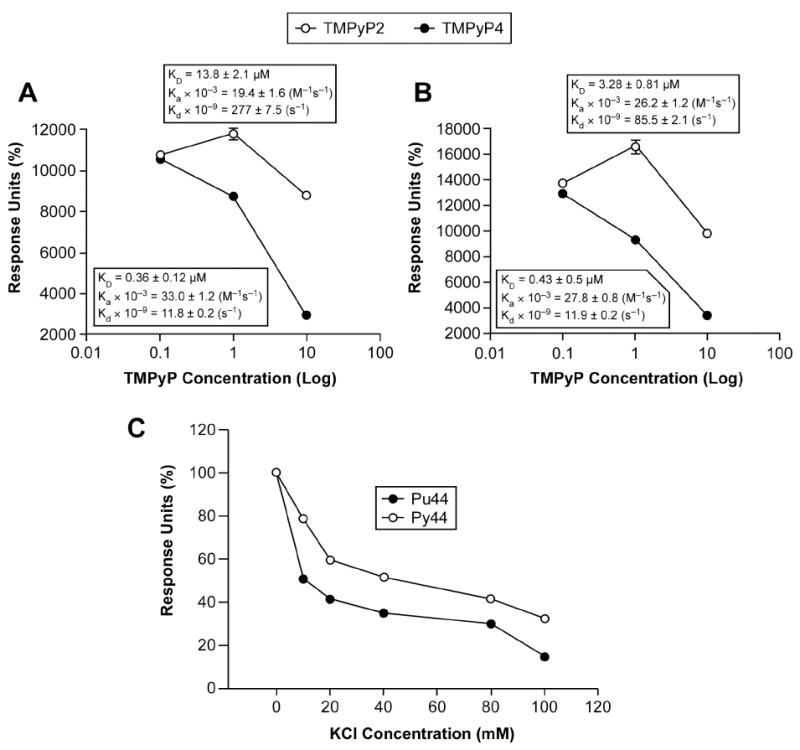
Inhibition of NM23-H2 DNA binding activity through the stabilization of the c-myc G-quadruplex and i-motif structures by TMPyP4. Quantitation of SPR sensorgrams for the binding of 300 nM NM23-H2 to the G-strand (Pu44) (A) and C-strand (Py44) (B) of the NHE III1 region of the c-myc promoter in the presence of increasing concentrations of TMPyP4 or TMPyP2. KD values were calculated using a 1:1 Langmuir model. (C) Effect of potassium concentration on binding of NM23-H2 to the G- and C-strands.
A Mutation within the Active Site of NM23-H2 Results in a Decrease in Single-Stranded DNA Binding
Prior studies have shown that various nucleotide substrates can inhibit the DNA binding activity of NM23-H2 (46). Inversely, single-stranded oligonucleotides have also been shown to be competitive inhibitors of the NDP kinase activity of NM23-H2 (58). Therefore, it has been suggested that one of the DNA bases within a given oligonucleotide may have a similar binding mode to that of a mononucleotide substrate (59). To determine whether the DNA and the nucleotide substrates involved in the catalytic reaction of NM23-H2 share a common binding site, we used an active site mutant protein that contained an arginine to alanine mutation at position 88 (R88A) within the primary amino acid sequence of NM23-H2 (provided by Dr. Edith Postel). The rationale for investigating this mutant was based on previous studies that have shown that the R88A mutation completely abolishes the NDP kinase activity, DNA binding activity, and nuclease activity of NM23-H2 (44, 50). The crystal structure of the R88A mutant described below confirms that the loss in activity is due to the loss of a critical contact between Arg88 and the phosphate group of the nucleotide. Using isothermal titration calorimetry, we also have shown that the R88A mutant was incapable of binding GDP, while the wild-type NM23-H2 convincingly bound GDP, giving a dissociation constant of 5.2 ± 0.8 μM (data not shown), a value nearly identical to that previously reported for NM23-H1 using a fluorescence anisotropy measurement (60). Therefore, because of the inability of the R88A mutant to interact with GDP, together with the suggestion that the binding sites for mononucleotides and polynucleotides within NM23-H2 have a common binding site, we reasoned that the R88A mutant would also be defective in DNA binding activity (50). The first clue to the low DNA binding affinity of the R88A mutant was seen from its low retention in a heparin affinity column during the initial purification of this protein. The structure and negative charge of heparin enable it to mimic DNA in its overall binding properties; thus, if the R88A mutant and the wild-type NM23-H2 have similar DNA binding affinity, then both should have similar retention times on a heparin column. However, while the wild-type protein was eluted at approximately 0.3 M NaCl, the R88A mutant protein was in one of the first fractions eluted from the column, which is indicative of its inability to bind to the negatively charged heparin (Figure 8A). The low DNA binding affinity associated with the R88A mutant was directly confirmed using a gel mobility shift assay (Figure 8B) and SPR measurements (Figure 8C). These results further confirm that the six corresponding active sites for the phosphotransferase reaction within NM23-H2 also serve as binding sites for single-stranded DNA.
Figure 8.
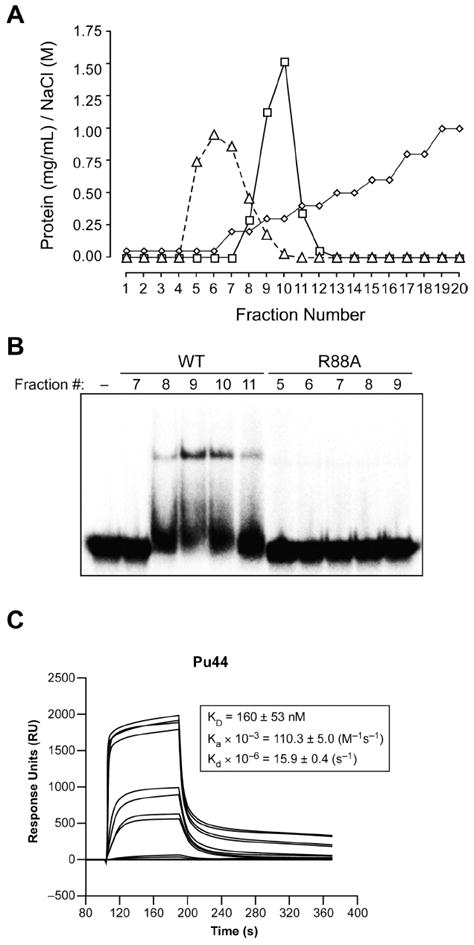
DNA binding activity of the wild-type NM23-H2 versus the R88A mutant. (A) Heparin affinity column elution profile of recombinant wild-type NM23-H2 (squares) and the R88A mutant (triangles). The NaCl concentration gradient is shown in diamonds. (B) Representative gel mobility shift assay of the protein-containing fractions eluted from the heparin affinity column binding to the Pu47 sequence. (C) SPR sensorgrams for the binding of increasing concentrations of R88A mutant NM23-H2 to the G-strand of the NHE III1 region of the c-myc promoter. The concentrations of the protein ranged from 1 nM for the lowest curve to 1000 nM for the top curve.
To further examine nucleotide binding to NM23-H2 in general, and the role of Arg88 in particular, we determined three crystal structures of the protein to resolutions between 1.3 and 1.7 Å (Table 1). First, we re-determined the structure of the complex with GDP but to a higher resolution than previously reported (52). The new structure was in good agreement with the previous structure, displaying Arg88 in close association with the β-phosphate of GDP and rotation of the helix-turn-helix domain (residues 42–71) by 13 degrees toward the binding pocket such that the GDP purine ring becomes sandwiched between Phe 60 and Val 112 (Figure 9A), which move toward one another by ~2.3 Å as compared with the unliganded subunits (described below). Magnesium coordination to oxygen of the α-phosphate occurs in all six active sites and displays octahedral coordination with five water molecules and an α-phosphate oxygen (Figure 9A). We also found that Cys 145 appears to be partially modified in all six subunits, apparently displaying a mixture of reduced cysteine, oxidized cysteine disulfide linked to dithiothreitol, and possibly oxidized cysteine disulfide linked to the neighboring subunit. Subunits A–F, B–D, and C–E are aligned such that Cys 145–Cys 145′ disulfide bonds could occur, suggesting that the protein may be under redox regulation.
We then examined the wild-type protein after soaking in the dinucleotide d(AG), which has been reported to bind tightly to NM23-H2, based on a competition assay (58). Weak binding of the dinucleotide was found in the structure, but the density for the dinucleotide was poorly defined, with only approximate positioning indicated. Interestingly, evidence for both nucleotides was found in two active sites (monomers C and E), and two active sites displayed only a single nucleotide, modeled as guanine (monomers B and F). The remaining two active sites did not appear to have any bound nucleotide. The mobile helix-turn-helix tracked the active site contents. In those subunits containing a single nucleotide, the mobile domain moved inward, similarly to that found in the GDP complex. In those subunits containing the dinucleotide, the domain moved outward, opening up the binding pocket and allowing the two stacked purine rings to fit between Phe 60 and Val 112 (Figure 9, B and C). Possibly a similar motion occurs upon interaction with single-stranded DNA. Since electron density for the dinucleotide appeared weak in the crystal, we directly examined binding by isothermal titration calorimetry. Dinucleotide binding was not detectable by this method, consistent with a weak (high micromolar to millimolar) interaction.
We also determined the R88A crystal structure in the absence (not shown) and presence (Table 1) of d(AG). The R88A mutant is nearly identical to the wild-type protein, with Ala88 well resolved in both structures. The overall RMS deviation in carbon alpha positions was 0.87 Å when fitting to the GDP-containing hexamer and 0.95 Å when fitting to the dinucleotide soaked hexamer; pairwise RMSD between individual subunits ranged between 0.4 and 1.0 Å, depending on the relative positions of the mobile helix-turn-helix subdomains. Water molecules fill the region previously occupied by the arginine side chain. No indication of nucleotide binding was found in the d(AG)-soaked crystal.
Molecular Modeling of Single-Stranded DNA Bound to NM23-H2
Two different models have been proposed for the binding of DNA to NM23-H2. The first describes the binding of a duplex DNA molecule at the equatorial region of NM23-H2, which was derived from mutational studies that identified three essential residues for DNA binding located on the surface of the hexamer at the 2-fold axis (i.e., Arg34, Asn69, and Lys135) (61). The second describes the binding of an 11-mer single-stranded oligonucleotide with the 4 and the A helices (also known as H4 and HA helices) (58), which was based on in vitro cross-linking experiments as well as clues taken from NM23-H2 crystal structures (52, 62, 63). In addition, this model suggested an active site-directed binding mode for the single-stranded DNA wherein a guanine at the 3′-end of the oligonucleotide binds to the active site (58) in a manner similar to that of the GDP in complex with NM23-H2 (52). Furthermore, it has been shown that this oligonucleotide containing a 3′-end guanine results in a 6-fold increase in binding affinity for NM23-H2 in comparison to an oligonucleotide without a guanine at the 3′-end (58). Indeed, several lines of evidence have demonstrated that the catalytic efficiency of NM23-H2 is dependent on the nucleotide of the substrate (64). For example, at least two independent studies have shown a relative specificity with an order of G > T = A > C (46, 58, 64), with an approximate difference between G and C of 50-fold (46). Supplementary support for this specificity has also been derived from the crystal structures of NDP kinases bound to different nucleotide substrates, wherein all the nucleobases have been shown to possess similar hydrophobic interactions with specific Phe and Val residues within the active site, while only guanine nucleobases form a specific hydrogen bond between their N2 amino group and the δ-carboxylate of a Glu residue (52, 65, 66). The latter model is in agreement with our observed preferential stability of NM23-H2 bound to the single-stranded guanine-rich strand of DNA and our studies with the active site R88A mutant protein. Thus, on the basis of available in-house binding data, we have expanded the latter model to describe the interaction of a 27-mer sequence corresponding to the guanine-rich strand of the c-myc NHE III1 with NM23-H2.
As revealed by the calculated electrostatic potential maps for NM23-H2 shown in Figure 10A, positive ion channels exist that link the individual active sites along the surface of each trimer. We reasoned that these paths of electropositive potential could accommodate the anionic backbone of DNA and thus allow a single-stranded DNA molecule to interact with all three active sites within a given trimer of NM23-H2. In addition, we envisage that the binding of a single base to each active site could be used to anchor the single-stranded DNA to the trimeric surface of the protein. On the basis of the preferred binding of guanine to the active sites of NM23-H2 and the optimal number of bases that could be accommodated by the electropositive channels, we have selected the three guanine residues positioned at the 3′-ends of the runs of four guanines within the guanine-rich strand of the c-myc NHE III1 region, which are equally spaced at intervals of eight bases, to interact with three active sites within a given trimer of NM23-H2 (Figure 10D).
Figure 10.
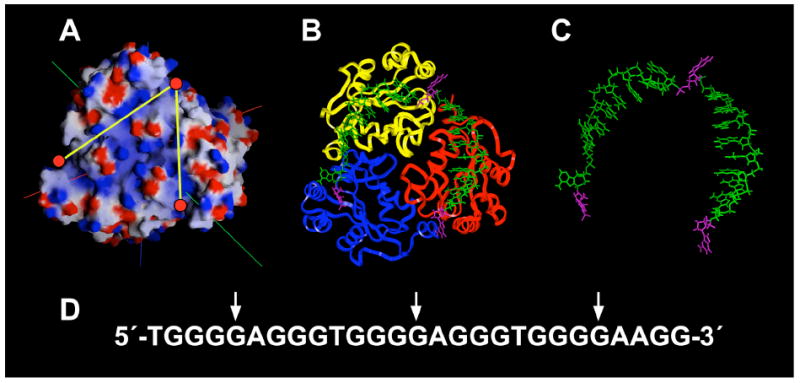
Molecular modeling of NM23-H2 and single-stranded DNA. (A) Calculated electrostatic potential maps for NM23-H2. Red circles indicate the active sites and yellow lines indicate channels of electropositivity. (B) Model of c-myc guanine-rich sequence with NM23-H2. The DNA strand is shown as capped sticks in green, with flipped guanines shown in magenta. A trimer of NM23-H2 is shown as a backbone ribbon (sidechains not shown). (C) DNA strand alone docked on the NM23-H2. (D) The c-myc guanine-rich sequence with arrows indicating the positions of the guanines docked within the active sites of NM23-H2.
Initially, the single-stranded 27-mer sequence corresponding to the c-myc NHE III1 was docked in a way analogous to the model proposed by Raveh et al. (58), in which the 5′-end of the DNA molecule entered NM23-H2 by interacting with the equatorial region or trimer interface. The DNA molecule was then extended between the 4 and A helices so as to allow the first guanine residue to interact with the active site of a given monomer unit. Following binding to the first active site, the DNA molecule was further extended to the second and third active sites along the surface of the trimer unit by way of the electropositive channels. Finally, the 3′-end of the single-stranded DNA molecule exited the protein through interactions with the residues of the α2 helix. This resulted in an increase in gap length of 15.7 Å between the 3′- and 5′-ends of the oligomer. This measurement represents the difference in gap length between the nearest adenines flanking the G-quadruplex at the 3′- and 5′-ends (21 Å) and the corresponding gap length after the quadruplex is unfolded on the face of the trimeric NM23-H2 (36.7 Å). The resulting model of the single-stranded guanine-rich strand docked onto a single trimer unit of NM23-H2 is shown in Figure 9B. This model reveals that the intervening bases stack readily with the deoxyribose backbone in the positive ion channels, with a favorable eight-base interval distance between the guanine bases, which were docked in the active sites (Figure 10, B and C). The significance of this unfolding of the G-quadruplex in transcriptional activation is described in the Discussion.
DISCUSSION
Of all the transcriptional factors involved in c-myc activation, NM23-H2 has been the most controversial. In this study, we set out to alleviate some of this controversy by evaluating and expanding on previous studies related to the biochemical mechanisms of how NM23-H2 activates c-myc transcription through the NHE III1 regulatory element. The first step was to confirm the regulatory role of NM23-H2, because some reports have demonstrated that NM23-H2 is capable of activating c-myc transcription via the NHE III1 region, while others have reported negative results. Using an RNAi approach, we discovered that knockdown of endogenous NM23-H2 expression results in approximately a 50% reduction in c-myc transcription in HeLa cells (Figure 2). The incomplete reduction in c-myc expression substantiates the previously described function of NM23-H2 as a possible accessory protein as opposed to a classic transcription factor. In addition, it is noteworthy that this level of reduction in c-myc expression has been shown to be sufficient to induce apoptosis and regression of tumors (67, 68).
It is well demonstrated that c-myc is overexpressed in the majority of colorectal and other malignancies (1, 69). However, until recently it has been less certain how NM23-H2 is involved in colorectal cancer. More recently there have been a number of studies (using tumors taken directly from patients) demonstrating increased nm23 expression as a predictor of metastasis and of poorer survival (70-73). Specifically, a study measuring protein levels in colon tumors and colon mucosa taken directly from patients using two-dimensional gel electrophoresis and SEIDI mass spectroscopy demonstrated that NM23-H2 was differentially expressed in colon cancer tissue versus normal colon mucosa (from the same patient) (73). Correspondingly, we have also shown that NM23-H2 gene expression is elevated in several human colon cancer cell lines and demonstrated that there is a direct correlation between NM23-H2 and c-myc gene expression within the same cell lines (Figure 3). Taken together, these studies corroborate previous findings and add to the growing evidence that NM23-H2 plays a role in the activation of c-myc transcription.
Most of the controversy surrounding NM23-H2 is attributable to the variable reports concerning NM23-H2–DNA substrate specificity and strand cleavage activity. We initially envisioned that the unprecedented mechanism involving reversible covalent attachment of NM23-H2 to DNA at six specific sites within the c-myc NHE III1 could result in unfolding of the G-quadruplex and i-motif structures for subsequent capture by DNA binding proteins involved in transcriptional activation. Unexpectedly, we discovered that this previously detected DNA cleavage activity was unassociated with NM23-H2 and could be related to a minor contaminant associated with the recombinant protein or to an accessory protein that is lost on more extensive purification or on mutation of NM23-H2 (44). Specifically, the results of this investigation demonstrated that the peak of DNA binding activity and that of DNA cleavage activity were not coincident during heparin affinity chromatography (Figure 5), which provides evidence against the former proposition that NM23-H2 possessed an inherent nuclease activity. If the protein responsible for the nuclease activity is tightly associated with NM23-H2, it may function under certain physiological conditions to cause strand breaks in DNA in this region. Selective cleavage of the NHE III1 could be an important event in apoptosis or even in transcriptional activation if the cleavage was reversible.
We next explored the DNA binding activity of NM23-H2 as a potential regulatory mechanism. Using the additionally purified NM23-H2, we observed that NM23-H2 bound equally well to both the single-stranded guanine- and cytosine-rich strands of the c-myc NHE III1, and to a lesser extent, to a random single-stranded oligomer, but it had no affinity to the duplex form (Figure 6A). In addition, we found that NM23-H2 binding to the guanine-rich strand could be easily reversed, based on the observation that the addition of unlabeled oligomer to the preformed complex reverses the binding, while a random sequence has no effect (Figure 6B). These findings suggest that NM23-H2 potentially functions in transcription through the recognition of DNA structural features within the target sequence (e.g., G-quadruplex and i-motif structures) and in subsequent remodeling and/or trapping-out of the c-myc NHE III1 region in a single-stranded or duplex form, which would allow single-strand-specific or duplex-specific transcription factors to recognize their DNA binding sites more efficiently. Importantly, we found that TMPyP4, and to a much lesser extent TMPyP2, reduced binding of NM23-H2 to the guanine-and cytosine-rich strands of the NHE III1 (Figure 7, A and B). Taken together, these results suggest that stabilization of the G-quadruplex and i-motif structures within the NHE III1 region hinder the recognition and remodeling functions of NM23-H2 in relation to the guanine- and cytosine-rich strands.
Prior to these studies, it was clear that the NHE III1 regulatory element in the promoter region of the c-myc gene was not a static matrix but was in equilibrium between at least three different DNA structural populations: two that cause activation and one that results in repression of c-myc transcription (74). One of the transcriptionally active complexes binds to the single-stranded binding proteins CNBP (purine-rich strand) and hnRNP K (pyrimidine-rich strand), whereas the second most likely binds to duplex binding transcriptional factors, such as Sp1 (74). The third and final transcriptionally silent form is an open, repressed form involving the formation of G-quadruplex structures, and possibly equivalent i-motif structures on the complementary strand within the NHE III1, leading to a quite distinct appearance from that of the active, attenuated, or vacant promoter forms described earlier (11). At present, one major question still remains to be addressed relating to this model: how does the NHE III1 region transition between the silenced form and the transcriptionally active forms? On the basis of the data presented herein, we have provided a potential answer to this question by inserting NM23-H2 into our working model, although other mechanisms involving topoisomerase I should also be considered. In either case, these enzymes assume the role of an accessory or transacting factor, functioning in the removal of repressive DNA secondary structures and subsequently preparing the DNA within the c-myc NHE III1 region for activating factors of c-myc transcription. Since the silencer element in the c-myc promoter is extremely stable (i.e., the G-quadruplex having a melting temperature of 85 °C) and is not the substrate for any known transcription factors, it is not surprising that there might be specific proteins that are required for transcriptional activation of c-myc, which must reorganize and resolve these structures.
The cartoon in Figure 11 illustrates the proposed transition from the nonnucleosomal form (A) of the NHE III1 region in the c-myc promoter to the silenced form (B), its subsequent remodeling by NM23-H2 to the unfolded form (C), and its ultimate capture by specific single-stranded DNA binding proteins to give a transcriptionally active form (D). Recent in vitro footprinting studies of a supercoiled plasmid containing the c-myc promoter region have revealed that the transition of the duplex DNA into secondary structures is accompanied by the local unwinding of the regions neighboring the NHE III1 region (submitted for publication). On the basis of these findings, we propose that NM23-H2 might initially recognize these contiguous single-stranded regions through interactions with the DNA binding domain located along the equatorial region of the protein. Subsequently, we speculate that the unfolding or trapping-out of the c-myc NHE III1 in a single-stranded form would occur in a stepwise manner wherein consecutive binding of the equally spaced guanine or cytosine residues occurs within each of the active sites of a given trimer unit of NM23-H2, resulting in a stepwise “pinning back” of the G-quadruplex and i-motif structures. Since the NM23-H2–DNA complex is highly reversible; we propose that the replacement by single-stranded or duplex binding transcriptional factors is facile (Figure 11D). Last, Figure 11 provides a structural basis for how compounds that stabilize the DNA secondary structures formed within the NHE III1 region of the c-myc gene promoter inhibit NM23-H2 from activating c-myc transcription (Figure 11E). Thus, without the intermediate involvement of NM23-H2 in remodeling the silencer element, it renders unnecessary all regulatory steps that follow.
Figure 11.
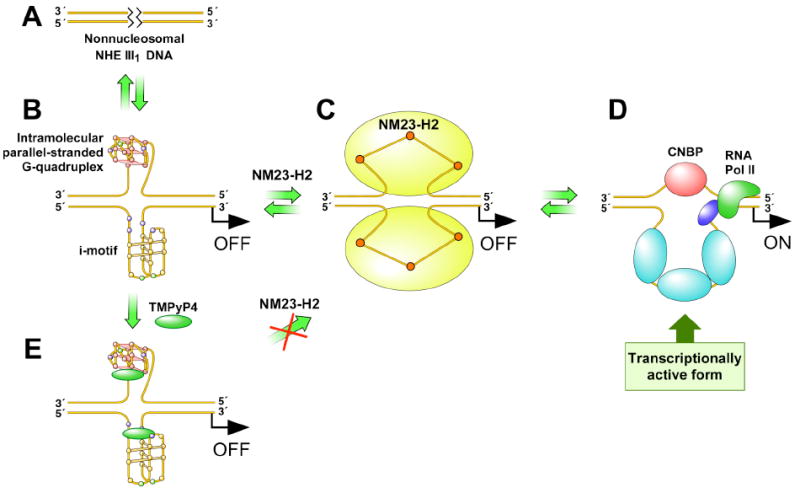
Proposed mechanism for the transcriptional activation of c-myc via the NM23-H2–DNA complex.
In conclusion, we believe that we have provided a working model of how NM23-H2 can activate c-myc transcription by recognition of the structural and functional features of the c-myc NHE III1, resulting in subsequent remodeling and trapping of the c-myc silencer element in an unfolded form. We have also established a new proof of principle that the c-myc-lowering effect can be achieved through drug stabilization of the G-quadruplex and consequent interference with the interaction of NM23-H2 with the silencer element in the c-myc promoter. However, further exploration into the interactions between the c-myc NHE III1 and NM23-H2 is of interest for the development of novel pharmacological agents.
Acknowledgments
We thank Edith Postel for her generous supply of materials, including protein for preliminary studies and clones and purification protocols for wild-type and R88A mutant NM23-H2. We also thank Jacquie Brailey for help with protein purification. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB entries 3BBB, 3BBF, and 3BBC). Finally, we are grateful to David Bishop for preparing, proofreading, and editing the final version of the manuscript and figures.
Financial Support: This work was supported by NIH grants HL62969 (to WRM) and CA94166 and CA95060 (to LHH). Diffraction measurements at BioCars Sector 14, Advanced Photon Source, Argonne National Laboratory, were supported by DOE Contract W-31-109-Eng-38 and NCRR Grant RR07707.
Abbreviations
- NHE
nuclease hypersensitive element
- NDP
nucleoside diphosphate
- SDS
sodium dodecyl sulfate
- siRNA
small interference RNA
References
- 1.Marcu KB, Bossone SA, Patel AJ. myc function and regulation. Annu Rev Biochem. 1992;61:809–860. doi: 10.1146/annurev.bi.61.070192.004113. [DOI] [PubMed] [Google Scholar]
- 2.Firulli AB, Maibenco DC, Kinniburgh AJ. The identification of a tandem H-DNA structure in the c-myc nuclease sensitive promoter element. Biochem Biophys Res Commun. 1992;185:264–270. doi: 10.1016/s0006-291x(05)80985-4. [DOI] [PubMed] [Google Scholar]
- 3.Postel EH, Mango SE, Flint SJ. A nuclease-hypersensitive element of the human c-myc promoter interacts with a transcription initiation factor. Mol Cell Biol. 1989;9:5123–5133. doi: 10.1128/mcb.9.11.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siebenlist U, Hennighausen L, Battey J, Leder P. Chromatin structure and protein binding in the putative regulatory region of the c-myc gene in Burkitt lymphoma. Cell. 1984;37:381–391. doi: 10.1016/0092-8674(84)90368-4. [DOI] [PubMed] [Google Scholar]
- 5.Davis TL, Firulli AB, Kinniburgh AJ. Ribonucleoprotein and protein factors bind to an H-DNA-forming c-myc DNA element: possible regulators of the c-myc gene. Proc Natl Acad Sci USA. 1989;86:9682–9686. doi: 10.1073/pnas.86.24.9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berberich SJ, Postel EH. PuF/NM23-H2/NDPK-B transactivates a human c-myc promoter-CAT gene via a functional nuclease hypersensitive element. Oncogene. 1995;10:2343–2347. [PubMed] [Google Scholar]
- 7.Boles TC, Hogan ME. DNA structure equilibria in the human c-myc gene. Biochemistry. 1987;26:367–376. doi: 10.1021/bi00376a006. [DOI] [PubMed] [Google Scholar]
- 8.Mathur V, Verma A, Maiti S, Chowdhury S. Thermodynamics of i-tetraplex formation in the nuclease hypersensitive element of human c-myc promoter. Biochem Biophys Res Commun. 2004;320:1220–1227. doi: 10.1016/j.bbrc.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 9.Phan AT, Modi YS, Patel DJ. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J Am Chem Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrus A, Chen D, Dai J, Jones RA, Yang D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization Biochemistry. 2005;44:2048–2058. doi: 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simonsson T, Pecinka P, Kubista M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998;26:1167–1172. doi: 10.1093/nar/26.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonsson T, Pribylova M, Vorlickova M. A nuclease hypersensitive element in the human c-myc promoter adopts several distinct i-tetraplex structures. Biochem Biophys Res Commun. 2000;278:158–166. doi: 10.1006/bbrc.2000.3783. [DOI] [PubMed] [Google Scholar]
- 14.Takimoto M, Tomonaga T, Matunis M, et al. Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J Biol Chem. 1993;268:18249–18258. [PubMed] [Google Scholar]
- 15.Tomonaga T, Levens D. Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J Biol Chem. 1995;270:4875–4881. doi: 10.1074/jbc.270.9.4875. [DOI] [PubMed] [Google Scholar]
- 16.Tomonaga T, Levens D. Activating transcription from single stranded DNA. Proc Natl Acad Sci USA. 1996;93:5830–5835. doi: 10.1073/pnas.93.12.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Postel EH, Berberich SJ, Flint SJ, Ferrone CA. Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science. 1993;261:478–480. doi: 10.1126/science.8392752. [DOI] [PubMed] [Google Scholar]
- 18.Michelotti EF, Tomonaga T, Krutzsch H, Levens D. Cellular nucleic acid binding protein regulates the CT element of the human c-myc protooncogene. J Biol Chem. 1995;270:9494–9499. doi: 10.1074/jbc.270.16.9494. [DOI] [PubMed] [Google Scholar]
- 19.Kolluri R, Torrey TA, Kinniburgh AJ. A CT promoter element binding protein: definition of a double-strand and a novel single-strand DNA binding motif. Nucleic Acids Res. 1992;20:111–116. doi: 10.1093/nar/20.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DesJardins E, Hay N. Repeated CT elements bound by zinc finger proteins control the absolute and relative activities of the two principal human c-myc promoters. Mol Cell Biol. 1993;13:5710–5724. doi: 10.1128/mcb.13.9.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majello B, De Luca P, Suske G, Lania L. Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Sp1-like proteins. Oncogene. 1995;10:1841–1848. [PubMed] [Google Scholar]
- 22.Hagen G, Muller S, Beato M, Suske G. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 1994;13:3843–3851. doi: 10.1002/j.1460-2075.1994.tb06695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filippova GN, Fagerlie S, Klenova EM, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsutsui H, Sakatsume O, Itakura K, Yokoyama KK. Members of the MAZ family: a novel cDNA clone for MAZ from human pancreatic islet cells. Biochem Biophys Res Commun. 1996;226:801–809. doi: 10.1006/bbrc.1996.1432. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama K, Tsutsui H, Fujita A. Negative repressor THZif-1 of protooncogene c-myc. Nippon Rinsho. 1995;53:2827–2836. [PubMed] [Google Scholar]
- 26.Sakatsume O, Tsutsui H, Wang Y, et al. Binding of THZif-1, a MAZ-like zinc finger protein to the nuclease-hypersensitive element in the promoter region of the c-MYC protooncogene. J Biol Chem. 1996;271:31322–31333. doi: 10.1074/jbc.271.49.31322. [DOI] [PubMed] [Google Scholar]
- 27.Zobel A, Kalkbrenner F, Vorbrueggen G, Moelling K. Transactivation of the human c-myc gene by c-Myb. Biochem Biophys Res Commun. 1992;186:715–722. doi: 10.1016/0006-291x(92)90805-u. [DOI] [PubMed] [Google Scholar]
- 28.Nakagoshi H, Kanei-Ishii C, Sawazaki T, Mizuguchi G, Ishii S. Transcriptional activation of the c-myc gene by the c-myb and B-myb gene products. Oncogene. 1992;7:1233–1240. [PubMed] [Google Scholar]
- 29.Lascu I, Gonin P. The catalytic mechanism of nucleoside diphosphate kinases. J Bioenerg Biomembr. 2000;32:237–246. doi: 10.1023/a:1005532912212. [DOI] [PubMed] [Google Scholar]
- 30.Janin J, Dumas C, Morera S, et al. Three-dimensional structure of nucleoside diphosphate kinase. J Bioenerg Biomembr. 2000;32:215–225. doi: 10.1023/a:1005528811303. [DOI] [PubMed] [Google Scholar]
- 31.Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO. The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembr. 2000;32:247–258. doi: 10.1023/a:1005584929050. [DOI] [PubMed] [Google Scholar]
- 32.Steeg PS, Bevilacqua G, Kopper L, et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988;80:200–204. doi: 10.1093/jnci/80.3.200. [DOI] [PubMed] [Google Scholar]
- 33.Stahl JA, Leone A, Rosengard AM, Porter L, King CR, Steeg PS. Identification of a second human nm23 gene, nm23-H2. Cancer Res. 1991;51:445–449. [PubMed] [Google Scholar]
- 34.Steeg PS, de la Rosa A, Flatow U, MacDonald NJ, Benedict M, Leone A. Nm23 and breast cancer metastasis. Breast Cancer Res Treat. 1993;25:175–187. doi: 10.1007/BF00662142. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama T, Ohtsuru A, Nakao K, et al. Expression in human hepatocellular carcinoma of nucleoside diphosphate kinase, a homologue of the nm23 gene product. J Natl Cancer Inst. 1992;84:1349–1354. doi: 10.1093/jnci/84.17.1349. [DOI] [PubMed] [Google Scholar]
- 36.Bevilacqua G. NM23 gene expression and human breast cancer metastases. Pathol Biol (Paris) 1990;38:774–775. [PubMed] [Google Scholar]
- 37.Rosengard AM, Krutzsch HC, Shearn A, et al. Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature. 1989;342:177–180. doi: 10.1038/342177a0. [DOI] [PubMed] [Google Scholar]
- 38.Boissan M, Wendum D, Arnaud-Dabernat S, et al. Increased lung metastasis in transgenic NM23-Null/SV40 mice with hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:836–845. doi: 10.1093/jnci/dji143. [DOI] [PubMed] [Google Scholar]
- 39.Ji L, Arcinas M, Boxer LM. The transcription factor, Nm23H2, binds to and activates the translocated c-myc allele in Burkitt’s lymphoma. J Biol Chem. 1995;270:13392–13398. doi: 10.1074/jbc.270.22.13392. [DOI] [PubMed] [Google Scholar]
- 40.Postel EH, Ferrone CA. Nucleoside diphosphate kinase enzyme activity of NM23-H2/PuF is not required for its DNA binding and in vitro transcriptional functions. J Biol Chem. 1994;269:8627–8630. [PubMed] [Google Scholar]
- 41.Pinon VP, Millot G, Munier A, et al. Cytoskeletal association of the A and B nucleoside diphosphate kinases of interphasic but not mitotic human carcinoma cell lines: specific nuclear localization of the B subunit. Exp Cell Res. 1999;246:355–367. doi: 10.1006/excr.1998.4318. [DOI] [PubMed] [Google Scholar]
- 42.Kraeft SK, Traincart F, Mesnildrey S, Bourdais J, Veron M, Chen LB. Nuclear localization of nucleoside diphosphate kinase type B (nm23-H2) in cultured cells. Exp Cell Res. 1996;227:63–69. doi: 10.1006/excr.1996.0250. [DOI] [PubMed] [Google Scholar]
- 43.Michelotti EF, Sanford S, Freije JM, MacDonald NJ, Steeg PS, Levens D. Nm23/PuF does not directly stimulate transcription through the CT element in vivo. J Biol Chem. 1997;272:22526–22530. doi: 10.1074/jbc.272.36.22526. [DOI] [PubMed] [Google Scholar]
- 44.Postel EH, Abramczyk BA, Gursky SK, Xu Y. Structure-based mutational and functional analysis identify human NM23-H2 as a multifunctional enzyme. Biochemistry. 2002;41:6330–6337. doi: 10.1021/bi025606+. [DOI] [PubMed] [Google Scholar]
- 45.Postel EH, Berberich SJ, Rooney JW, Kaetzel DM. Human NM23/nucleoside diphosphate kinase regulates gene expression through DNA binding to nuclease-hypersensitive transcriptional elements. J Bioenerg Biomembr. 2000;32:277–284. doi: 10.1023/a:1005541114029. [DOI] [PubMed] [Google Scholar]
- 46.Agou F, Raveh S, Mesnildrey S, Veron M. Single strand DNA specificity analysis of human nucleoside diphosphate kinase B. J Biol Chem. 1999;274:19630–19638. doi: 10.1074/jbc.274.28.19630. [DOI] [PubMed] [Google Scholar]
- 47.Hildebrandt M, Lacombe ML, Mesnildrey S, Veron M. A human NDP-kinase B specifically binds single-stranded poly-pyrimidine sequences. Nucleic Acids Res. 1995;23:3858–3864. doi: 10.1093/nar/23.19.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nosaka K, Kawahara M, Masuda M, Satomi Y, Nishino H. Association of nucleoside diphosphate kinase nm23-H2 with human telomeres. Biochem Biophys Res Commun. 1998;243:342–348. doi: 10.1006/bbrc.1997.8097. [DOI] [PubMed] [Google Scholar]
- 49.Postel EH. Cleavage of DNA by human NM23-H2/nucleoside diphosphate kinase involves formation of a covalent protein-DNA complex. J Biol Chem. 1999;274:22821–22829. doi: 10.1074/jbc.274.32.22821. [DOI] [PubMed] [Google Scholar]
- 50.Postel EH, Abramczyk BM, Levit MN, Kyin S. Catalysis of DNA cleavage and nucleoside triphosphate synthesis by NM23-H2/NDP kinase share an active site that implies a DNA repair function. Proc Natl Acad Sci USA. 2000;97:14194–14199. doi: 10.1073/pnas.97.26.14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 52.Morera S, Lacombe ML, Xu Y, LeBras G, Janin J. X-ray structure of human nucleoside diphosphate kinase B complexed with GDP at 2 Å resolution. Structure. 1995;3:1307–1314. doi: 10.1016/s0969-2126(01)00268-4. [DOI] [PubMed] [Google Scholar]
- 53.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401–423. doi: 10.1146/annurev.med.56.082103.104606. [DOI] [PubMed] [Google Scholar]
- 56.Zhang L, Zhou W, Velculescu VE, et al. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
- 57.Cashman DJ, Buscaglia R, Freyer MW, Dettler J, Hurley LH, Lewis EA. Molecular modeling and biophysical analysis of the c-MYC NHE-III1 silencer element. J Mol Model. 2008;14:93–101. doi: 10.1007/s00894-007-0254-z. [DOI] [PubMed] [Google Scholar]
- 58.Raveh S, Vinh J, Rossier J, Agou F, Veron M. Peptidic determinants and structural model of human NDP kinase B (Nm23-H2) bound to single-stranded DNA. Biochemistry. 2001;40:5882–5893. doi: 10.1021/bi001085j. [DOI] [PubMed] [Google Scholar]
- 59.Agou F, Raveh S, Veron M. The binding mode of human nucleoside diphosphate kinase B to single-strand DNA. J Bioenerg Biomembr. 2000;32:285–292. doi: 10.1023/a:1005593130867. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Gallois-Montbrun S, Schneider B, et al. Nucleotide binding to nucleoside diphosphate kinases: X-ray structure of human NDPK-A in complex with ADP and comparison to protein kinases. J Mol Biol. 2003;332:915–926. doi: 10.1016/j.jmb.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 61.Postel EH, Weiss VH, Beneken J, Kirtane A. Mutational analysis of NM23-H2/NDP kinase identifies the structural domains critical to recognition of a c-myc regulatory element. Proc Natl Acad Sci USA. 1996;93:6892–6897. doi: 10.1073/pnas.93.14.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reed JC, Kitada S, Takayama S, Miyashita T. Regulation of chemoresistance by the bcl-2 oncoprotein in non-Hodgkin’s lymphoma and lymphocytic leukemia cell lines. Ann Oncol. 1994;1(5 Suppl):61–65. doi: 10.1093/annonc/5.suppl_1.s61. [DOI] [PubMed] [Google Scholar]
- 63.Webb PA, Perisic O, Mendola CE, Backer JM, Williams RL. The crystal structure of a human nucleoside diphosphate kinase, NM23-H2. J Mol Biol. 1995;251:574–587. doi: 10.1006/jmbi.1995.0457. [DOI] [PubMed] [Google Scholar]
- 64.Schaertl S, Konrad M, Geeves MA. Substrate specificity of human nucleoside-diphosphate kinase revealed by transient kinetic analysis. J Biol Chem. 1998;273:5662–5669. doi: 10.1074/jbc.273.10.5662. [DOI] [PubMed] [Google Scholar]
- 65.Morera S, Chiadmi M, LeBras G, Lascu I, Janin J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry. 1995;34:11062–11070. [PubMed] [Google Scholar]
- 66.Williams RL, Oren DA, Munoz-Dorado J, Inouye S, Inouye M, Arnold E. Crystal structure of Myxococcus xanthus nucleoside diphosphate kinase and its interaction with a nucleotide substrate at 2.0 A resolution. J Mol Biol. 1993;234:1230–1247. doi: 10.1006/jmbi.1993.1673. [DOI] [PubMed] [Google Scholar]
- 67.Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev. 2004;14:37–42. doi: 10.1016/j.gde.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 68.Felsher DW. Oncogenes as therapeutic targets. Semin Cancer Biol. 2004;14:1. doi: 10.1016/j.semcancer.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 69.Spencer CA, Groudine M. Control of c-myc regulation in normal and neoplastic cells. Adv Cancer Res. 1991;56:1–48. doi: 10.1016/s0065-230x(08)60476-5. [DOI] [PubMed] [Google Scholar]
- 70.Sarris M, Lee CS. nm23 protein expression in colorectal carcinoma metastasis in regional lymph nodes and the liver. Eur J Surg Oncol. 2001;27:170–174. doi: 10.1053/ejso.2000.1070. [DOI] [PubMed] [Google Scholar]
- 71.Giarnieri E, Alderisio M, Valli C, et al. Overexpression of NDP kinase nm23 associated with ploidy image analysis in colorectal cancer. Anticancer Res. 1995;15:2049–2053. [PubMed] [Google Scholar]
- 72.Berney CR, Yang JL, Fisher RJ, Russell PJ, Crowe PJ. Overexpression of nm23 protein assessed by color video image analysis in metastatic colorectal cancer: correlation with reduced patient survival. World J Surg. 1998;22:484–490. doi: 10.1007/s002689900421. [DOI] [PubMed] [Google Scholar]
- 73.Melle C, Osterloh D, Ernst G, Schimmel B, Bleul A, von Eggeling F. Identification of proteins from colorectal cancer tissue by two-dimensional gel electrophoresis and SELDI mass spectrometry. Int J Mol Med. 2005;16:11–17. [PubMed] [Google Scholar]
- 74.Michelotti GA, Michelotti EF, Pullner A, Duncan RC, Eick D, Levens D. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol Cell Biol. 1996;16:2656–2669. doi: 10.1128/mcb.16.6.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]