

Abstract
The drug addiction process shares many commonalities with normal learning and memory. Addictive drugs subvert normal synaptic plasticity mechanisms, and the consequent synaptic changes underlie long-lasting modifications in behavior that accrue during the progression from drug use to addiction. Supporting this hypothesis, it was recently shown that nicotine administered to freely moving mice induces long-term synaptic potentiation of the perforant path connection to granule cells of the dentate gyrus. The perforant path carries place and spatial information that links the environment to drug taking. An example of that association is the nicotine-induced synaptic potentiation of the perforant path that was found to underlie nicotine-conditioned place preference. The present study examines the influence of nicotine over local GABAergic inhibition within the dentate gyrus during the drug-induced synaptic potentiation. In vivo recordings from freely moving mice suggested that both feedforward and feedback inhibition onto granules cells were diminished by nicotine during the induction of synaptic potentiation. In vitro brain slice studies indicated that nicotine altered local circuit inhibition within the dentate gyrus leading to disinhibition of granule cells. These changes in local inhibition contributed to nicotine-induced in vivo synaptic potentiation, thus, likely contributed to drug-associated memories. Through this learning process, environmental features become cues that motivate conditioned drug-seeking and drug-taking behaviors.
Introduction
Evidence supports that addictive drugs subvert normal mechanisms of neuronal adaptation, learning, and memory, leading to long-lasting changes in behavior that accrue with the ongoing progression of addiction (Mansvelder and McGehee, 2000, 2002; Everitt et al., 2001; Bonson et al., 2002; Kauer, 2004; Robinson and Kolb, 2004; Dani and Harris, 2005; Jones and Bonci, 2005; Paterson et al., 2005; Kenny and Markou, 2006; Kauer and Malenka, 2007). Events and processes linked to the acquisition and delivery of drugs (for example, the environment during drug taking) become learned associations. Over time, these associations become salient cues that prompt continued drug use.
Recently, it was shown that nicotine administration to freely moving mice produces in vivo synaptic potentiation of the medial perforant path to the dentate gyrus (Tang and Dani, 2009). The medial perforant path carries place, context, and spatial information from the entorhinal cortex (Hargreaves et al., 2005), which receives convergent inputs from the neocortex (Deadwyler et al., 1979; Lavenex and Amaral, 2000). Because the medial perforant path carries such pertinent information, the nicotine-induced synaptic potentiation correlated with the learned (or conditioned) place preference to nicotine (Tang and Dani, 2009). These finding spurred the question, how does nicotine act on the local circuits of the dentate gyrus to enable in vivo synaptic potentiation that underlies drug-associated memory?
Nicotinic acetylcholine receptors (nAChRs) are densely expressed especially in the inhibitory interneurons of the hippocampus, including the dentate gyrus (Jones and Yakel, 1997; Alkondon et al., 1999; Ji and Dani, 2000; Alkondon and Albuquerque, 2001, 2005; Yakel and Shao, 2004). In addition, inhibitory interneurons exert powerful control over circuit activity through feedforward and feedback inhibition (Freund and Buzsáki, 1996). Therefore, we hypothesized that nicotine administration has influences over local inhibition within the dentate gyrus that contribute to the nicotine-induced synaptic potentiation of the perforant path.
To investigate this hypothesis, we conducted in vivo field recordings from the dentate gyrus while stimulating the medial perforant path in the absence or presence of nicotine at concentrations that induced synaptic potentiation. Guided by previous studies performed on anesthetized rodents (Andersen et al., 1966; Buzsáki, 1984; Sloviter, 1991; Rosenblum et al., 1999), we applied various stimulation protocols to examine feedforward and feedback inhibition in freely moving mice. Then we cut brain slices that included the dentate gyrus to examine more directly the action of nicotine on dentate gyrus circuitry. The results indicate that nicotine alters local inhibition leading to disinhibition of granule cells. This process enhances perforant path transmission that contributes to nicotine-induced in vivo synaptic potentiation of the kind that underlies drug-associated memory.
Materials and Methods
In vivo surgery, handling, and habituation.
C57BL/6J mice (3–4 months) were studied. They were housed individually with food and water ad libitum in a temperature-controlled room (23 ± 0.5°C) with a 12 h light/dark cycle (light on at 9:00 P.M.). All experiments were performed during the dark phase of the cycle. The Institutional Animal Care and Use Committee in accordance with federal guidelines approved all procedures.
Animal surgery, postsurgical handling, and habituation were as described previously (Tang and Dani, 2009). After sodium pentobarbital (80 mg/kg, i.p.) anesthesia and mounting in stereotaxic apparatus, concentric bipolar stimulating electrodes were positioned to the medial part of the perforant path (anteroposterior, −0.2 mm posterior and 2.8–3.0 mm lateral of lambda; 1.0–1.3 mm below dura). The recording electrode (Teflon-coated tungsten wire; bare diameter, 50 μm) was targeted ipsilaterally to the hilus of the dentate (1.8–2.0 mm posterior, 1.4–1.6 mm lateral of bregma, and 2.2–2.3 mm below the skull) (Franklin and Paxinos, 1997). Mice were given at least 2 weeks to recover. Each animal was handled and habituated to needle injections (saline, 0.3 ml, i.p.) as well as the recording apparatus daily for 4–6 d before taking measurements (Tang and Dani, 2009).
In vivo examination of feedforward inhibition using the frequency-dependent inhibition index.
Monophasic square pulses (100 μs) were delivered to the medial perforant path (pClamp8 and 1320A; Molecular Devices). Signals were amplified (100×), filtered (bandpass, 0.1–5 kHz), digitized at 10 kHz, and stored on disk for off-line analysis. The frequency-dependent inhibition (FDI) was determined as described previously for anesthetized animals (Sloviter, 1991; Rosenblum et al., 1999). Briefly, 10 stimuli at 0.1 Hz and at 1.0 Hz were applied at 40–50% of the maximal population (pop) spike amplitude, and this pattern was repeated twice. The pop spike amplitude of responses 2–4 at 0.1 Hz was averaged and compared with that of the averaged last three responses at 1.0 Hz in each set (see Fig. 1A). The results of the two sets were averaged. Inhibition was expressed as a ratio of the averaged response at 0.1 Hz to that at 1.0 Hz. Saline and nicotine (0.5 mg/kg, i.p.) sessions were counterbalanced across animals. Basic synaptic transmission (see Fig. 1A, test transmission) was collected between FDI determinations using single stimuli at 0.033 Hz.
Figure 1.
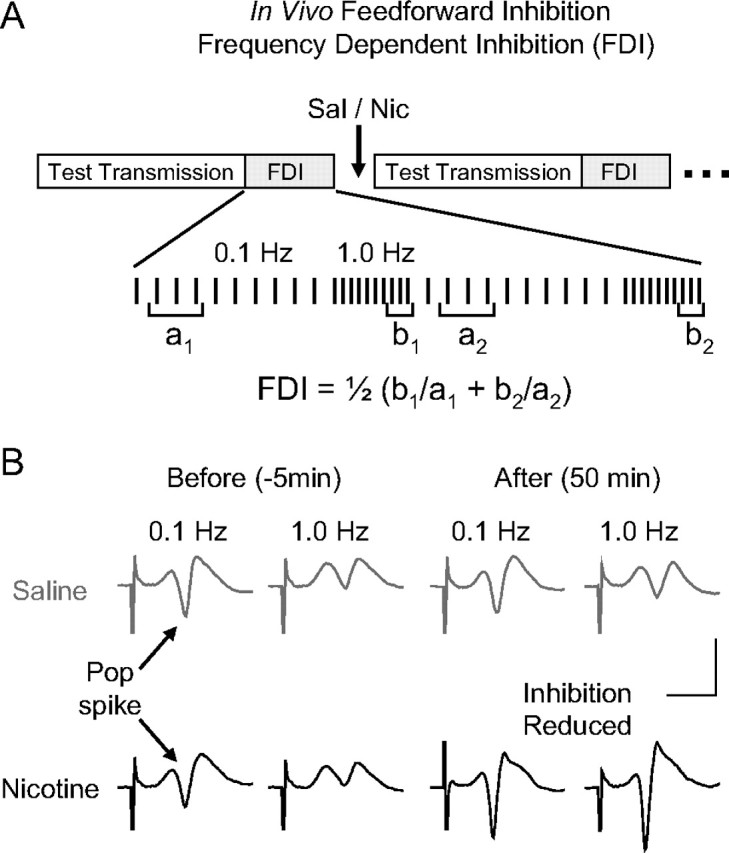
In vivo evaluation of feedforward inhibition arising from perforant path activity within the dentate gyrus. A, Didactic representation of the stimulus protocols used to determine FDI. The stimulus protocols for FDI and synaptic transmission were delivered alternately. To assess FDI, 10 pulses were delivered at 0.1 Hz followed by 10 pulses at 1.0 Hz. This pattern was repeated twice. The population spikes from responses 2–4 at 0.1 Hz (a1, a2) were averaged and compared with that of the averaged last three responses at 1.0 Hz (b1, b2) in each set. The results of the two sets were averaged to give the FDI. Basic synaptic transmission was evaluated by 10 stimuli delivered at 0.033 Hz between the FDI protocols. B, Representative example population spikes (Pop spike; arrows) before and after saline or nicotine administration. Note that the amplitude of the pop spikes is larger after nicotine administration, indicating synaptic potentiation. Calibration: 4 mV, 4 ms.
In vivo examination of feedback inhibition using a paired-pulse protocol.
Guided by previous studies applied to anesthetized animals (Andersen et al., 1966; Sloviter, 1991), paired pulses with interstimulus interval of 15, 30, 50, 100, or 150 ms were delivered at 0.025 Hz to the medial perforant path (see Fig. 3A). The strength of the conditioning pulse (see Fig. 3A, cond.) was adjusted to evoke 70–80% inhibition of the second pop spike when using a 15 ms interpulse interval (i.e., 20–30% of the test response). The strength of the test pulse was adjusted to evoke a pop spike that was 70% of the maximal response (see Fig. 3A, test). Paired pulses induced feedback inhibition that was expressed as the amplitude of the second pop spike divided by the amplitude evoked by the test pulse applied alone (see Fig. 3A, test). To evaluate the nicotine effect on feedback inhibition, saline- and nicotine (0.5 mg/kg, i.p.)-treated sessions were counterbalanced across animals. Basic synaptic transmission tested by 0.033 Hz single stimuli (evoked at 40–50% of the maximal responses) was evaluated between the paired-pulse protocols applied with an interpulse interval of 15 or 30 ms (see Fig. 4A).
Figure 3.

In vivo evaluation of feedback inhibition arising from perforant path activity within the dentate gyrus. A, Field potential recordings (average of 10 traces collected at 0.025 Hz) from the dentate in response to the paired stimulation of the medial perforant path. The conditioning pulse (Cond.) was set to produce a second response of 20–30% when using a 15 ms interpulse interval. The test pulse (Test) was set to evoke 70% of the maximal response to a single stimulation. The examples show the responses evoked by the second (test) pulse after the conditioning stimulation at the following interstimulus intervals (in milliseconds): 15, 30, 50, 100, and 150 ms. B, Summary (±SEM) of the data shown in A and normalized to the test response alone. The pop spike responses were significantly inhibited at the interstimulus intervals of 15 and 30 ms, and one-way ANOVA with repeated measures showed the significant differences [F(5,15) = 14.58, p < 0.01; n = 4; *p < 0.05, Tukey's honestly significant difference (HSD) test].
Figure 4.
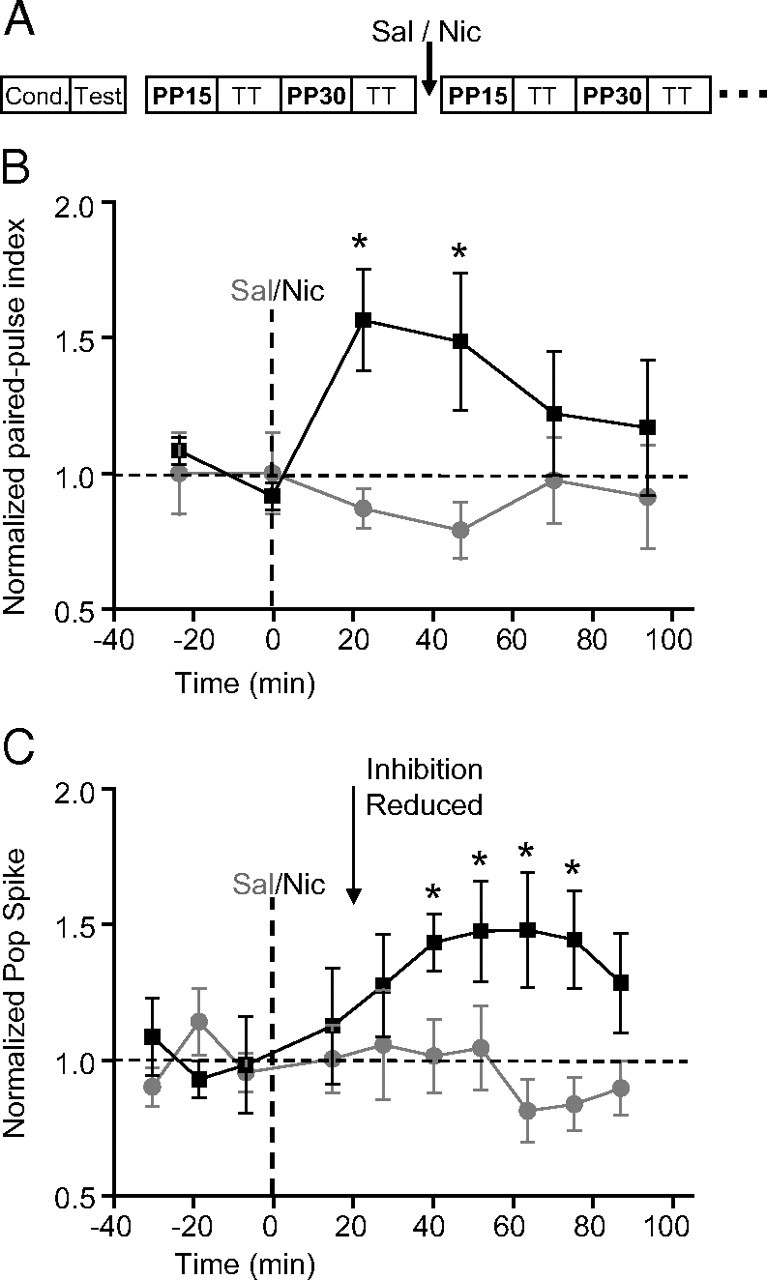
Systemic nicotine reduces feedback inhibition before in vivo induction of nicotine-induced synaptic potentiation. A, Didactic representation of the protocol to assess the effects of nicotine on feedback inhibition. After determining the appropriate stimulus amplitude for the conditioning and test pulses, 10 paired stimulus pulses were delivered at 0.025 Hz. PP15 represents paired pulses that were given at a 15 ms interspike interval, and PP30 represents paired pulses that were give at 30 ms interspike intervals. In between paired pulse tests, synaptic transmission (TT) was evaluated with 10 single pulses delivered at 0.033 Hz. B, Summary (±SEM) of paired-pulse index: the amplitude of the pop spike measured from the second stimulation coming 30 ms after the conditioning stimulus was normalized to baseline before nicotine. The feedback inhibition was significantly reduced (i.e., the index increased) 22 min after nicotine (0.5 mg/kg, i.p.; black squares; n = 6) when compared with saline treatment (gray circles; n = 6). ANOVA showed the significant differences (group by time: F(5,50) = 2.73, p < 0.05; n = 6; *p < 0.05 vs saline, Tukey's HSD test). C, Summary (±SEM) of pop spike amplitudes normalized to baseline. The synaptic transmission of the perforant path recorded in the dentate was potentiated 40 min after nicotine (0.5 mg/kg, i.p.; black squares; n = 6) when compared with saline treatment (gray circles; n = 6). ANOVA showed the significant differences [group: F(1,10) = 7.43, p < 0.05; group by time: F(9,90) = 1.90, p = 0.06; *p < 0.05 vs saline, least significant difference (LSD) test].
In vivo drug administration and histology.
(−)-Nicotine hydrogen tartrate salt (calculated as the free base) was dissolved in saline and adjusted for 0.1 ml per 10 g body weight (intraperitoneal). Mice were killed with an overdose of isoflurane at the end of the experiments. An anodal current (30 μA; 10 s) was passed through the tungsten wire for identification of the electrode placements. Frozen 30 μm coronal sections were cut and stained with 0.4% cresyl violet (Tang and Dani, 2009).
Analysis of the in vivo data.
The amplitude of the population spikes was measured for each evoked response, and the values from 10 consecutive recordings were averaged. Data were expressed as the mean ± SEM and analyzed with one-way or two-way ANOVA with repeated measurements.
In vitro brain slices and electrophysiology.
C57BL/6 mice (17–25 d of age) were anesthetized with a mixture of ketamine (42.8 mg/ml), xylazine (8.6 mg/ml), and acepromazine (1.4 mg/ml) at a dosage of 0.05 ml/10 g and were decapitated, and horizontal slices (220 μm thick) were cut following published procedures (Ji et al., 2001; Ge and Dani, 2005). The ice-cold cutting solution was the following (in mm): 120 NaCl, 25 NaHCO3, 3.3 KCl, 1.23 NaH2PO4, 4 MgSO4, 0.5 CaCl2, 10 dextrose, oxygenated with 95% O2/5% CO2. After a 20 min recovery at 32°C, slices were maintained at room temperature and were used for recording within the following 5 h.
The recording chamber was continuously perfused with artificial CSF (ACSF) buffer at 32–34°C. Tight-seal whole-cell recordings were used in the voltage-clamp or current-clamp mode. Recording electrodes were made from thin-walled borosilicate glass (TW150F-4; WPI; 1.2–2.2 MΩ) and filled with the following (in mm): 130 KCl or CsCl, 10 HEPES, 0.5 EGTA, 1 MgCl2, 2 ATP (magnesium salt), 0.3 GTP (sodium salt), adjusted to pH 7.3–7.4. For the in vitro experiments examining feedback inhibition, QX-314 (lidocaine N-ethyl bromide) (5 mm) was usually included in the intracellular solution to suppress action potentials from the granule cell that was being patch clamped. Data were collected using a 200B amplifier and acquired via a Digidata 1322A device (Molecular Devices) at 20 kHz. The series resistance was uncompensated and continuously monitored during the course of all experiments. If the access resistance changed by >20%, the recording was terminated and not analyzed.
Constant-current pulses (100 μs duration, evoked every 60 s) were applied through a stimulus isolation unit driven by a Master-8 stimulator (A.M.P.I.). For accessing feedback inhibition, a bipolar electrode was positioned in the distal mossy fiber pathway in the CA3 area. In paired-pulse ratio experiments, a train of two identical constant-current pulses with interstimulus intervals of 300 ms was delivered once every 60 s (Colino and Malenka, 1993). For drug application, after at least 6–10 min of stable baseline recording, ACSF was switched to the drug-containing solution, and the baseline response was monitored for at least 15–20 min to allow the drug to reach a stable concentration within the slice.
To decrease variability among the patch-clamped cells, we chose granule cells from the upper blade of the dorsal hippocampus, and interneurons with fusiform soma shape from the border of the granule cell layer and the hilus. Interneurons were filled with 0.3% Neurobiotin or biocytin during whole-cell recordings, and slices were fixed in 4% paraformaldehyde overnight at 4°C before processing (Nissl staining) to examine the structure. HIPP cells (i.e., hilar interneuron with perforant pathway-associated axon terminals) were distinguished from inhibitory basket cells in this region by structure analysis and neuron firing properties. Shortly after achieving the whole-cell recording condition, the current-clamp recording mode was used to apply currents ranging from −0.25 to 0.5 nA (2 s duration) with a 0.05 nA step increment to access the electrophysiological behavior of the cell. Frequency adaptation was measured by comparing the maximal firing rates on injection of depolarizing currents, and the sag ratio was measured on injection of hyperpolarizing currents under whole-cell conditions. The sag ratio was defined as follows: (voltage at the end of hyperpolarizing step minus baseline voltage before current injection)/(voltage at the beginning of hyperpolarizing step minus baseline voltage before current injection).
In vitro determination of the AMPA receptor/NMDA receptor ratio.
To assess nicotine-induced long-term potentiation (LTP) by determining the AMPA receptor (AMPAR)/NMDA receptor (NMDAR) ratios, mice were injected with either saline or nicotine (1 mg/kg, i.p.) 1 h before cutting brain slices. EPSCs were evoked by a bipolar electrode positioned in the medial perforant path of the dentate molecular layer, and the granule cell was voltage clamped at +40 mV to relieve the magnesium blockade of NMDA receptors. A recording of a glutamatergic EPSC containing both AMPAR and NMDAR components was made first in the presence of the GABAA antagonist picrotoxin (100 μm). Then, the AMPA component of the EPSC was isolated by bath application of the NMDA receptor antagonist (dl)-AP-5 (100 μm). The NMDA component of the EPSC was calculated by subtracting the AMPA component from the total EPSC (Ungless et al., 2001; Saal et al., 2003).
In vitro measurement of spontaneous IPSCs.
GABAA receptor-mediated spontaneous IPSCs (sIPSCs) were pharmacologically isolated in the presence of DNQX (10 μm) and (dl)-AP-5 (50 μm) to prevent glutamatergic postsynaptic currents. The sIPSCs were screened by using the template search of Clampfit (version 9.2; Molecular Devices). The template was created by selecting at least 30 typical events during baseline recordings, and remained unchanged for data analysis after drug application within each cell. Cells with nonstable baselines (>20% changes in amplitude or frequency during 6 min baseline recordings) were not included in the analysis. After obtaining at least 6–10 min of stable baseline, the recording solution was switched to the solution containing nicotine (1 μm), and sIPSCs were continuously recorded for at least 15 min. The percentage change in mean IPSC amplitude and frequency after drug application were assessed via Student's t test or one-way ANOVA with p < 0.05 considered statistically significant.
Results
Decreased feedforward inhibition precedes nicotine-induced in vivo synaptic potentiation
To examine the influence of nicotine over feedforward inhibition in the dentate gyrus, we used previously established in vivo stimulus protocols to determine the FDI index (Sloviter, 1991; Rosenblum et al., 1999). The procedure exploits the knowledge that some perforant path afferent fibers directly excite GABAergic interneurons that feedforward to inhibit granule cells (Buzsáki, 1984). Altering the frequency of perforant path stimulation (Fig. 1A) changes the amplitude of the evoked population spike generated by granule cells (Fig. 1B, pop spike). As the stimulation frequency of the perforant path was varied between 0.1 and 1.0 Hz, the FDI index was calculated from the pop spike amplitudes at the two stimulus frequencies (Sloviter, 1991; Rosenblum et al., 1999). Ten pulses at 0.1 Hz were delivered to the perforant path followed by 10 pulses at 1.0 Hz, and in between FDI determinations, the basic strength of synaptic transmission was monitored from the amplitude of the pop spike as usual (Fig. 1A, test transmission).
Representative field recordings before and after saline injection (Fig. 1B, gray traces) and nicotine injection (Fig. 1B, black traces) are shown at the two stimulation frequencies. Saline injection did not change the FDI index (Fig. 2A, gray data): the FDI index equaled 0.60 ± 0.13 before and 0.68 ± 0.11 after saline (n = 6; p > 0.05). In contrast, nicotine injection (0.5 mg/kg, i.p.) decreased inhibition, which increased the FDI index (Fig. 2A, black data): the FDI index equaled 0.72 ± 0.10 before and 1.50 ± 0.31 after nicotine (n = 6; p < 0.05). An increase in the FDI index indicates a reduction of the feedforward inhibition occurring ∼35 min after nicotine injection (Fig. 2A).
Figure 2.
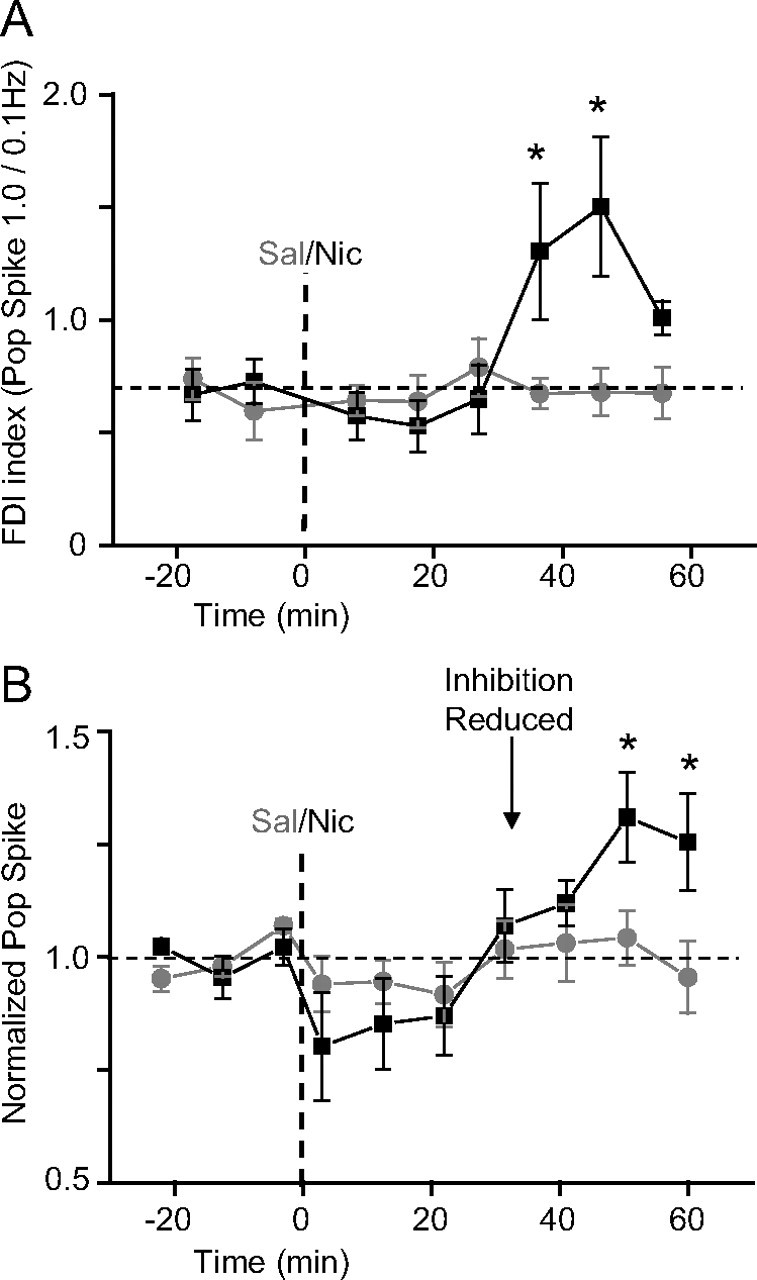
Systemic nicotine reduces feedforward inhibition before in vivo induction of nicotine-induced synaptic potentiation. A, Summary (±SEM) of FDI index of pop spike amplitudes. After nicotine administration (0.5 mg/kg, i.p.; black squares; n = 6), the feedforward inhibition was reduced (i.e., the FDI index increased) after the 35 min time point when compared with saline treatment (gray circles; n = 6). Two-way ANOVA with repeated measures showed the significant effect of the treatment (group by time: F(7,70) = 3.35, p < 0.01). Post hoc analysis showed a significant difference between nicotine- and saline-treated sessions as indicated (*p < 0.05 vs saline, Duncan's test). B, Summary (±SEM) of pop spike amplitude normalized to the baseline showing nicotine-induced synaptic potentiation (Tang and Dani, 2009). Synaptic transmission of the perforant path recorded in the dentate was potentiated 50 min after nicotine administration (0.5 mg/kg, i.p.; black squares; n = 6) when compared with saline treatment (gray circles; n = 6). ANOVA showed the significant effect of treatment (group by time: F(9,90) = 2.21, p < 0.05), and post hoc analysis showed significance as indicated (*p < 0.05 vs saline, Duncan's test).
After the reduced inhibition, the nicotine injection (0.5 mg/kg, i.p.), but not the saline injection, induced in vivo synaptic potentiation (Fig. 2B), as indicated by an increase in the amplitude of the granule cell population spike (Tang and Dani, 2009). Approximately 50 min after nicotine injection (Figs. 1B, bottom right; 2B), synaptic strength was significantly increased (130.94 ± 9.98% of baseline; n = 6; p < 0.05) compared with saline control (104.26 ± 6.01% of baseline; n = 6). The nicotine-induced reduction of feedforward inhibition (Fig. 2A) occurred before there was significant synaptic potentiation (Fig. 2B).
Decreased feedback inhibition also precedes nicotine-induced in vivo synaptic potentiation
To further understand the in vivo action of nicotine during the induction of in vivo synaptic potentiation, we examined feedback inhibition within the dentate gyrus. Feedback inhibition arises from recurrent granule cell collaterals that activate inhibitory interneurons that feed back onto the granule cells, including those granule cells that initially activated the interneurons (Andersen et al., 1966; Buzsáki, 1984; Wang, 2008). Stimulation of the perforant path produces a population spike from the granule cells that induces feedback inhibition. That inhibition decreases the amplitude of a second stimulated pop spike relative to the first. The feedback inhibition depends on the amplitude of the first stimulated spike (Andersen et al., 1966; Sloviter, 1991).
In the present study using freely moving mice, the conditioning pulse (Fig. 3A, cond.) was determined using the 15 ms interpulse interval. The stimulus strength of the conditioning pulse was adjusted to produce a second pop spike that was 20–30% of the test response (Fig. 3A, test). The test response was determined separately from a single stimulation that produced a pop spike that was 70% of the maximum. In that way, only a small number of granular cells were excited by the conditioning pulse. By delivering a second stimulus at various interstimulus intervals (Fig. 3A, 15–150 ms), the intensity of feedback inhibition was observed based on the amplitude of the second pop spike relative to the amplitude of the separately obtained test pop spike (Fig. 3A, test). The greatest feedback inhibition was observed in freely moving mice when the interstimulus interval was 15 or 30 ms (Fig. 3A,B). When compared with the test pulse alone, the amplitude of the pop spike was 18.5 ± 3.9% for a 15 ms interval and 36.0 ± 12.4% for a 30 ms interval (n = 4; p < 0.05). In anesthetized animals, paired-pulse-induced inhibition can last up to 100 ms (Andersen et al., 1966). Similarly, we found that in vivo paired stimulations separated by ∼100 ms or more produced little inhibition of the second pop spike (Fig. 3B).
After determining that interstimulus intervals of 15 and 30 ms maximized feedback inhibition, we used those stimulus protocols to examine how systemically administered nicotine influenced local dentate inhibition. Paired pulses at 15 ms (Fig. 4A, PP15) and 30 ms (Fig. 4A, PP30) interstimulus intervals were alternately delivered to the perforant path in between tests of synaptic transmission strength (Fig. 4A, TT). The paired-pulse index was calculated by dividing the amplitude of the second pop spike by the amplitude of the test pop spike alone. Whereas saline injection did not change the paired-pulse index, nicotine (0.5 mg/kg, i.p.) treatment increased the paired-pulse index, which indicated decreased feedback inhibition. The reduced inhibition was not significant at 15 ms, but at a 30 ms interspike interval, feedback inhibition was significantly reduced (group by time: F(5,50) = 2.73, p < 0.05) beginning <23 min after nicotine injection (Fig. 4B): paired-pulse index, 156.5 ± 18.7% of baseline (n = 6; p < 0.05).
Nicotine injection (0.5 mg/kg, i.p.) also induced significant potentiation of synaptic transmission during this stimulus paradigm (Fig. 4C) beginning 40 min after nicotine injection: 143.4 ± 10.5% of baseline (n = 6; p < 0.05). As seen with feedforward inhibition, significant nicotine-induced reduction of feedback inhibition (Fig. 4B) occurred before there was synaptic potentiation (Fig. 4C).
In vivo synaptic potentiation induced by nicotine administration can be detected as an increase in the AMPA/NMDA receptor ratio measured in vitro
It has been shown that LTP induction involves AMPA receptor trafficking or changes in AMPA receptor conductance or phosphorylation (Malinow and Malenka, 2002; Boehm et al., 2006). Synaptic potentiation of the kind observed after injection of nicotine (Tang and Dani, 2009) commonly requires trafficking of AMPA receptors into the postsynaptic membrane and/or changes in AMPA receptor conductance. In that case, potentiation would cause an increase in the ratio of AMPA to NMDA current at the potentiated synapses (Ungless et al., 2001; Saal et al., 2003).
To determine whether nicotine-induced in vivo synaptic potentiation of the perforant path could be examined with in vitro brain slices, we injected freely moving mice with either saline or nicotine (1 mg/kg) and waited for 1 h before preparing ex vivo brain slices from the hippocampus. That dose of nicotine produces reliable in vivo LTP that reaches its maximum by 1 h and is maintained for at least 5 h in vivo (Tang and Dani, 2009). In the brain slices, we electrically stimulated the medial perforant path and measured EPSCs from dentate granule cells. The glutamatergic EPSCs were measured at a membrane holding potential of +40 mV to relieve the magnesium blockade of NMDA receptors, and GABAA receptor currents were inhibited by picrotoxin (100 μm). First, glutamatergic EPSCs containing both the AMPA and NMDA components were measured. Then, the NMDA receptor antagonist dl-AP-5 (100 μm) was bath applied to isolate the AMPA component of the EPSC. The NMDA component of the EPSC was obtained by subtracting the AMPA component from the total (Ungless et al., 2001; Saal et al., 2003).
After nicotine was injected, the ratio of the AMPA receptor current component of the EPSC to the NMDA receptor component (i.e., the AMPA/NMDA ratio) was 54% larger than that measured after saline injection (Fig. 5): 1.28 ± 0.07 after nicotine; 0.83 ± 0.05 after saline (p < 0.01; n = 10–12 cells from 5–6 mice for each group). The result indicates that the nicotine dose that induces in vivo synaptic potentiation also induces synaptic change that can be detected using in vitro brain slices. The results suggest that LTP has occurred because of AMPA receptor insertion into the postsynaptic membranes after nicotine treatment (Ungless et al., 2001; Saal et al., 2003), and we can study the nicotine-induced synaptic changes using ex vivo brain slices.
Figure 5.

A single nicotine injection given to a freely moving mouse significantly increases the ratio of AMPA/NMDA components of the EPSCs recorded from dentate granule cells. Brain slices were cut from mice after intraperitoneal injection of saline (A) or nicotine (B). EPSC recordings (average of 5 traces) are shown from granule cells after perforant path stimulation at a holding potential of +40 mV. Stimulus artifacts have been omitted for clarity. The ratio of AMPA/NMDA EPSCs was significantly increased from 0.8 ± 0.05 after saline injection to 1.3 ± 0.07 after nicotine injection (p < 0.01; n = 10–12 cells from 5–6 mice for each group), suggesting the induction of synaptic potentiation.
We examined that conclusion further by showing that the increased amplitude of the EPSCs resides postsynaptically after nicotine-induced potentiation. In brain slices, the medial perforant path is known to exhibit paired-pulse depression in contrast to the lateral perforant path, which exhibits paired-pulse facilitation (Colino and Malenka, 1993). Consistent with in vivo recordings (Tang and Dani, 2009), the EPSCs showed paired-pulse depression (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), as expected for the medial perforant path. Furthermore, nicotine administration increased the amplitude of the EPSCs, but it did not change paired-pulse depression (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material): paired-pulse ratio equals 0.83 ± 0.01, n = 3 after saline injection, and 0.86 ± 0.03, n = 5 after nicotine injection (p > 0.05). This in vitro result suggests that the presynaptic probability of transmitter release is not changed by nicotine-induced potentiation.
Using freely moving mice, we also measured the in vivo presynaptic fiber volley before and after nicotine-induced potentiation and found the fiber volley was unchanged (supplemental Fig. S1C,D, available at www.jneurosci.org as supplemental material): 99.9 ± 3.4% (n = 4; p > 0.05). This result suggests that the presynaptic afferent excitation is not changing. Both the in vitro and in vivo results support that the nicotine-induced potentiation resides postsynaptically, as would be expected from insertion of AMPA receptors into the postsynaptic membrane.
Nicotine influences local inhibition within the dentate gyrus leading to disinhibition of granule cells
The in vivo experiments indicated that nicotine decreased inhibition onto granule cells. To examine the inhibitory effects further, we cut brain slices from the dentate gyrus and measured sIPSCs from granule cells (Fig. 6). The amplitude (Fig. 6C) and the frequency (Fig. 6D) of sIPSCs were decreased 15 min after bath application of nicotine (1 μm). The amplitude decreased to 90 ± 3% of the baseline (n = 8; p < 0.05), and the frequency decreased to 85 ± 7% of the baseline (n = 8; p < 0.05).
Figure 6.

Nicotine reduced synaptic inhibition onto dentate granule cells in a hippocampal brain slice. Representative recordings of sIPSCs made from granule cells in the dentate gyrus before (A) and 15 min after (B) bath application of nicotine (1 μm). sIPSCs were pharmacologically isolated by applying DNQX (10 μm) and dl-APV (50 μm) to block glutamate receptors. Cumulative probability plots are shown for the normalized IPSC amplitude (C) and the normalized interevent interval (ISI) (D) to represent the frequency of IPSCs. Nicotine decreased both the frequency (0.8 ± 0.07 of baseline; p < 0.05) and amplitude (0.9 ± 0.03 of baseline; p < 0.05) of sIPSCs onto granule cells (n = 8).
The activity of nAChRs has been shown previously to decrease GABAergic current amplitudes in the CA1 region of the hippocampus (Wanaverbecq et al., 2007). To verify that similar mechanisms may occur in the dentate gyrus, we used a puffer pipette to pressure apply exogenous GABA onto granule cells, inducing GABAergic currents (Fig. 7A, gray trace). When ACh (1 mm) was applied by pressure immediately before the GABA application, small nAChR currents were observed (ACh) (Fig. 7A, black trace), and the subsequent GABA-induced currents were decreased. GABA-induced currents also were decreased by bath application of 1.0 μm nicotine (Fig. 7B): GABAergic amplitude was 70 ± 6% of baseline (n = 4; p < 0.05). Thus, the same nicotine concentration that decreased sIPSCs (Fig. 6) also decreased the amplitude of GABAergic currents activated by exogenous application of GABA.
Figure 7.

Activation of nAChRs on granule cells by ACh pressure application decreased GABAA currents. The internal patch pipette solution contained only 0.2 mm EGTA. The bath contained atropine (0.5 μm) and DNQX (20 μm) to inhibit muscarinic and glutamatergic receptors, respectively. A, Puffer-applied ACh (1 mm; 1 s; black bar) activated a small nAChR current and decreased GABA-induced current (puffer applied at 0.2 mm; 0.3 s; upward arrow; black trace) when compared with the control GABA-induced current taken from the same cell just before the ACh treatment. B, Time course of the amplitude of GABA-induced currents in the absence (open circles; n = 5) or presence of bath-applied nicotine (0.5 μm; filled circles; n = 7). Error bars indicate SEM.
To understand how nicotine application decreased the frequency of GABAergic sIPSCs onto granule cells (Fig. 6D), we examined the hilar interneurons that project to the granule cell distal dendrites near the perforant path afferents, called HIPP cells (Han et al., 1993; Soriano and Frotscher, 1993; Sik et al., 1997). These interneurons are positioned to play a major role in feedback inhibition onto granule cells, particularly at the site where the perforant path synapses onto the granule cells in the distal dendrites (Katona et al., 1999).
Because the HIPP cells are physiologically heterogeneous (Scharfman, 1995), we restricted our recordings to those cells with pyramidal soma located at the granule cell layer/hilus border and with major dendritic processes projecting perpendicular to the granule cell layer (Fig. 8A). HIPP cells were backfilled with biocytin during recordings for later structural verification, and their firing properties were examined. HIPP cells were distinguished from inhibitory basket cells in this region because HIPP cells displayed moderate frequency adaptation on injection of depolarizing current (56.9 ± 5.2 Hz for HIPP cells vs 82.6 ± 7.3 Hz for basket cells; p < 0.02) and a voltage sag on injection of hyperpolarizing current (sag ratio, 0.85 ± 0.02, vs 0.96 ± 0.02; p < 0.02; n = 19 for HIPP cells and n = 16 for basket cells) (Fig. 8B). Basket cells have a higher firing rate, lack frequency adaptation, and show no voltage sag (Lübke et al., 1998).
Figure 8.
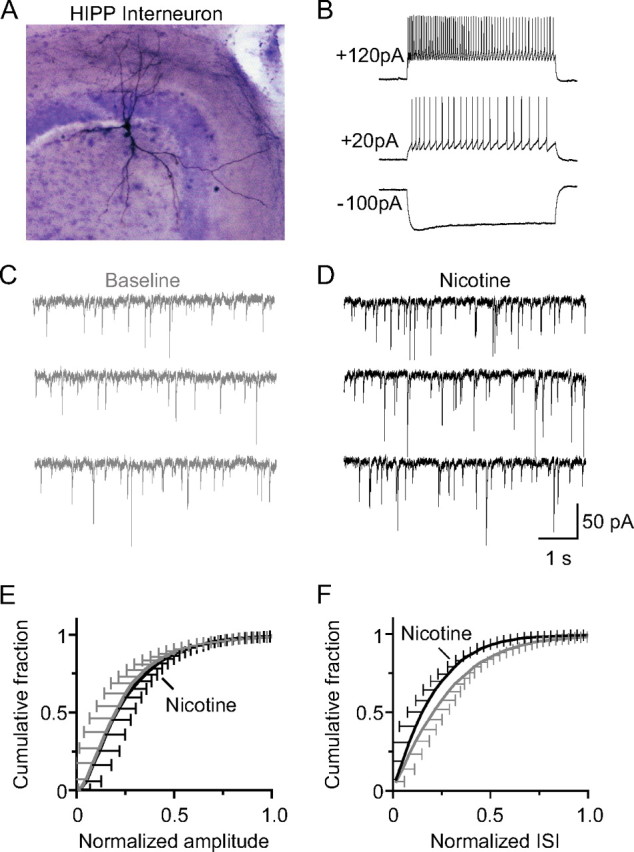
Nicotine increased GABAergic sIPSC inhibition onto HIPP-type interneurons in the dentate gyrus. A, Low magnification (10× objective) of biocytin-filled HIPP interneuron in a horizontal hippocampal slice. The cell has a large soma and is located on the border of the hilus and granule cell layer. The dendrites of this cell extend into both the molecular layer and hilus, and the axons ramify extensively in the outer molecular layer. Biocytin (0.3%) was included in the intracellular recording solution. After recording, slices were fixed with formaldehyde and stained in avidin–horseradish peroxidase solution (Vectastain ABC Elite kit). B, Membrane responses to current injection from the same cell as shown in A at its resting potential (−60 mV) under current clamp. Current injection (20–120 pA) induced high-frequency firing of action potentials that showed modest frequency adaptation, indicating HIPP cells. Representative recordings of sIPSCs onto HIPP cells in the dentate gyrus before (C) and after (D) nicotine (1 μm) application to the bath. Cumulative probability plot for the normalized amplitude (E) and interevent interval (F) of sIPSCs onto HIPP neurons.
Bath application of nicotine (1 μm) increased the frequency of sIPSCs onto HIPP cells (Fig. 8C,D,F). The frequency increased to 133 ± 10% of baseline (n = 9; p < 0.01), but the amplitude did not significantly change, 93 ± 8% of baseline (n = 9; p > 0.05). These results suggest that nAChR activation increases inhibition onto HIPP cells, thereby decreasing inhibition onto granule cells (i.e., disinhibition of granule cells).
Nicotine decreases feedback inhibition onto granule cells directly measured using in vitro brain slices
Granule cells are known to receive potent feedback inhibition from hilar interneurons (Acsády et al., 1998), and nicotine decreased inhibition from HIPP cells (Figs. 6, 8). Therefore, we devised an in vitro experiment to test directly whether nicotine administration could reduce feedback inhibition. A bipolar electrode was placed near the CA3 stratum lucidum to stimulate granule cells antidromically via their mossy fibers (Fig. 9A). The antidromic spike reached the granule cells and also followed collaterals to reach feedback interneurons. The antidromic spike observed in granule cells was followed by a synaptic response (Fig. 9B, baseline) that was blocked by the GABAA receptor antagonist, picrotoxin (Fig. 9B, PTX), or by the AMPA receptor antagonist, DNQX (Fig. 9B). The synaptic response onto the granule cells had the same reversal potential as GABAA currents, consistent with the inhibitory feedback circuitry depicted in Figure 9A (Patel et al., 2004). Bath application of nicotine (1 μm) decreased these feedback inhibitory currents to 60 ± 5% of baseline (n = 12; p < 0.01) (Fig. 9B, Nic). The nicotine effects were blocked by application of the β2*-nAChR antagonist dihydro-β-erythroidine (DHβE) (91 ± 8%; n = 7; p > 0.05) or by the nonselective nAChR antagonist mecamylamine (MEC) (98 ± 7%; n = 5; p > 0.05), but not by the selective α7*-nAChR antagonist methyllycaconitine (MLA) (67 ± 7%; n = 7; p < 0.01) (Fig. 9C). When MLA or DHβE was applied alone without nicotine, the feedback currents were not affected (94 ± 6 and 97 ± 6%; p > 0.05 in both cases). These data suggest that β2*-nAChRs are particularly important for reduced feedback inhibition after nicotine application.
Figure 9.
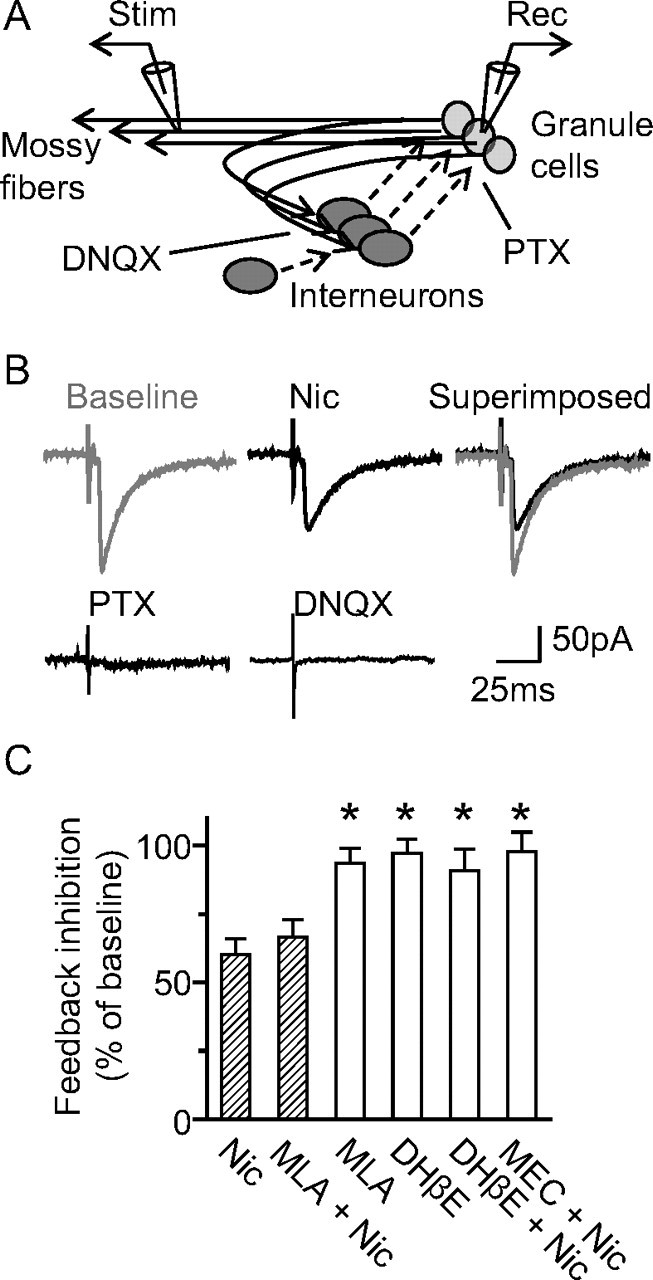
Nicotine decreases feedback inhibition onto granule cells. A, Didactic representation of the recording paradigm for inducing feedback inhibition onto dentate granule cells. The stimulating electrode was placed in the CA3 mossy fiber pathway. Whole-cell recordings were made from dentate granule cells with high chloride internal solution. Granule cells form synapses onto hilar interneurons that feedback and inhibit granule cells. Hilar interneurons also synapse onto other interneurons. The sites where DNQX and picrotoxin (PTX) inhibit GluRs and GABAARs, respectively, are shown. B, Representative recordings depicting feedback inhibitory synaptic transmission onto granule cells, which can be blocked by either PTX or DNQX. Bath application of nicotine (Nic) (1 μm) reduced feedback inhibition onto granule cells. C, Cumulative bar graph showing that nicotine-induced reduction of feedback inhibition onto granule cells was blocked by β2* nAChR-selective antagonist, DHβE, but not by the α7* nAChR-selective antagonist, MLA. Error bars indicate SEM. *p < 0.05.
Discussion
Nicotine-induced synaptic potentiation of the perforant path to the dentate granule cells involves diminished inhibition and other synaptic processes
The addictive drug, nicotine, induces in vivo synaptic potentiation that underlies learning and memory, and this associative memory can be quantified as nicotine-induced preference for a place (Tang and Dani, 2009). In this study, we examined how nicotine acts on the local circuits of the hippocampal dentate gyrus to induce in vivo synaptic potentiation. Various stimulus protocols applied in vivo to the perforant path probed the local inhibitory circuits that exert powerful control over granule cells (Freund and Buzsáki, 1996). These in vivo experiments suggested that nicotine decreased both feedforward and feedback inhibition onto the granule cells, thereby contributing to the nicotine-induced synaptic potentiation.
Using in vitro brain slices, we were able to position a stimulating electrode within the mossy fiber efferents from the granule cells. Stimulation evoked retrograde action potentials along granule cell axons and collaterals. The collaterals excited local GABAergic interneurons that provided feedback inhibition to the granule cells (Andersen et al., 1966; Buzsáki, 1984; Wang, 2008). The experiments showed that nicotine acted to disinhibit the granule cells by actively increasing inhibition onto GABAergic HIPP interneurons. The HIPP interneurons are ideally suited for this action because they project near to the perforant path afferents in the granule cell distal dendrites (Han et al., 1993; Soriano and Frotscher, 1993; Sik et al., 1997). Decreased GABAergic inhibition at that site permits greater coupling between the perforant path input and the postsynaptic response of the granule cell. This enhanced coincidence underlies the synaptic potentiation (Brown and Milner, 2003) examined in this study.
In contrast, we could not examine feedforward inhibition in the slice with a simple stimulus protocol like that used to study feedback inhibition. When we attempted to stimulate in a manner aimed at isolating feedforward inhibition, the complex interactions within the circuit prevented us from interpreting the results only with respect to feedforward inhibition. Part of this problem arose because the perforant path into the dentate gyrus was not perfectly and completely preserved within the slice. Therefore, we could not move extremely far away from the granule cells to stimulate exclusively the perforant path and no other circuitry. Furthermore, the perforant path innervates broadly, producing a response that is a combination of excitatory and inhibitory interactions. Since we could not block either GABAergic or glutamatergic transmission for these experiments, there were indirect currents confounding our interpretations. Consequently, we could not examine the details of feedforward inhibition in the same manner that we examined feedback inhibition in the slice.
As shown in hippocampal brain slices, other nicotine-induced cellular mechanisms have the ability to influence synaptic plasticity (McGehee, 2002; Alkondon and Albuquerque, 2004; Yakel and Shao, 2004; Dani and Bertrand, 2007; Albuquerque et al., 2009). Nicotinic receptors have been found in all the major cell types of the hippocampus, but nAChRs are most highly expressed in GABAergic interneurons (Jones and Yakel, 1997; Alkondon and Albuquerque, 2001; Yakel and Shao, 2004). The activation of nAChRs on GABAergic interneurons produces potent effects, causing inhibition or disinhibition of pyramidal neurons (Ji and Dani, 2000; Alkondon and Albuquerque, 2001). In addition, in vitro results from the ventral midbrain suggest that low levels of nicotine can reduce tonic inhibition by desensitizing particularly β2* nAChRs on interneurons (Mansvelder and McGehee, 2002; Mansvelder et al., 2002; Pidoplichko et al., 2004). β2* nAChRs have a high affinity for nicotine resulting in greater desensitization (Fenster et al., 1999; Wooltorton et al., 2003), and the pharmacology observed in our experiments (Fig. 9) is consistent with this possibility. However, the main process we observed in these experiments is that active β2* nAChRs increased inhibition onto other GABAergic inhibitory neurons, consequently producing disinhibition of granule cells.
Nicotinic receptors also occupy cellular locations where they may alter the coincidence between presynaptic and postsynaptic activity that underlies synaptic plasticity (Mansvelder and McGehee, 2000, 2002; Ji et al., 2001; Mansvelder et al., 2002; Pidoplichko et al., 2004; Ge and Dani, 2005; Albuquerque et al., 2009). The induction of long-term potentiation commonly requires pairing of a presynaptic stimulation with a postsynaptic depolarization. Nicotinic receptors are capable of enhancing this coincidence in multiple ways. Presynaptic nAChRs enhance the release of many neurotransmitters, and this mechanism has been most extensively documented for nAChR-enhanced release of glutamate (McGehee et al., 1995; Alkondon et al., 1996; Pidoplichko et al., 1997). Postsynaptic and nonsynaptic nAChRs also can contribute to the depolarization and calcium signal caused by excitatory neurotransmitter release (Rathouz and Berg, 1994; Ji et al., 2001; Shoop et al., 2001; Khiroug et al., 2003; Sharma and Vijayaraghavan, 2003; Fayuk and Yakel, 2005; Ge and Dani, 2005). Nicotinic receptors presynaptically modulate glutamate release and alter the amplitude and timing of the postsynaptic depolarization and subsequent intracellular calcium signal that underlie the induction of synaptic plasticity (Jones et al., 1999; Ji et al., 2001; Mansvelder and McGehee, 2002; Ge and Dani, 2005; Dani and Bertrand, 2007; Albuquerque et al., 2009).
Although nicotinic plasticity mechanisms have been carefully documented in slice preparations, very little has been done in vivo. The in vivo evidence presented here indicates that systemic nicotine administration potently influences inhibitory circuitry in the dentate gyrus. The nicotine-induced reduction of inhibition occurs before there is significant synaptic potentiation, suggesting a causal relationship between the decreased inhibition and potentiation. When GABAergic inhibition within hippocampal brain slices was kept intact, we were not able to induce synaptic potentiation of the perforant path to the dentate gyrus by simply bath applying biologically reasonable concentrations of nicotine (0.1–10 μm). Long-term depression could be induced, but we did not observe consistent long-term potentiation as was observed during in vivo nicotine administration. However, when we decreased inhibition by applying bicuculline and/or picrotoxin to the slice, then bath applied nicotine was able to induce synaptic potentiation. Although a proof of principle has been demonstrated for many nicotinic synaptic mechanisms in slices, the experimental connection between in vivo circuit measurements and in vivo synaptic mechanisms of this study more clearly demonstrates the causal effects of addictive nicotine.
Another issue that bears discussion is the timing of synaptic potentiation after nicotine administration. Although nicotinic receptors are expected to respond within minutes to in vivo administration of nicotine (intraperitoneal injection), synaptic potentiation could take much longer to achieve statistical significance (Fig. 2B). This delay depends on the nicotine dose and the stimulus protocols that accompany nicotine administration. For example, when 0.5 mg/kg nicotine was administered alone, without any accompanying stimulus protocol, significant LTP was achieved rapidly, as shown in the study by Tang and Dani (2009), their supplemental Figure 1. However, when 0.5 mg/kg of nicotine was administered during stimulus protocols at 0.1 and 1.0 Hz, then statistically significant LTP was delayed ∼50 min (Fig. 2B). Those low-frequency stimulus protocols on their own would produce synaptic depression (Bear and Malenka, 1994). Therefore, those stimulus protocols are pushing the synaptic connections toward long-term depression, whereas the nicotine administration is pushing toward LTP. Consequently, statistically significant LTP is delayed. At the higher nicotine dose of 1 mg/kg, synaptic potential is delayed even when nicotine is administered alone. At that concentration, nicotine decreases the core body temperature of mice (Tapper et al., 2004), and that decrease in temperature slows the induction of synaptic potentiation (Moser et al., 1993). It should therefore be expected that nicotinic receptors can act rapidly on inhibitory circuits to influence synaptic potentiation, but their action has to be considered within the overall conditions impinging on the circuitry.
Synaptic plasticity, learning, and memory within the addiction process
Based on continually growing evidence, there is general acceptance that addictive drugs alter and/or produce synaptic plasticity that underlies learning and memory (Mansvelder and McGehee, 2000, 2002; Kauer, 2004; Kelley, 2004; Dani and Harris, 2005; Hyman et al., 2006; Dani and Bertrand, 2007; Kauer and Malenka, 2007). When given systemically, addictive drugs, including nicotine, produce long-term synaptic potentiation in dopamine neurons of the ventral midbrain (Mansvelder and McGehee, 2000; Ungless et al., 2001; Saal et al., 2003; Chen et al., 2008). Although many areas of the brain participate, dopamine signaling within the mesocorticolimbic system serves a vital role in the acquisition of behaviors that progress to addiction (Dani and Heinemann, 1996; Corrigall, 1999; Di Chiara, 2000; Mansvelder and McGehee, 2002; Balfour, 2004; Dani and Harris, 2005). In addition, a dopamine signal arriving in the dentate gyrus from the ventral tegmental area (VTA) was shown to be necessary for the induction of in vivo nicotine-induced LTP in the dentate (Tang and Dani, 2009). As nicotine acts locally on the circuitry of the dentate gyrus, it also induces a long-range dopamine signal from the VTA (Grenhoff et al., 1986; Mameli-Engvall et al., 2006; Zhang et al., 2009). Both the local action of nicotine within the dentate and the arrival of the dopamine signal are required to produce the in vivo LTP (Tang and Dani, 2009), consistent with in vitro work indicating that dopamine modulates LTP (Jay, 2003).
In this study, we focused on the hippocampus because it has an important role in the development of learned or conditioned associations between the environment and the drug experience (Volkow et al., 2002; Biala et al., 2005). In addition, we monitored and controlled the perforant path, which relays neocortical information from the entorhinal cortex to the hippocampus (Deadwyler et al., 1979; Lavenex and Amaral, 2000). The study focused particularly on the medial perforant path because it specifically carries place and spatial information (Hargreaves et al., 2005) that is important for drug-associated memories. An emerging view is that memories associated with addictive behaviors become internal motivational drives contributing to relapse (Everitt et al., 2001; Kilts et al., 2001; Bonson et al., 2002; Balfour, 2004; Kenny and Markou, 2005; Dani and Montague, 2007). That is, drug-conditioned associations become environmental cues that elicit internal bodily states capable of motivating drug use. Under this hypothesis, addictive drugs act on synaptic plasticity mechanisms within brain systems that normally function to help us learn how to exploit our environments successfully. Normally, these systems associate environmental situations with behavioral sequences that produce success (Schultz et al., 1997), but cues associated with the drug experience motivate addictive behaviors.
In addition to requiring the long-range dopamine signal arriving from the midbrain (Tang and Dani, 2009), in vivo nicotine-induced LTP in the dentate gyrus involves (at least) actions on local inhibitory circuitry. Among the actions by nicotine, it alters the tone and strength of GABAergic inhibition, likely leading to disinhibition of some granule cells. That action by nicotine is an active process; it is not caused just via desensitization of nAChRs because inhibition of nAChRs with antagonists prevented nicotine-induced LTP (Tang and Dani, 2009). Because this nicotine-induced time window of enhanced synaptic plasticity occurs while the medial perforant path activity is linked to drug-associated behaviors, conditioning to the drug use is reinforced by this memory process. For example, nicotine conditions preference to the place where the drug was administered, and normal mechanisms of synaptic plasticity participate to reinforce the learning of drug-conditioned behaviors (Grabus et al., 2006; Tang and Dani, 2009).
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant NS21229 and National Institutes of Health–National Institute on Drug Abuse Grant DA09411. This study was also supported by the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine. We thank Dr. Carsten T. Wotjak for his suggestion to calculate the frequency-dependent index.
References
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol. 2001;86:3043–3055. doi: 10.1152/jn.2001.86.6.3043. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi: 10.1016/S0079-6123(03)45007-3. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic receptor subtypes in rat hippocampal slices are differentially sensitive to desensitization and early in vivo functional up-regulation by nicotine and to block by bupropion. J Pharmacol Exp Ther. 2005;313:740–750. doi: 10.1124/jpet.104.081232. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Rocha ES, Maelicke A, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat brain. V. alpha-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther. 1996;278:1460–1471. [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand. 1966;66:448–460. doi: 10.1111/j.1748-1716.1966.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Balfour DJ. The neurobiology of tobacco dependence: a preclinical perspective on the role of the dopamine projections to the nucleus. Nicotine Tob Res. 2004;6:899–912. doi: 10.1080/14622200412331324965. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic d-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci. 2005;21:3089–3096. doi: 10.1111/j.1460-9568.2005.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Bonson KR, Grant SJ, Contoreggi CS, Links JM, Metcalfe J, Weyl HL, Kurian V, Ernst M, London ED. Neural systems and cue-induced cocaine craving. Neuropsychopharmacology. 2002;26:376–386. doi: 10.1016/S0893-133X(01)00371-2. [DOI] [PubMed] [Google Scholar]
- Brown RE, Milner PM. The legacy of Donald O. Hebb: more than the Hebb synapse. Nat Rev. 2003;4:1013–1019. doi: 10.1038/nrn1257. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Feed-forward inhibition in the hippocampal formation. Prog Neurobiol. 1984;22:131–153. doi: 10.1016/0301-0082(84)90023-6. [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colino A, Malenka RC. Mechanisms underlying induction of long-term potentiation in rat medial and lateral perforant paths in vitro. J Neurophysiol. 1993;69:1150–1159. doi: 10.1152/jn.1993.69.4.1150. [DOI] [PubMed] [Google Scholar]
- Corrigall WA. Nicotine self-administration in animals as a dependence model. Nicotine Tob Res. 1999;1:11–20. doi: 10.1080/14622299050011121. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Dani JA, Montague PR. Disrupting addiction through the loss of drug-associated internal states. Nat Neurosci. 2007;10:403–404. doi: 10.1038/nn0407-403. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, West M, Lynch G. Activity of dentate granule cells during learning: differentiation of perforant path input. Brain Res. 1979;169:29–43. doi: 10.1016/0006-8993(79)90371-8. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol. 2000;393:295–314. doi: 10.1016/s0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW. The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev. 2001;36:129–138. doi: 10.1016/s0165-0173(01)00088-1. [DOI] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL. Ca2+ permeability of nicotinic acetylcholine receptors in rat hippocampal CA1 interneurones. J Physiol. 2005;566:759–768. doi: 10.1113/jphysiol.2005.089789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface α4β2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci. 1999;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. London: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Ge S, Dani JA. Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci. 2005;25:6084–6091. doi: 10.1523/JNEUROSCI.0542-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabus SD, Martin BR, Brown SE, Damaj MI. Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology (Berl) 2006;184:456–463. doi: 10.1007/s00213-006-0305-7. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand. 1986;128:351–358. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- Han ZS, Buhl EH, Lörinczi Z, Somogyi P. A high degree of spatial selectivity in the axonal and dendritic domains of physiologically identified local-circuit neurons in the dentate gyrus of the rat hippocampus. Eur J Neurosci. 1993;5:395–410. doi: 10.1111/j.1460-9568.1993.tb00507.x. [DOI] [PubMed] [Google Scholar]
- Hargreaves EL, Rao G, Lee I, Knierim JJ. Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science. 2005;308:1792–1794. doi: 10.1126/science.1110449. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–2690. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–25. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Conditioned nicotine withdrawal profoundly decreases the activity of brain reward systems. J Neurosci. 2005;25:6208–6212. doi: 10.1523/JNEUROSCI.4785-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology. 2006;31:1203–1211. doi: 10.1038/sj.npp.1300905. [DOI] [PubMed] [Google Scholar]
- Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL. Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci. 2003;23:9024–9031. doi: 10.1523/JNEUROSCI.23-27-09024.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F, Ely TD, Hoffman JM, Drexler KP. Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry. 2001;58:334–341. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- Lavenex P, Amaral DG. Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus. 2000;10:420–430. doi: 10.1002/1098-1063(2000)10:4<420::AID-HIPO8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lübke J, Frotscher M, Spruston N. Specialized electrophysiological properties of anatomically identified neurons in the hilar region of the rat fascia dentata. J Neurophysiol. 1998;79:1518–1534. doi: 10.1152/jn.1998.79.3.1518. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, Faure P. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–921. doi: 10.1016/j.neuron.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- McGehee DS. Nicotinic receptors and hippocampal synaptic plasticity. It's all in the timing. Trends Neurosci. 2002;25:171–172. doi: 10.1016/s0166-2236(00)02127-5. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Moser E, Mathiesen I, Andersen P. Association between brain temperature and dentate field potentials in exploring and swimming rats. Science. 1993;259:1324–1326. doi: 10.1126/science.8446900. [DOI] [PubMed] [Google Scholar]
- Patel LS, Wenzel HJ, Schwartzkroin PA. Physiological and morphological characterization of dentate granule cells in the p35 knock-out mouse hippocampus: evidence for an epileptic circuit. J Neurosci. 2004;24:9005–9014. doi: 10.1523/JNEUROSCI.2943-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Froestl W, Markou A. Repeated administration of the GABAB receptor agonist CGP44532 decreased nicotine self-administration, and acute administration decreased cue-induced reinstatement of nicotine-seeking in rats. Neuropsychopharmacology. 2005;30:119–128. doi: 10.1038/sj.npp.1300524. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem. 2004;11:60–69. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathouz MM, Berg DK. Synaptic-type acetylcholine receptors raise intracellular calcium levels in neurons by two mechanisms. J Neurosci. 1994;14:6935–6945. doi: 10.1523/JNEUROSCI.14-11-06935.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):S33–S46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Maroun M, Richter-Levin G. Frequency-dependent inhibition in the dentate gyrus is attenuated by the NMDA receptor blocker MK-801 at doses that do not yet affect long-term potentiation. Hippocampus. 1999;9:491–494. doi: 10.1002/(SICI)1098-1063(1999)9:5<491::AID-HIPO1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Electrophysiological diversity of pyramidal-shaped neurons at the granule cell layer/hilus border of the rat dentate gyrus recorded in vitro. Hippocampus. 1995;5:287–305. doi: 10.1002/hipo.450050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–781. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sik A, Penttonen M, Buzsáki G. Interneurons in the hippocampal dentate gyrus: an in vivo intracellular study. Eur J Neurosci. 1997;9:573–588. doi: 10.1111/j.1460-9568.1997.tb01634.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Feedforward and feedback inhibition of hippocampal principal cell activity evoked by perforant path stimulation: GABA-mediated mechanisms that regulate excitability in vivo. Hippocampus. 1991;1:31–40. doi: 10.1002/hipo.450010105. [DOI] [PubMed] [Google Scholar]
- Soriano E, Frotscher M. GABAergic innervation of the rat fascia dentata: a novel type of interneuron in the granule cell layer with extensive axonal arborization in the molecular layer. J Comp Neurol. 1993;334:385–396. doi: 10.1002/cne.903340305. [DOI] [PubMed] [Google Scholar]
- Tang J, Dani JA. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron. 2009;63:673–682. doi: 10.1016/j.neuron.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol Learn Mem. 2002;78:610–624. doi: 10.1006/nlme.2002.4099. [DOI] [PubMed] [Google Scholar]
- Wanaverbecq N, Semyanov A, Pavlov I, Walker MC, Kullmann DM. Cholinergic axons modulate GABAergic signaling among hippocampal interneurons via postsynaptic α7 nicotinic receptors. J Neurosci. 2007;27:5683–5693. doi: 10.1523/JNEUROSCI.1732-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ. Decision making in recurrent neuronal circuits. Neuron. 2008;60:215–234. doi: 10.1016/j.neuron.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel JL, Shao Z. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat hippocampal interneurons. Prog Brain Res. 2004;145:95–107. doi: 10.1016/S0079-6123(03)45006-1. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci. 2009;29:4035–4043. doi: 10.1523/JNEUROSCI.0261-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]