

Abstract
Objective
To report a rare case of diabetes caused by type B insulin resistance due to development of insulin receptor autoantibodies during treatment of hepatitis C with interferon alpha and ribavirin.
Methods
Clinical and laboratory findings in the case are presented. Literature on type B insulin resistance and interferon-induced autoimmunity is reviewed.
Results
A 55 year old African American man with Hepatitis C was treated with interferon and ribavirin. Eight months later he presented with rapid onset of hyperglycemia, profound weakness, and weight loss. Severe hyperglycemia persisted in spite of insulin infusion rates as high as 120 units per hour. Presence of insulin receptor autoantibodies was confirmed by immunoprecipitation of recombinant human insulin receptor with patient serum. Assays for autoantibodies to islet cell antigens and glutamic acid decarboxylase were negative. The interferon and ribavirin were discontinued. His insulin requirement spontaneously fell to low levels and his blood glucose measurements normalized over a six month period. Two years later, insulin receptor autoantibodies could no longer be demonstrated in his serum. He remains euglycemic and is no longer taking insulin.
Conclusion
This case demonstrates that type B insulin resistance can occur as a complication of interferon alpha therapy. To our knowledge, this is the first case in the United States of type B insulin resistance with insulin receptor autoantibodies during treatment with interferon alpha.
Keywords: Type B Insulin Resistance, Interferon alpha, Autoimmunity, Insulin Receptor
Introduction
Type B insulin resistance is a rare syndrome caused by insulin receptor autoantibodies. These antibodies were initially described in patients with diabetes and extreme insulin resistance (1). However, it is now apparent that anti-insulin receptor antibodies can cause abnormalities of glucose homeostasis ranging from profound insulin resistance to life-threatening hypoglycemia (2). Most patients with insulin receptor autoantibodies have an underlying connective tissue disorder, most commonly systemic lupus erythematosus. Autoimmune hypoglycemia with insulin receptor autoantibodies has been described as a paraneoplastic syndrome in Hodgkin’s lymphoma (3, 4). There is also a case report of autoimmune hypoglycemia arising after heterologous bone marrow transplantation (5). In a series of 24 patients with type B insulin resistance or autoimmune hypoglycemia evaluated at the National Institutes of Health, 83% were women and 88% were African Americans (2). Most patients with type B insulin resistance develop acanthosis nigricans (6), and women of reproductive age usually have ovarian hyperandrogenism (2).
Case Report
A 55 year old African American male was diagnosed with hepatitis C, genotype 1b. A liver biopsy revealed chronic hepatitis with minimal activity and mild fibrosis. He started treatment with pegylated interferon α-1a and ribavirin. His hepatitis C viral RNA titer at the start of treatment was 3950 KIU/mL. He had no personal history of diabetes mellitus. A fasting plasma glucose before interferon treatment was 112 mg/dL. He developed anemia two months later which was managed with erythropoietin α and a reduction of his ribavirin. Six months after starting therapy, his weight had fallen 16 kg and viral RNA was not detectable. Two months later, he presented with polyuria, polydipsia, weakness, blurred vision, and fatigue. His weight had fallen 11 kg over the preceding month, and his weakness was so profound that he was unable to tie his shoes. He was admitted to the hospital. His serum glucose was 405 mg/dl, CO2 24 mmol/L, creatinine 1.5 mg/dl, and anion gap 10 mmol/L. Urine ketones were 1+ and hemoglobin A1c was 9.3%. The creatinine fell to 0.8 mg/dl with aggressive hydration. Bilirubin, AST, ALT, alkaline phosphatase, amylase, and lipase were normal. Interferon and ribavirin were discontinued. Subcutaneous insulin was started and increased over three days to a daily dose of 180 units, but blood glucoses still ranged from 300 to over 600 mg/dL. An insulin infusion was started and titrated over two days to 52 units per hour, but glucoses were still 230–300 mg/dL.
He was transferred to our institution. Physical exam revealed a thin, African American male in no acute distress. Weight was 68 kg, height 170 cm, and blood pressure 114/72 mm Hg. His sclerae were anicteric, and his abdomen was soft and nondistended. There was mild right upper quadrant tenderness and a palpable liver edge 4 cm below the costal margin. He did not have acanthosis nigricans, spider angiomas, palmar erythema, or splenomegaly. The insulin infusion was increased to 125 units/hour, but blood glucoses still ranged from 170 to 430 mg/dL, with lower values after fasting overnight and higher readings through the day. Type B insulin resistance was suspected. After a week on intravenous insulin, he was transitioned to U500 regular insulin, 300 units QID. Blood glucoses ranged from 110 to 300 mg/dL. During the third hospital week, he developed bilateral facial weakness, greater on the right, right-sided facial numbness, and weakness of the right lateral rectus. Brain MRI was normal. Examination of his cerebrospinal fluid revealed no white blood cells, nonreactive VDRL, negative viral and bacterial cultures, angiotensin converting enzyme activity 3 units (reference range < 10), glucose 114 mg/dL (reference interval 40–70), and protein 70 mg/dL (reference interval 15–45). His polycranial neuropathy was treated with prednisone, 40 mg daily for 5 days, and acyclovir. Partial improvement of the neuropathy was noted one week later. He was switched back to an insulin infusion with frequent glucose monitoring during the prednisone treatment because of concern that his insulin requirements might decline dramatically, but his glucoses and insulin requirement only increased. After 4 weeks, with glucoses ranging from 70 to 260 mg/dL, he was discharged home on U500 insulin, 400 units QID.
The patient’s serum was assayed for anti-insulin receptor antibodies using an immunoprecipitation assay (2). An intense band corresponding to insulin receptor α-subunit was present in antibody precipitates formed with patient serum and recombinant human insulin receptor (Figure 1A). Additional laboratory measurements during the hospitalization included islet cell antibodies (Smith-Kline Labs, St. Louis, Missouri) and glutamic acid decarboxylase antibodies (Quest-Nichols, Chantilly, Virginia), both of which were negative. His C-peptide was 2.1 ng/mL and insulin antibodies were 4.2 units/mL (reference range < 5.0). Anti-nuclear antibodies and screens for antibodies to extractable nuclear antigens (SM, RNP, Ro/SS-A, La/SS-B, Scl-70, and Jo-1), single stranded DNA, and double stranded DNA were all negative. An erythrocyte sedimentation rate was 32 mm/hr (reference range < 20). Serum protein electrophoresis was normal. A TSH was 1.45 μIU/mL. Free T4 and free T3 were normal. Several early morning measurements of cortisol and ACTH did not suggest any pituitary-adrenal abnormality. A fasting lipid panel two weeks after admission showed cholesterol 206 mg/dL, triglyceride 68 mg/dL, HDL 58 mg/dL, and LDL 135 mg/dL.
Figure 1. Assay of Patient Serum for anti-Insulin Receptor Antibodies.
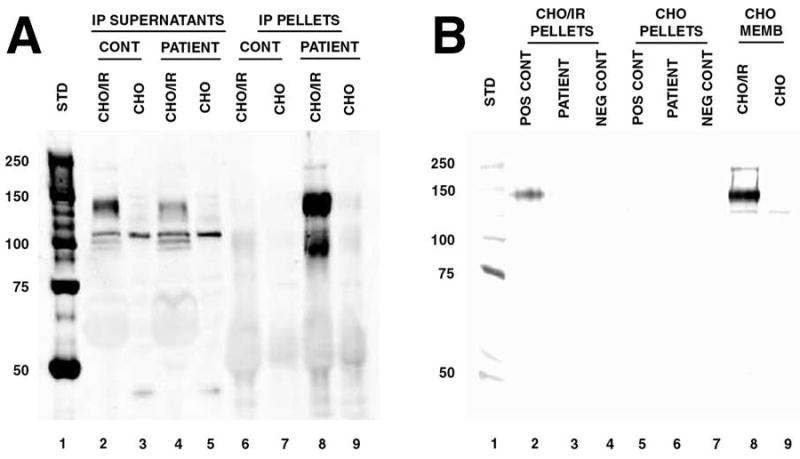
Serum was assayed for anti-insulin receptor antibodies using recombinant human insulin receptor in an immunoprecipitation assay (2). Detergent-solubilized membranes of CHO/IR cells expressing human insulin receptor (IR) were the source of insulin receptor used. The CHO/IR cells and parent CHO cells, used as controls, were obtained from Dr. Jeffrey Pessin, University of Iowa College of Medicine (7). Parallel control immunoprecipitations with pooled healthy donor serum (Irvine Scientific, Santa Ana, California) were performed in all cases. Standard protocols for immunoprecipitation and western blotting were employed (8,9). Serum samples were incubated with CHO/IR or CHO membrane lysates. Patient or control serum (50 μl) was added to the solubilized membranes and incubated at 4°C overnight, followed by addition of protein G sepharose for 1 hour at 4°C. Antibody complexes were collected by centrifugation. Precipitated antibody complexes (pellets) and residual soluble proteins (IP supernatants) were analyzed by western blotting with rabbit anti-insulin receptor α (N-20: sc-710 pAb, Santa Cruz Biotechnology, Inc.). Panel A: Assay of patient serum obtained during the presenting hospitalization. 1, molecular weight standards; 2 & 3, supernatants of CHO/IR and CHO membranes after precipitation with pooled human serum; 4 & 5, supernatants of CHO/IR and CHO membranes after precipitation with patient serum; 6 & 7, pelleted antibody complexes from solubilized CHO/IR and CHO membranes using pooled human serum; 8 & 9, pelleted antibody complexes from solubilized CHO/IR and CHO membranes using patient serum. An intense band corresponding to insulin receptor α-subunit was present in antibody precipitates formed with patient serum and solubilized CHO/IR membranes (lane 8), but not in precipitates with control CHO membranes (lane 9) or precipitates with pooled human serum (lanes 6 & 7). Depletion of insulin receptor from the CHO/IR supernatants by antibodies in the patient’s serum, but not pooled human serum from healthy donors, is also evident (lanes 2 & 4). Panel B: Assay of patient serum obtained two years after presentation. 1, molecular weight standards; 2, 3 & 4, pelleted antibody complexes from solubilized CHO/IR membranes using positive control serum (patient serum from his initial presentation), patient serum, and pooled human sera; 5, 6 & 7, pelleted antibody complexes from solubilized CHO membranes using positive control serum, patient serum, and pooled human sera; 8 & 9, CHO/IR and CHO membranes loaded directly onto the gel without immunoprecipitation to confirm insulin receptor detection.
Over the first three months after discharge, he reported glucoses fluctuating between 60 and 300 mg/dL, and his insulin was reduced to 50 units QID. Four months after discharge, he experienced two episodes of severe hypoglycemia, one of which required treatment in an emergency room. His insulin was reduced to 30 units/day. Six months after discharge, he was taking 15 units of insulin/day and had returned to his pre-interferon weight of 96 kg. Nine months after discharge, he reported glucoses between 90 and 190 mg/dL and a hemoglobin A1c was 5.7%. Two years after discharge, insulin had been discontinued and a hemoglobin A1c was 5.9%. Glucoses ranged from 90–160 mg/dL. Anti-insulin receptor antibodies could no longer be demonstrated in his serum (Figure 1B). His cranial neuropathies resolved over a one year period. A hepatitis C viral RNA titer eighteen months after discharge had returned to pretreatment levels.
Discussion
Interferon α is used in the treatment of chronic viral hepatitis and some malignancies. It is associated with a variety of autoimmune complications, among which, thyroid autoimmunity is most common (10). The average incidence of hyper- or hypothyroidism in several recent studies of hepatitis C patients treated with interferon α was 6% (11). Some patients have destructive thyroiditis without evidence of autoimmunity, but the majority have autoimmune thyroid dysfunction (11,12). About 50% of patients with interferon-induced autoimmune hypothyroidism and a smaller percentage of those with hyperthyroidism remit after interferon discontinuation (11). Other autoimmune conditions reported with interferon α include type 1 diabetes mellitus, systemic lupus erythematosus, myasthenia gravis, celiac disease, autoimmune hepatitis, psoriasis, vitiligo, hemolytic anemia, thrombocytopenia, and sarcoidosis (10,13,14). Interferon α-induced autoimmune disease tends to occur in individuals at higher baseline risk. Patients genetically predisposed to thyroid or islet autoimmunity and patients with thyroid or islet cell autoantibodies at commencement of therapy are much more likely to develop overt disease while taking interferon α (11,13).
The majority of patients developing diabetes on interferon therapy have type 1 diabetes with autoantibodies to islet cell antigens (13). Chronic hepatitis C infection also causes insulin resistance, and people with hepatitis C have a higher prevalence of type 2 diabetes and impaired fasting glucose (15). Moreover, hepatitis C patients who experience a sustained virologic response to interferon therapy have a lower incidence of glucose abnormalities than non-responders (16). Hepatitis C does not cause insulin resistance of the magnitude seen in this case. Development of type B insulin resistance during interferon therapy is distinctly unusual. There is a report from Japan of a man who developed diabetes with insulin receptor autoantibodies during interferon-α treatment for hepatitis C (17). Screens for islet autoantibodies were negative. His clinical course was characterized by frequent hypoglycemia on relatively low doses of insulin. Insulin receptor antibodies could no longer be detected a few months after interferon was discontinued, but the diabetes did not resolve. Given this, the contribution of these antibodies to his diabetes is not clear. There is also a brief report from India of a woman with type 2 diabetes developing marked hyperglycemia during interferon treatment for chronic hepatitis C (18). The hyperglycemia did not respond to treatment with insulin doses as high as 700 units/day. The extreme insulin resistance remitted after discontinuation of interferon therapy. Although measurements of insulin receptor and other autoantibodies were not reported, hyperglycemia refractory to such high insulin doses is consistent with type B insulin resistance.
Insulin receptor antibodies can cause both hyperglycemia and hypoglycemia. The variability in clinical presentation is seen because insulin receptor antibodies can act as either agonists or antagonists of the insulin receptor (2,19). Insulin receptor antagonism can manifest as severe hyperglycemia refractory to massive doses of insulin. Insulin receptor stimulation causes hypoglycemia, which can be life-threatening (2). There are a few reports of patients with hyperglycemia due to type B insulin resistance developing severe hypoglycemia later in the course of their illness (2). There are also reports of insulin receptor autoantibodies in patients with systemic autoimmune disease and no obvious abnormality of glucose metabolism (20). Whether these simply represent antibodies that bind insulin receptors without altering insulin signaling, or whether these patients are at risk of developing autoimmune hyperglycemia or hypoglycemia is unknown.
In the United States, type B insulin resistance and autoimmune hypoglycemia are much more common in women and African Americans than other groups (2). Most reported cases have occurred in people with other autoimmune diseases such as systemic lupus erythematosus (2, 19). Cranial neuropathies can occur in systemic autoimmunity, and there are case reports of Bell’s palsy arising during interferon and ribavirin therapy (21–23). There is also an increased incidence of cranial neuropathies in diabetes (24). Whether our patient’s polycranial neuropathy was caused by autoimmunity, diabetes, or another etiology is unknown. Most patients with type B insulin resistance have acanthosis nigricans, which was not present in our patient. Treatments for the underlying autoimmunity in type B insulin resistance have included steroids and other immunosuppressive therapies such as azathioprine, cyclophosphamide, cyclosporine, mycophenolate, and rituximab, sometimes in combination with plasmapheresis (2, 19, 25–27). Type B insulin resistance has a high spontaneous remission rate, and it is difficult to conclusively attribute improvements in these reports to the therapies (2). There was initial reluctance to use immunosuppressive therapy or plasmapheresis in our patient because of concern that his response to the interferon might be compromised and because his insulin resistance appeared to be improving on its own. He did eventually receive steroids for cranial polyneuropathy, but his glycemic control only worsened during that period. Some patients with type B insulin resistance develop autoimmune hypoglycemia later in the course of their illness. The two episodes of severe hypoglycemia in our patient reflected resolution of his insulin resistance. They did not recur after a sharp reduction of his insulin dose. In this case, the severe insulin resistance remitted spontaneously over a 6 month period after interferon was discontinued.
Conclusion
This case demonstrates that insulin receptor autoantibodies and type B insulin resistance can occur as a complication of therapy with interferon α. The possibility of insulin receptor autoantibodies should be considered in patients receiving interferon α who develop new hyperglycemia or hypoglycemia with no evidence for autoimmunity to islet antigens.
Footnotes
Disclosure
The authors have no conflicts of interest to disclose.
References
- 1.Flier JS, Kahn CR, Roth J, Bar RS. Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science. 1975;190:63–65. doi: 10.1126/science.170678. [DOI] [PubMed] [Google Scholar]
- 2.Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical Course of the Syndrome of Autoantibodies to the Insulin Receptor (Type B Insulin Resistance) Medicine. 2002;81:87–100. doi: 10.1097/00005792-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Khokher MA, Avasthy N, Taylor AM, Fonseca VA, Dandona P. Insulin-receptor antibody and hypoglycemia associated with Hodgkin’s disease [Letter] Lancet. 1987;1:693–694. doi: 10.1016/s0140-6736(87)90467-3. [DOI] [PubMed] [Google Scholar]
- 4.Walters EG, Tavaré JM, Denton RM, Walters G. Hypoglycaemia due to an insulin-receptor antibody in Hodgkin’s disease [Letter] Lancet. 1987;1:241–243. doi: 10.1016/s0140-6736(87)90064-x. [DOI] [PubMed] [Google Scholar]
- 5.Rochet N, Blanche S, Carel JC, et al. Hypoglycemia induced by antibodies to insulin receptor following a bone marrow transplantation in an immunodeficient child. Diabetologia. 1989;32:167–172. doi: 10.1007/BF00265089. [DOI] [PubMed] [Google Scholar]
- 6.Fareau GG, Maldonado M, Oral E, Balasubramanyam A. Regression of acanthosis nigricans correlates with disappearance of anti-insulin receptor autoantibodies and achievement of euglycemia in type B insulin resistance syndrome. Metabolism. 2007;56:670–675. doi: 10.1016/j.metabol.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Yamauchi K, Pessin JE. Enhancement or inhibition of insulin signaling by insulin receptor substrate 1 is cell context dependent. Mol Cell Biol. 1994;14:4427–4434. doi: 10.1128/mcb.14.7.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boniface JS, Dell’Angelica EC, Springer TA. Immunoprecipitation. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Somerset, NJ: John Wiley & Sons, Inc; 1999. pp. 10.16.1–10.16.29. [Google Scholar]
- 9.Gallagher S, Winston SE, Fuller SA, Hurrell JGR. Immunoblotting and Immunodetection. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Somerset, NJ: John Wiley & Sons, Inc; 2004. pp. 10.16.1–10.16.29. [Google Scholar]
- 10.Borg FA, Isenberg DA. Syndromes and complications of interferon therapy. Curr Opin Rheumatol. 2007;19:61–66. doi: 10.1097/BOR.0b013e328010c547. [DOI] [PubMed] [Google Scholar]
- 11.Prummel MF, Laurberg P. Interferon-α and autoimmune thyroid disease. Thyroid. 2003;6:547–551. doi: 10.1089/105072503322238809. [DOI] [PubMed] [Google Scholar]
- 12.Mandac JC, Chaudhry S, Sherman KE, Tomer Y. The clinical and physiological spectrum of interferon-alpha induced thyroiditis: toward a new classification. Hepatology. 2006;43:661–672. doi: 10.1002/hep.21146. [DOI] [PubMed] [Google Scholar]
- 13.Fabris P, Floreani A, Tositti G, Vergani D, de Lalla F, Betterle C. Type 1 diabetes mellitus in patients with chronic hepatitis C before and after interferon therapy. Ailment Pharmacol Ther. 2003;18:549–558. doi: 10.1046/j.1365-2036.2003.01681.x. [DOI] [PubMed] [Google Scholar]
- 14.Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology. 2002;36:S237–S244. doi: 10.1053/jhep.2002.36810. [DOI] [PubMed] [Google Scholar]
- 15.Lecube A, Hernández C, Genescà J, Simó R. Proinflammatory cytokines, insulin resistance, and insulin secretion in chronic hepatitis C patients. Diabetes Care. 2006;29:1096–1101. doi: 10.2337/diacare.2951096. [DOI] [PubMed] [Google Scholar]
- 16.Simó R, Lecube A, Genescà J, Esteban JI, Hernández C. Sustained virological response correlates with reduction in the incidence of glucose abnormalities in patients with chronic hepatitis C virus infection. Diabetes Care. 2006;29:2462–2466. doi: 10.2337/dc06-0456. [DOI] [PubMed] [Google Scholar]
- 17.Uto H, Matsuoka H, Murata M, et al. A case of chronic hepatitis C developing insulin-dependent diabetes mellitus associated with various autoantibodies during interferon therapy. Diabetes Res Clin Pract. 2000;49:101–106. doi: 10.1016/s0168-8227(00)00143-1. [DOI] [PubMed] [Google Scholar]
- 18.Chatterjee S. Massive increase of insulin resistance in a patient with chronic hepatitis C after treatment with interferon [Letter] J Assoc Physicians India. 2004;52:514. [PubMed] [Google Scholar]
- 19.Bao S, Root C, Jagasia S. Type B insulin resistance syndrome associated with systemic lupus erythematosus. Endocr Pract. 2007;13:51–55. doi: 10.4158/EP.13.1.51. [DOI] [PubMed] [Google Scholar]
- 20.Rosenstein ED, Advani S, Reitz RE, Kramer N. The prevalence of insulin receptor antibodies in patients with systemic lupus erythematosus and related conditions. J Clin Rheumatol. 2001;7:371–373. doi: 10.1097/00124743-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Servioli L, Pérez C, Consani S, et al. Prevalence and characteristics of immunomediated neuropathies in a group of patients with autoimmune diseases. J Clin Neuromuscul Dis. 2007;9:285–290. doi: 10.1097/CND.0b013e318157614b. [DOI] [PubMed] [Google Scholar]
- 22.Hwang I, Calvit TB, Cash BD, Holtzmuller KC. Bell’s palsy: a rare complication of interferon therapy for hepatitis C [Letter] Dig Dis Sci. 2004;49:619–620. doi: 10.1023/b:ddas.0000026389.56819.0c. [DOI] [PubMed] [Google Scholar]
- 23.Hoare M, Woodall T, Alexander GJ. Bell’s palsy associated with IFN-alpha and ribavirin therapy for hepatitis C virus infection. J Interferon Cytokine Res. 2005;25:174–176. doi: 10.1089/jir.2005.25.174. [DOI] [PubMed] [Google Scholar]
- 24.Boulton AJM, Malik RA, Arezzo JC, Sosenko JM. Diabetic somatic neuropathies. Diabetes Care. 2004;27:1458–1486. doi: 10.2337/diacare.27.6.1458. [DOI] [PubMed] [Google Scholar]
- 25.Coll AP, Thomas S, Mufti GJ. Rituximab therapy for the type B syndrome of severe insulin resistance [Letter] N Engl J Med. 2004;350:310–11. doi: 10.1056/NEJM200401153500324. [DOI] [PubMed] [Google Scholar]
- 26.Page KA, Dejardin S, Kahn CR, Kulkarni RN, Herold KC, Inzucchi SE. A patient with type B insulin resistance syndrome, responsive to immune therapy. Nat Clin Pract Endocrinol Metab. 2007;3:835–840. doi: 10.1038/ncpendmet0693. [DOI] [PubMed] [Google Scholar]
- 27.Eriksson JW, Bremell T, Eliasson B, Fowelin J, Fredriksson L, Yu ZW. Successful treatment with plasmapheresis, cyclophosphamide, and cyclosporine A in type B syndrome of insulin resistance. Diabetes Care. 1998;21:1217–1220. doi: 10.2337/diacare.21.8.1217. [DOI] [PubMed] [Google Scholar]