

Abstract
Variability in cognitive functioning increases markedly with age, as does cognitive vulnerability to physiological and psychological challenges. Exploring the basis of this vulnerability may provide important insights into the mechanisms underlying aging-associated cognitive decline. As we have previously reported, the cognitive abilities of aging (24-month-old) F344 × BN rats are generally good, but are more vulnerable to the consequences of a peripheral immune challenge (an intraperitoneal injection of live Escherichia coli) than those of their younger (3-month-old) counterparts. Four days after the injection, the aging, but not the young rats show profound memory deficits, specific to the consolidation of hippocampus-dependent memory processes. Here, we have extended these observations, using hippocampal slices to examine for the first time the combined effects of aging and a recent infection on several forms of synaptic plasticity. We have found that the specific deficit in long-lasting memory observed in the aged animals after infection is mirrored by a specific deficit in a form of long-lasting synaptic plasticity. The late-phase long-term potentiation induced in area CA1 using theta-burst stimulation is particularly compromised by the combined effects of aging and infection—a deficit that can be ameliorated by intra-cisterna magna administration of the naturally occurring antiinflammatory cytokine IL-1Ra (interleukin-1 receptor antagonist). These data support the idea that the combination of aging and a negative life event such as an infection might produce selective, early-stage failures of synaptic plasticity in the hippocampus, with corresponding selective deficits in memory.
Introduction
Although it is not clear that a decline in the ability to learn and remember is a normal feature of aging, it is clear that variability in cognitive functioning increases with aging in humans (Laursen, 1997; Unverzagt et al., 2001) and animals (Gage et al., 1984; Barnes and McNaughton, 1985; Deupree et al., 1991; Gallagher et al., 2003). An intriguing clue about sources of this variability comes from the observation that aging increases cognitive vulnerability to challenging life events such as infection (Wofford et al., 1996), surgery (Bekker and Weeks, 2003), heart attack, and psychological stress (VonDras et al., 2005).
Because little is known about the mechanisms that mediate aging-associated increases in cognitive vulnerability, we have developed a rodent model to study them (Barrientos et al., 2006). Twenty-four-month-old Fischer 344/Brown Norway rats generally do not display significant physical or cognitive impairments before a brief infection produced by an intraperitoneal injection of Escherichia coli. However, after recovering from the active infection, the aged animals show significant impairment in hippocampus-dependent memory tasks (e.g., contextual fear and place learning); the young animals generally do not (Barrientos et al., 2006).
Data from conventional aging models examining variability in cognitive functioning with aging per se suggest that, when age-related deficits in hippocampus-dependent learning occur, they do not arise from a loss of hippocampal neurons or synapses (Rapp and Gallagher, 1996; Geinisman et al., 2004), but rather from more subtle alterations in synaptic efficacy (Rapp et al., 1999; Smith et al., 2000). Not surprisingly, some age-related neurodegenerative disorders (e.g., Alzheimer's disease) first manifest themselves as disorders of synaptic plasticity, before the onset of overt cellular pathology (Selkoe, 2002). Thus, synaptic plasticity, particularly long-term potentiation (LTP), has been extensively studied in aging and disease models (Barnes and McNaughton, 1985; Deupree et al., 1991; Diana et al., 1995; Bach et al., 1999; Martin et al., 2000; Tombaugh et al., 2002; Bliss et al., 2003).
These previous studies have reinforced the idea that synaptic plasticity has multiple forms: short-term forms (e.g., early-phase LTP or E-LTP), involving the covalent modification of existing proteins, and long-lasting forms (e.g., late-phase LTP or L-LTP) that require transcription and translation (Bliss et al., 2007). It has also become apparent that different stimulation paradigms can evoke similarly sized, and similarly enduring, manifestations of synaptic plasticity—for example, L-LTP—that nonetheless arise from distinct biochemical processes and may reflect different information storage processes with differential vulnerabilities to disruption (Kang et al., 1997; Patterson et al., 2001). Thus, examining the impact of aging and immune challenge on these processes should ultimately provide mechanistic insights into aging-associated cognitive vulnerability.
In the present study, we have examined several forms of synaptic plasticity in hippocampal slices from young and aged rats, with and without a recent history of E. coli infection as a first step in using the slice system to examine the cellular and molecular mechanisms underlying the memory deficits evoked by immune challenge in aged animals.
Materials and Methods
The experimental animals
The rats were 3- and 24-month-old male Fischer 344/Brown Norway F1 crosses from the National Institute on Aging Aged Rodent Colony. Animals were pair housed, on a 12 h light/dark cycle, with ad libitum access to food and water, and were allowed to acclimate to the animal facility for 2 weeks before experiments were begun. All experiments were conducted in accordance with protocols approved by the University of Colorado Animal Care and Use Committee.
The infection model
Preparing the bacteria.
One day before the start of experimentation, stock E. coli cultures (ATCC 15746; American Type Culture Collection) were thawed and cultured overnight (15–20 h) in 40 ml of brain–heart infusion (Difco) in an incubator (37°C; 95% air plus 5% CO2). The number of bacteria in individual cultures was quantified by extrapolating from previously determined growth curves. Cultures were then centrifuged for 15 min at 3000 rpm, the supernatants were discarded, and the bacteria were resuspended in sterile PBS, to achieve a final dose of 2.5 × 109 CFU in 250 μl.
Producing the infection.
All animals received an intraperitoneal injection of 250 μl of either E. coli or the vehicle (sterile PBS).
Blocking CNS consequences of the peripheral infection.
Cisterna magna rather than intrahippocampal or intracerebroventricular injections were used because this procedure does not require surgery, which can itself produce memory impairments in aging animals. Twenty-four-month-old rats were briefly anesthetized using halothane, and a 27-gauge needle connected to a 25 μl Hamilton syringe via PE50 tubing was inserted into the cisterna magna. Interleukin-1-specific receptor antagonist (IL-1Ra) or vehicle (endotoxin-free saline from Abbott Laboratories) was then injected into the cisterna magna. The IL-1Ra (112 μg; Amgen) was administered in a total volume of 3 μl. Immediately after this procedure, the rats received an intraperitoneal injection of either E. coli or vehicle.
Slice preparations
Physiology experiments were performed 4–5 d after the initial infection. This time point was chosen based on several observations: (1) All of the animals have completely recovered from the acute infection after 4 d (symptoms such as fever are gone within 3 d); (2) the 24-month-old rats, but not the 3-month-old rats show a significant impairment in long-term hippocampus-dependent memory 4 d after the E. coli infection (Barrientos et al., 2006); and (3) levels of interleukin-1 (IL-1) protein in the hippocampus are still significantly elevated in the 24-month-old rats, but not in the 3-month-old rats, 4–5 d after the infection (Barrientos et al., 2009).
Experiments on hippocampi from young and aged, saline- or E. coli-injected animals were interleaved. Hippocampi were collected from rats after decapitation. Transverse hippocampal slices (400 μm) were prepared using conventional techniques (Patterson et al., 1992, 1996). Slices were maintained in an interface chamber at 28°C and perfused with an oxygenated saline solution (in mm: 124.0 NaCl, 4.4 KCl, 26.0 NaHCO3, 1.0 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, 10 glucose). Slices were permitted to recover for at least 90 min before recording. Field EPSPs (fEPSPs) were recorded from Schaffer collateral–CA1 synapses by placing both stimulating and recording electrodes in the stratum radiatum. All stimuli were delivered at intensities that evoked fEPSP slopes equal to one-third of the maximum in each slice. Test stimuli were delivered once every minute, and test responses were recorded for 15–30 min before beginning the experiment to assure stability of the response.
Stimulation protocols
Slices were tetanized using one of three protocols: 1, 1 s train at 100 Hz; 4, 1 s trains, at 100 Hz, delivered 5 min apart; or 12 bursts, of four pulses at 100 Hz, delivered 200 ms apart (theta frequency). The one-train protocol was used to induce E-LTP. The four-train and theta-burst protocols were used to induce L-LTP. The same stimulus intensity was used for tetanization and evoking test responses.
Statistical analysis
Data were analyzed using factorial ANOVA, followed by Fisher's PLSD post hoc tests.
Results
Input–output curves did not differ across groups
We began seeking functional deficits associated with aging and or a recent history of infection by examining basal synaptic transmission at the Schaffer collateral–CA1 synapse in hippocampal slices (Fig. 1A). To provide an initial indication of possible differences in the response to stimuli of a given intensity, we generated input–output curves from slices from young and aged rats injected 4 d earlier with either E. coli or saline. We found that these curves were not significantly altered by aging, or infection.
Figure 1.
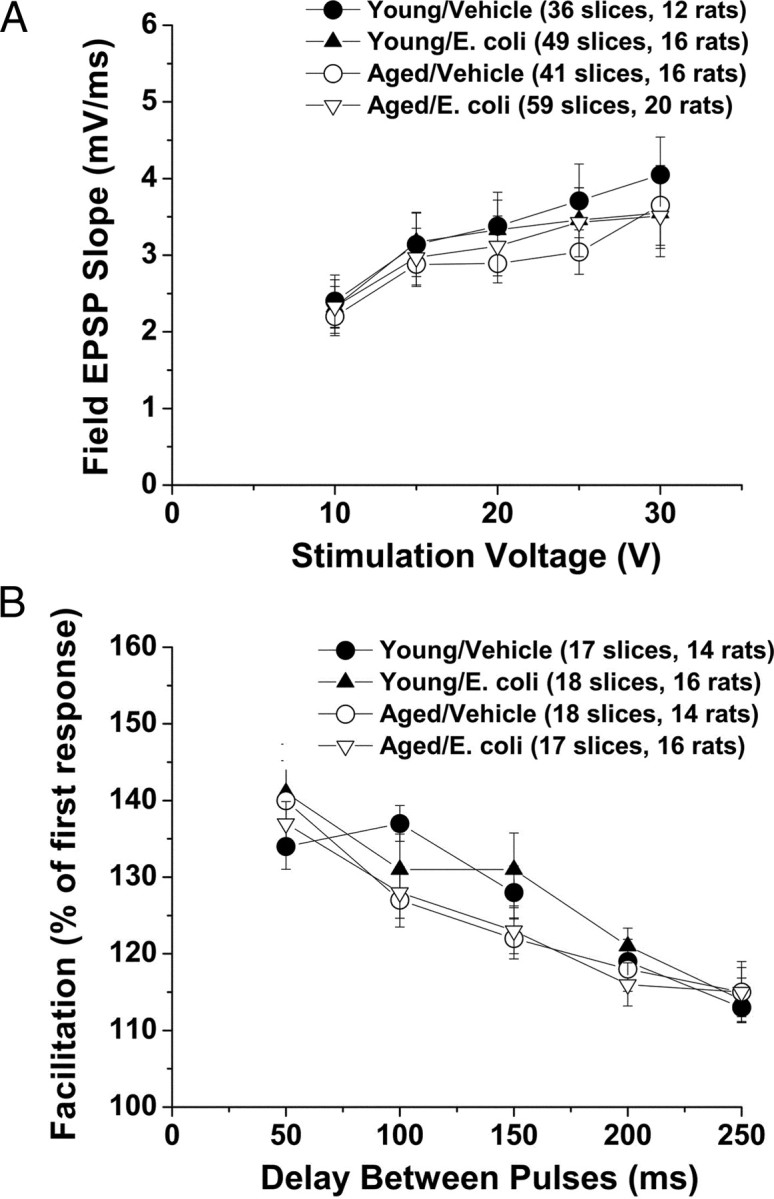
Slices from young and aged rats, with and without a history of infection, respond similarly to test stimuli. A, Stimulus–response curves are not altered by age or a history of infection. Plots of fEPSP slopes (in millivolts per millisecond) at various stimulation intensities for hippocampal slices from young and aged rats with and without a recent history of infection (4 d after injection of E. coli or saline) show no significant differences in basal synaptic transmission in area CA1. B, Paired-pulse facilitation is also not significantly altered by age or a history of infection. Percentage facilitation, calculated from the ratio of the second fEPSP slope to the first fEPSP slope, is shown at interpulse intervals ranging from 50 to 250 ms. No significant differences in PPF were observed across the groups at any of the interpulse intervals examined. Error bars indicate SEM.
Paired-pulse facilitation was not significantly different across groups
We next examined paired-pulse facilitation (PPF) (Fig. 1B), a presynaptic form of short-term plasticity in which the synaptic response to the second of a pair of closely spaced stimuli is increased. This is thought to reflect residual Ca2+ in the presynaptic nerve terminal from the first stimulus adding to the influx of Ca2+ evoked by the second stimulus, with a resulting increase in presynaptic neurotransmitter release (Katz and Miledi, 1968). Neither aging nor a history of infection had a significant effect on PPF across a range of interstimulus intervals.
Short-term synaptic plasticity was not significantly altered by aging or a history of infection
To examine possible alterations in short-term synaptic plasticity, we tetanized the slices using one high-frequency stimulus train: 1 s, at 100 Hz—a protocol frequently used to induce early-phase LTP lasting ∼1–2 h in slices from naive rats (Fig. 2). Neither age nor a history of infection had a significant effect on posttetanic potentiation (percentage baseline, immediately after the stimulus train: young/vehicle, 235 ± 20%; young/E. coli, 214 ± 17%; aged/vehicle, 220 ± 24%; and aged/E. coli, 223 ± 16%; page = 0.581 and pinfection = 0.290), or one-train E-LTP (percentage baseline, measured 90 min after the stimulus train: young/vehicle, 163 ± 23%; young/E. coli, 134 ± 13%; aging/vehicle, 143 ± 13%; and aging/E. coli, 141 ± 5%; page = 0.373 and pinfection = 0.250).
Figure 2.

One-train E-LTP is not greatly altered by age or infection. Mean fEPSP slopes are plotted as a percentage of pretetanus baseline values. E-LTP evoked by a relatively weak stimulus protocol (a single, 1 s, 100 Hz stimulus train) at the Schaffer collateral–CA1 synapse in hippocampal slices from aged and young rats, with and without a previous history of infection. Slices were prepared from animals 4 d after injection of E. coli or vehicle. Neither age nor a history of infection had a significant effect on E-LTP in area CA1. The insets show representative traces. Error bars indicate SEM.
Four-train L-LTP and theta-burst L-LTP, two distinct types of long-lasting synaptic plasticity, are differentially affected by age and infection
Different stimulus protocols produce long-lasting forms of synaptic plasticity with somewhat different molecular requirements (Kang et al., 1997; Patterson et al., 2001). Thus, for the experiments reported here, slices were tetanized using one of two protocols: either four trains of high-frequency stimulation or theta-burst stimulation. Both of these protocols induce late-phase LTP in animals of the hybrid strain used here. The high-frequency four-train protocol produces a robust activation of many, although not all, plasticity-related signaling cascades (for review, see Bliss et al., 2007). The theta-burst protocol is more naturalistic—designed to mimic the burst firing of CA1 pyramidal cells at theta frequency recorded in vivo from awake behaving animals during spatial exploration (for review, see O'Keefe, 2007)—and has proven to be a sensitive indicator of alterations in mnemonic processes associated with aging (for review, see Lynch et al., 2006) or pharmacological or genetic manipulation of the substrates for memory.
When we examined the effects of aging and infection on the L-LTP evoked by the four-train protocol (Fig. 3A), we found no significant effects on posttetanic potentiation (percentage baseline: young/vehicle, 247 ± 23%; young/E. coli, 247 ± 18%; aged/vehicle, 253 ± 42%; and aged/E. coli, 280 ± 27%; page = 0.857 and pinfection = 0.420) or 3 h after tetanus (percentage baseline: young/vehicle, 204 ± 20%; young/E. coli, 178 ± 13%; young/vehicle, 202 ± 32%; and young/E. coli, 184 ± 16%; page = 0.970 and pinfection = 0.408).
Figure 3.
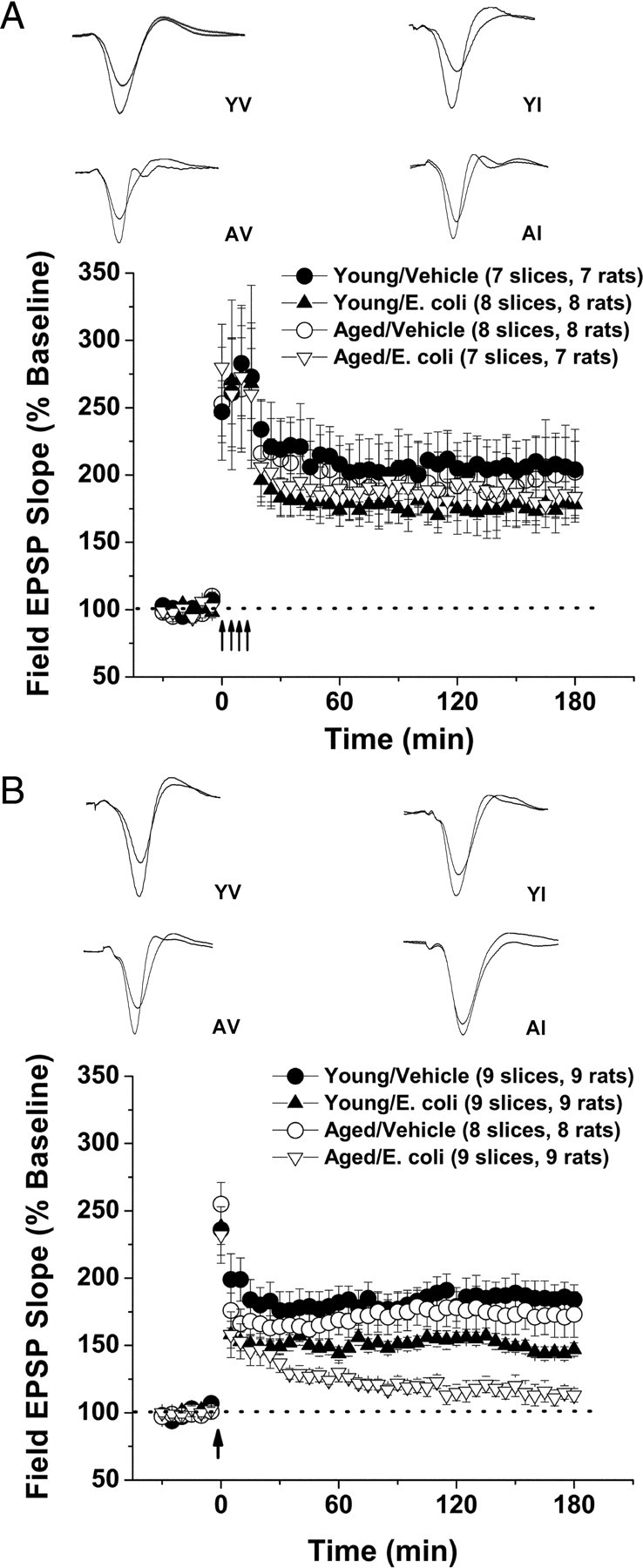
Age and infection differentially affect distinct types of L-LTP. Experiments examining two forms of L-LTP in aged and young rats, with and without a previous history of infection. Slices were prepared from animals 4 d after the E. coli or vehicle injections. L-LTP was elicited at the Schaffer collateral–CA1 synapse using one of two distinct stimulus protocols: a four-train protocol (four 1 s, 100 Hz stimulus trains, delivered 5 min apart) and a theta-burst protocol (12 bursts of 4 pulses at a 100 Hz, delivered 200 ms apart). A, The L-LTP evoked by the intense, second-long bouts of high-frequency stimulation (the 4-train protocol) was not significantly affected by age or infection. B, In contrast, infection suppressed the full expression of theta-burst L-LTP, and aging greatly exacerbated this effect. The insets show representative traces. Error bars indicate SEM.
The effects of the theta-burst stimulation were more complex (Fig. 3B). Under the conditions used, age did not have a significant effect on posttetanic potentiation (page = 0.703) or L-LTP 3 h after the tetanus (page = 0.307). In contrast, E. coli infection had no effect on posttetanic potentiation (pinfection = 0.534%; baseline: young/vehicle, 236 ± 19%; young/E. coli, 238 ± 13%; aged/vehicle, 255 ± 14%; and aged/E. coli, 232 ± 21%) but resulted in significantly smaller L-LTP in slices from young rats (pinfection in young rats = 0.010) and profoundly reduced L-LTP in slices from aged animals (pinfection in aged rats = 0.006; percentage baseline, 3 h after tetanus: young/vehicle, 184 ± 11%; young/E. coli, 147 ± 5%; aged/vehicle, 169 ± 12%; and aged/E. coli, 113 ± 5%).
Central administration of the antiinflammatory cytokine IL-1Ra ameliorates the aging-associated, infection-induced impairment of theta-burst L-LTP
The proinflammatory cytokine IL-1β is a major mediator of inflammatory responses in the brain. We have previously found that IL-1β is elevated in the hippocampi of aged rats with a recent history of peripheral E. coli infection (Barrientos et al., 2006). This elevation parallels the E. coli-evoked deficits in hippocampus-dependent long-term memory (Barrientos et al., 2006), and blocking IL-1 signaling in the brain with the naturally occurring interlukin-1 receptor antagonist (Dinarello, 1997) blocks the memory deficit (Frank et al., 2010). We therefore set out to determine whether blocking hippocampal IL-1β signaling with IL-1Ra would also block the E. coli-evoked deficit in theta-burst L-LTP in aged animals (Fig. 4).
Figure 4.

Blocking the actions of the IL-1β in the CNS ameliorates the effect of peripheral infection on theta-burst L-LTP in aged rats. The antiinflammatory cytokine IL-1Ra or vehicle was injected into the cisternae magna of aged rats, and immediately afterward the rats received an intraperitoneal injection of either E. coli or vehicle. Hippocampal slices were prepared from the animals 4 d after the injections, and Schaffer collateral–CA1 synapses were stimulated using the theta-burst protocol. Infusion of exogenous IL-1Ra into the brain greatly reduced the E. coli-induced impairment in theta-burst L-LTP. The insets show representative traces. Error bars indicate SEM.
As before, E. coli infection greatly reduced theta-burst L-LTP in the aged animals (pperipheral E. coli = 0.002), but we found that this reduction could be blocked by central administration of IL-1Ra (pperipheral E. coli + CNS IL-1Ra = 0.001), which had no significant effect on L-LTP in the absence of infection (pperipheral vehicle + CNS IL-1Ra = 0.69; percentage baseline 3 h after tetanus: vehicle/vehicle, 156 ± 15%; E. coli/vehicle, 104 ± 3%; E. coli/IL-1Ra, 143 ± 3%; and vehicle/IL-1Ra, 166 ± 17%). We found no significant effects of E. coli or IL-Ra on posttetanic potentiation (percentage baseline: E. coli/vehicle, 199 ± 13%; E. coli/IL-1Ra, 237 ± 26%; vehicle/vehicle, 241 ± 16%; and vehicle/IL-1Ra, 228 ± 22%; pperipheral injection = 0.417 and pCNS injection = 0.476).
Discussion
In the experiments presented here, we examined for the first time the effects of aging combined with a secondary experimental insult—a peripheral immune challenge—on synaptic function in area CA1 of the hippocampus. Our principal findings are that the E. coli infection (1) had no significant effects on basal synaptic transmission or short-term synaptic plasticity in slices from young or aged rats, (2) had no significant effects on a form of late-phase LTP evoked by high-frequency stimulation in slices from young or aged rats, but (3) significantly reduced a form of L-LTP evoked by theta-burst stimulation in slices from young rats and essentially abolished it in slices from aged rats. Interestingly, we were able to block the reduction in theta-burst L-LTP in aged animals by blocking IL-1 signaling in the brain with the antiinflammatory cytokine IL-Ra.
These results are consistent with the results of previous behavioral studies indicating that the E. coli infection does not compromise the initial learning of the test tasks, or the formation of short-term memories in any of the animals, but instead produces profound deficits specific to the consolidation of hippocampus-dependent memory in aged, but not in young rats (Barrientos et al., 2006). The physiology experiments presented here add support to the idea that the infection does not produce large-scale, nonspecific disruptions in hippocampal function. Instead, they suggest that limited and relatively subtle synaptic deficits might give rise to the selective memory deficits associated with the combined effects of aging and infection.
A number of studies have examined the impact of aging alone on learning and memory and synaptic plasticity—often with mixed result. As noted previously, the range of cognitive and synaptic function grows wider with increasing age—an observation consistent with the idea that age is not the only important variable in aging-associated cognitive decline. Aging is often, but not always, associated with some cognitive impairment, and with deficits in the induction and or maintenance of hippocampal LTP (Gage et al., 1984; Barnes and McNaughton, 1985; Deupree et al., 1991; Gallagher et al., 2003). At Schaffer collateral–CA1 synapses, the available data suggest that the basic mechanisms for producing LTP remain intact into old age but are somewhat less likely to be recruited by naturalistic patterns of stimulation or by patterns of afferent activity associated with normal behavior—in contrast, age-related impairments tend to be masked by high-frequency stimulation protocols (for review, see Lynch et al., 2006).
Our results are generally in line with these previous findings but may also offer insight into secondary events that can interfere with production of long-lasting plasticity in aging. In the absence of immune challenge, the aging Fischer/Brown Norway rats did not show overt cognitive deficits or impairments in synaptic function. This is not particularly surprising, as we elected to use the aging, but presenescent 24-month-old F344 × BN rats to minimize basal differences in memory functions between young and aged rats. However, after the immune challenge, the aged animals showed dramatic deficits in consolidation of hippocampus-dependent memories (Barrientos et al., 2006), and, as reported here, in theta-burst L-LTP. The association of these deficits is intriguing since it has been suggested that the formation of stable spatial memories may require selective strengthening of synapses in hippocampal area CA1 in response to short bursts of action potentials at theta frequency (Buzsáki, 2002)—an idea supported by the observation that deficits in theta-frequency LTP in area CA1 distinguish cognitively impaired from unimpaired aged Fischer 344 rats (Tombaugh et al., 2002).
How might aging render hippocampal memory processes vulnerable to the deleterious effects of a peripheral infection? One possibility is suggested by the fact that products of peripheral immune activation can communicate with the brain both via circulatory and neural routes, leading to a cascade of CNS effects including microglial activation and subsequent production of proinflammatory cytokines such as interleukin-1β (for review, see Maier et al., 2001; Konsman et al., 2002). Numerous studies have provided evidence that elevated levels of proinflammatory molecules such as IL-1β may sometimes impair cognition and synaptic plasticity. Conditions or treatments likely to lead to aberrant increases in brain levels of proinflammatory cytokines (e.g., autoimmune diseases) are intermittently associated with problems in memory, learning, and concentration. Experimentally elevated levels of IL-1β in the hippocampus impair the formation of long-lasting memory in hippocampus-dependent tasks (Oitzl et al., 1993; Gibertini et al., 1995; Pugh et al., 1999; Barrientos et al., 2002; Yirmiya et al., 2002) and inhibit LTP in several regions of the hippocampus (Katsuki et al., 1990; Bellinger et al., 1993; Coogan and O'Connor, 1997) in young adult animals. Thus, individuals with exaggerated brain inflammatory responses to peripheral immune challenge might be more vulnerable to challenge-evoked disruptions of hippocampal memory systems.
Interestingly, aging sensitizes the hippocampal inflammatory response to peripheral E. coli (Frank et al., 2006; Chen et al., 2008). We have previously reported that basal levels of IL-1β protein in the hippocampus are low in our F344 × BN rats and do not differ significantly between 3- and 24-month-old animals (Barrientos et al., 2006). However, when levels of hippocampal IL-1β protein were examined 4 d after infection with E. coli, IL-1β was markedly increased in the older animals, but not in the younger (Barrientos et al., 2006). This is not because the dose of E. coli used failed to induce an inflammatory response in the younger animals. Rather, both the magnitude and the duration of the inflammatory response were increased in the older animals (Barrientos et al., 2009). Not surprisingly, we have very recently found that blunting this response in the brain using the IL-1 receptor antagonist IL-1Ra mostly prevents the E. coli-induced impairment in hippocampus-dependent memory in the aged rats (Frank et al., 2010) and, as shown here, blocks the deficit in theta-burst L-LTP.
It is not yet clear how aberrant elevation of IL-1β impairs synaptic plasticity and learning and memory. Potential mechanisms may involve activation of p38MAPK, JNK (c-Jun N-terminal kinase), caspase 1, and NF-κB (nuclear factor kappa B) (Vereker et al., 2000a,b; Curran et al., 2003; Kelly et al., 2003), and downregulation of BDNF (brain-derived neurotrophic factor) (Guan and Fang, 2006).
The aging and immune challenge model provides an excellent system for exploring these questions. The relatively physiological E. coli infection has been shown to produce selective deficits in hippocampus-dependent memory. Here, we extend these results, demonstrating that the interaction between aging and peripheral infection also produces selective effects on synaptic plasticity. Since these behavioral and physiological deficits occur in a predictable time frame and are not confounded by genetic manipulation, the aging-induced vulnerability model may be especially tractable for examining the cellular and molecular basis of the initial events (e.g., early failures of synaptic plasticity) giving rise to a form of memory disruption that mimics many aspects of human pathology.
Footnotes
This work was supported by an Innovative Seed Grant Award from the University of Colorado (S.L.P.) and National Institute on Aging Grants 1R21AG031467 (S.L.P.) and 1R01AG02827 (R.M.B., S.F.M.). We thank A. M. Hein, N. M. Kim, and J. Hoover for skillful technical assistance.
References
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci U S A. 1999;96:5280–5285. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, McNaughton BL. An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behav Neurosci. 1985;99:1040–1048. doi: 10.1037//0735-7044.99.6.1040. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/s0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker AY, Weeks EJ. Cognitive function after anaesthesia in the elderly. Best Pract Res Clin Anaesthesiol. 2003;17:259–272. doi: 10.1016/s1521-6896(03)00005-3. [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR. Interleukin 1beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- Bliss T, Collingridge G, Morris RG. Synaptic plasticity in the hippocampus. In: Andersen P, Morris RG, Amaral D, O'Keefe J, editors. The hippocampus book. Oxford, New York: Oxford UP; 2007. pp. 343–475. [Google Scholar]
- Bliss TV, Collingridge GL, Morris RG. Introduction. Long-term potentiation and structure of the issue. Philos Trans R Soc Lond B Biol Sci. 2003;358:607–611. doi: 10.1098/rstb.2003.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan A, O'Connor JJ. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1beta. Neuroreport. 1997;8:2107–2110. doi: 10.1097/00001756-199707070-00004. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O'Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118:347–357. doi: 10.1016/s0306-4522(02)00941-7. [DOI] [PubMed] [Google Scholar]
- Deupree DL, Turner DA, Watters CL. Spatial performance correlates with in vitro potentiation in young and aged Fischer 344 rats. Brain Res. 1991;554:1–9. doi: 10.1016/0006-8993(91)90164-q. [DOI] [PubMed] [Google Scholar]
- Diana G, Domenici MR, Scotti de Carolis A, Loizzo A, Sagratella S. Reduced hippocampal CA1 Ca2+-induced long-term potentiation is associated with age-dependent impairment of spatial learning. Brain Res. 1995;686:107–110. doi: 10.1016/0006-8993(95)00440-2. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–265. doi: 10.1016/s1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344×BN F1 rats. Brain Behav Immun. 2010;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Dunnett SB, Björklund A. Spatial learning and motor deficits in aged rats. Neurobiol Aging. 1984;5:43–48. doi: 10.1016/0197-4580(84)90084-8. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Bizon JL, Hoyt EC, Helm KA, Lund PK. Effects of aging on the hippocampal formation in a naturally occurring animal model of mild cognitive impairment. Exp Gerontol. 2003;38:71–77. doi: 10.1016/s0531-5565(02)00159-6. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Ganeshina O, Yoshida R, Berry RW, Disterhoft JF, Gallagher M. Aging, spatial learning, and total synapse number in the rat CA1 stratum radiatum. Neurobiol Aging. 2004;25:407–416. doi: 10.1016/j.neurobiolaging.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Gibertini M, Newton C, Friedman H, Klein TW. Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav Immun. 1995;9:113–128. doi: 10.1006/brbi.1995.1012. [DOI] [PubMed] [Google Scholar]
- Guan Z, Fang J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun. 2006;20:64–71. doi: 10.1016/j.bbi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1beta on long term potentiation in rat dentate gyrus. J Biol Chem. 2003;278:19453–19462. doi: 10.1074/jbc.M301938200. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- Laursen P. The impact of aging on cognitive functions. An 11 year follow-up study of four age cohorts. Acta Neurol Scand Suppl. 1997;172:7–86. [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. Synaptic plasticity in early aging. Ageing Res Rev. 2006;5:255–280. doi: 10.1016/j.arr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LD, Nance DM. Multiple routes of action of interleukin-1 on the nervous system. In: Ader R, Felton DL, Cohen N, editors. Psychoneuroimmunology. Ed 3. New York: Academic; 2001. pp. 563–583. [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, van Oers H, Schöbitz B, de Kloet ER. Interleukin-1beta, but not interleukin-6, impairs spatial navigation learning. Brain Res. 1993;613:160–163. doi: 10.1016/0006-8993(93)90468-3. [DOI] [PubMed] [Google Scholar]
- O'Keefe J. Neurophysiology in the behaving animal. In: Andersen P, Morris RG, Amaral D, O'Keefe J, editors. The hippocampus book. Oxford, New York: Oxford UP; 2007. pp. 475–549. [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Nguyen KT, Gonyea JL, Fleshner M, Wakins LR, Maier SF, Rudy JW. Role of interleukin-1beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res. 1999;106:109–118. doi: 10.1016/s0166-4328(99)00098-4. [DOI] [PubMed] [Google Scholar]
- Rapp PR, Gallagher M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci U S A. 1996;93:9926–9930. doi: 10.1073/pnas.93.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp PR, Stack EC, Gallagher M. Morphometric studies of the aged hippocampus: I. Volumetric analysis in behaviorally characterized rats. J Comp Neurol. 1999;403:459–470. [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Smith TD, Adams MM, Gallagher M, Morrison JH, Rapp PR. Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci. 2000;20:6587–6593. doi: 10.1523/JNEUROSCI.20-17-06587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Chow AR, Michael TH, Rose GM. Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats. J Neurosci. 2002;22:9932–9940. doi: 10.1523/JNEUROSCI.22-22-09932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unverzagt FW, Gao S, Baiyewu O, Ogunniyi AO, Gureje O, Perkins A, Emsley CL, Dickens J, Evans R, Musick B, Hall KS, Hui SL, Hendrie HC. Prevalence of cognitive impairment: data from the Indianapolis Study of Health and Aging. Neurology. 2001;57:1655–1662. doi: 10.1212/wnl.57.9.1655. [DOI] [PubMed] [Google Scholar]
- Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000a;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- Vereker E, O'Donnell E, Lynch MA. The inhibitory effect of interleukin-1β on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci. 2000b;20:6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDras DD, Powless MR, Olson AK, Wheeler D, Snudden AL. Differential effects of everyday stress on the episodic memory test performances of young, mid-life, and older adults. Aging Ment Health. 2005;9:60–70. doi: 10.1080/13607860412331323782. [DOI] [PubMed] [Google Scholar]
- Wofford JL, Loehr LR, Schwartz E. Acute cognitive impairment in elderly ED patients: etiologies and outcomes. Am J Emerg Med. 1996;14:649–653. doi: 10.1016/S0735-6757(96)90080-7. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78:379–389. doi: 10.1006/nlme.2002.4072. [DOI] [PubMed] [Google Scholar]