

Abstract
Mortality in children with sepsis is most often related to diminished cardiac output with cardiovascular collapse, resulting in impaired oxygen delivery and ultimately, end-organ failure. Although cardiovascular “collapse” is commonly observed in individuals with septic shock, the hemodynamic causes of this differ greatly. In children, intrinsic myocardial dysfunction is most commonly present, while the systemic vascular resistance is typically high. This pattern is distinct from adults with sepsis where the principle hemodynamic profile shows elevated cardiac output, but substantially reduced systemic vascular resistance. Various studies support the concept that myocardial dysfunction, as occurs in pediatric septic patients, is due to intrinsic abnormalities in cardiomyocyte function and is not related to hypoperfusion as a result of low systemic vascular resistance. Importantly, when examined more closely, data from adults with septic shock also reveal that intrinsic myocardial dysfunction may play a larger role then previously appreciated. As a result, cardiovascular support, especially in pediatric sepsis requires a treatment strategy directed at the underlying mechanism(s) responsible for this dysfunction. Thus, it is imperative to gain a better understanding of the myocardial derangements that occur during sepsis in order to identify targets that will ultimately influence the management of children with septic shock and favorably alter the associated mortality. We hypothesize that key signaling pathways that control myocardial calcium flux, regulated to key kinases and phosphatases, influence myocyte contractility in sepsis. Thus, we review the data relevant to the sepsis-induced intracellular alterations in calcium flux in the cardiomyocyte, with an emphasis on changes in the phosphorylation state of the contractile proteins regulated by the balance between kinases and phosphatases. We believe therapies modulating the activity of these key proteins may provide an improvement in calcium handling and myocardial contractility and alter the clinical outcomes in sepsis.
Keywords: Phosphoinositide 3-kinase, Protein Phosphatse 2A, Protein kinase A, Sepsis, Myocardial dysfunction
Burden of Pediatric Sepsis
Perhaps one of the most devastating diseases faced by pediatricians is severe sepsis, as this condition has had a profound impact on child mortality throughout the world. Fortunately, improvements in therapeutic strategies over the past 40 years have dramatically decreased the mortality rate of children with sepsis. In 1963 a study of 900 infants at the University of Minnesota revealed a 97% mortality rate in children with gram negative sepsis and septic shock (1). In stark contrast, recent data, reflecting the use of therapeutic approaches recommended in the 2002 American College of Critical Care Medicine (ACCM) Clinical Practice Parameters for Hemodynamic Support of Pediatric and Neonatal Patients in Septic Shock, has reported mortality to be as low as 10% in children with sepsis (2) However, despite medical advances leading to improved survival, the case load and mortality rate combine to place sepsis among the leading causes of death in pediatric patients with more children dying with severe sepsis than from cancer (3). The estimated annual health care cost of pediatric sepsis is $4 billion in the United States (3). This health care burden resulting from its prevalence, devastating consequences, and high costs necessitates a more compehensive understanding of sepsis pathobiology in order to identify novel targets for improved therapies.
Hemodynamics in Sepsis: Adult versus Pediatric Profiles
It has been established that the cause of death in adults with septic shock is vasomotor paralysis (4). Contrary to the adult experience, pediatric patients with sepsis have a hemodynamic profile most often characterized by low cardiac output that results in impaired oxygen delivery and cardiovascular collapse (5, 6). Thus, the cardiovascular collapse most often identified in septic children is related to intrinsic myocardial dysfunction. In contrast to adult patients with sepsis, the myocardial dysfunction found in most pediatric patients is unrelated to changes in their systemic vascular resistance and, therefore, requires a different physiologic and pharmacologic approach to treatment (7). Both a heightened awareness of the prevalence and molecular mechanisms of the intrinsic myocardial damage induced by sepsis could ultimately impact medical management decisions and favorably alter mortality rates in children.
Mechanistic Theories of Sepsis-Induced Myocardial Dysfunction
With the observation that the systemic inflammatory response triggered by sepsis can result in myocardial dysfunction, investigators have continued to pursue the mechanistic causes of this pathobiology. Numerous studies have supported the concept that damage to the cardiomyocyte, as occurs in pediatric patients, is not solely explained by hypoperfusion related to low systemic vascular resistance. In a series of seminal studies, Parrillo et al showed that the serum of septic patients, containing high concentrations of pro-inflammatory cytokines (notably TNF-α and IL-1β), resulted in abnormalities in myocyte contraction and relaxation when added to isolated cardiomyocytes (8, 9). Accumulating evidence indicates that cytokines are not only important mediators of sepsis-induced heart failure, but also play a role in other various models of heart failure (10). While cytokines, such as TNF-α and IL-1β, have been shown to disturb cardiomyocyte function, treatment modalities targeting their receptors have not changed the outcome of septic patients. Furthermore, although many cytokines have been implicated as a cause of myocardial dysfunction, the intracellular mechanisms by which they mediate this response have not been elucidated. As a result, researchers have employed a number of models in order to further advance our understanding of the mechanisms of myocardial dysfunction.
Sepsis-induced myocardial dysfunction remains a difficult topic to study due to the limitations of animal models in correlating with the human disease state in large part because of the shear complexity of modeling this systemic inflammatory response (11). Animal models, have been developed with a goal of mimicking the two distinct phases of sepsis that are commonly observed in humans (12). Early in the disease process there is a hyperdynamic phase characterized by an increased ejection fraction and systemic vasodilation. In contrast, later in sepsis, a hypodynamic state develops during which the heart becomes dilated, contractility is impaired, and as a global result, oxygen delivery is inadequate. Fortunately the phenotype of the second phase, characterized by a decrease in isovolumic contraction, is similar to many other recognized forms of heart failure so that additional molecular insight gained from these models may be applied to sepsis (13, 14). Because there is a substantial amount of research being performed at the present time to evaluate the mechanisms leading to intrinsic myocardial disease, these data, though not specifically focused on sepsis-induced heart failure, may still be applicable in guiding sepsis-related research.
Signaling in Myocardial Dysfunction
In various models of heart failure, it has been shown that complex intracellular signaling pathways are activated. As occurs in a myriad of complex cellular functions, a number of key signaling pathways are regulated by phosphorylation events. As a result, these pathways are dependent on the activation of one or more kinases mediating phosphorylation and/or phosphatases mediating de-phosphorylation with subsequent alterations in downstream mediators as a consequence of their phosphorylation state (15). The phosphorylation state of downstream proteins specific to the cardiomyocyte has attracted the attention of many researchers interested in heart failure—though less so in models of sepsis. Although the basis of cardiac dysfunction in sepsis is likely to be multifactorial, there is strong evidence that intrinsic impairment of the cardiac contractile apparatus plays a major role. The phosphorylation state of the myocardial contractile proteins are fundamentally involved in regulating both contraction (inotropic) and relaxation (lusitropic) function of the heart. In sepsis, alterations in the phosphorylation state of a number of proteins by various kinases and phosphatases can affect changes in calcium sensitivity and flux within the myocyte. As a result, both kinases and phosphatases are attractive targets for drug therapies due to their central role in myocyte signaling.
Despite the logic in considering the role of kinases and phosphatases in mediating sepsis-induced myocardial dysfunction, their ubiquitous nature and involvement in multiple, complex signaling pathways make them very difficult enzymes to study. Early work in this field focused on the physiologic role of the cAMP-dependent kinase, protein kinase A (PKA) in mediating contractility. More recently, the role of other kinases, such as I kappa kinase (IKK), protein kinase C (PKC), members of the mitogen-activated protein kinases (MAPK) family of kinases (e.g. extracellularly regulated kinases) in regulating myocardial function have been examined (16-18). Insight into the regulation of signaling pathways by additional kinases (e.g. Phosphoinositide 3-kinase, PI3K) and now phosphatases (e.g. PP1 and PP2A) afforded by other fields of biomedical research has created an exciting avenue for novel investigations.
Protein kinase A
The best studied kinase, protein kinase A (PKA), is a cAMP-dependent protein kinase whose activity has been shown to increase in the hypodynamic phase of sepsis (19). Many contractile proteins are phosphorylated by PKA, which together coordinate a significant increase in calcium-mediated inotropy. Downstream proteins activated by PKA include phospholamban, troponin, ryanodine receptor and the L-type Ca2+ channel (20). Phospholamban, a negative regulator of the sarcoplasmic reticulum Ca −ATPase2 (SERCA2), has been studied extensively as a prospective target for treatment modalities in the failing heart (21). PKA-mediated phosphorylation of phospholamban at Ser16 causes its disassociation from SERCA2 permitting maximal calcium ATPase activity and sarcoplasmic reticulum calcium loading which, in turn, generates larger action potentials during systole (Figure 1) (21). In a canine model of endotoxemia, a substantial decrease in SERCA2 calcium uptake was observed and correlated with the predicted impairment of cardiac contractility (22). To further study this, Wu et al. took advantage of the observation that cecal ligation and puncture in an intact animal model of sepsis triggered two distinct hemodynamic phases: an early hyperdynamic phase and later hypodynamic phase (23). During the hyperdynamic phase in this model of sepsis there were no alterations in the SERCA2 calcium uptake, but in the hypodynamic phase the SERCA2 calcium uptake was reduced by half (23). The phosphorylation of phospholamban was also examined and shown to significantly increase in the hyperdynamic phase; however, this did not correlate with increased SERCA2 calcium uptake. Conversely, during the hypodynamic phase, phospholamban was significantly dephosphorylated and correlated with a decrease in myocardial contractility. The β-agonist, isoproterenol, expected to act through cAMP-activated PKA to cause phosphorylation of phospholamban did not ameliorate this effect (12). Together these data show that during sepsis there may be attenuated sarcoplasmic reticulum calcium uptake related to the phosphorylation state of phospholamban. This modulatory effect of phosphorylated phospholamban on SERCA2 activity may influence the development of myocardial dysfunction in sepsis. Therefore, more completely understanding the interaction of all proteins responsible for phosphorylating and dephosphorylating phospholamban is critical for identifying potential novel therapeutic targets.
Figure 1. Phospholamban (PLB) phosphorylation.
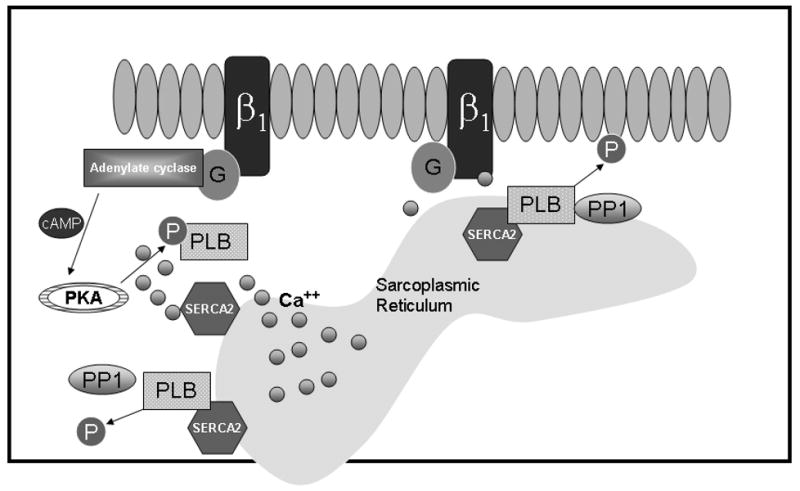
In sepsis, β-adrenergic receptors are activated by endogenous catecholamines leading to an increase in cAMP and PKA activation. PKA phosphorylates PLB, leading to separation of PLB from the SERCA2 and subsequently the uptake of calcium into the sarcoplasmic reticulum (SR) allowing for diastolic relaxation and reloading of calcium into the SR for the next contraction.
PKA-mediated phosphorylation of additional targets can similarly influence myocyte contractility. PKA-dependent phosphorylation of contractile proteins, such as troponin I and myosin binding protein C, not only enhance relaxation cycling of myofilaments but also reduce myofilament calcium sensitivity (24). In the phosphorylated state, troponin I inhibits calcium binding, prevents exposure of actin to myosin and limits the force generated during contraction so that phosphorylated troponin I has a negative inotropic effect (25). Interestingly, it was shown that PKA-dependent phosphorylation of troponin at Ser23/24 was upregulated in the myocardium of endotoxemic rats when compared to control animals. This is consistent with many studies showing that myofilament calcium responsiveness is altered in sepsis (26-28). In transgenic mice with cardiac-specific replacement of troponin I by the slow skeletal isoform, PKA-sensitive phosphorylation sites are eliminated and consequently, resistant to PKA-induced reduction in myofilament calcium sensitivity (29). An important proof of concept was achieved using these transgenic mice in a model of sepsis. These studies demonstrated that the inability to phosphorylate troponin I in the genetically altered mice substantially protected these animals from endotoxemia-induced contractile dysfunction (29).
Protein Kinase C
Although it has received less attention in the realm of sepsis investigation, protein kinase C (PKC) has been espoused to be a key inducer of pathologic cardiac hypertrophy via its activation of the AP-1 signal transduction cascade. It is believed that activation of the c-jun and fos heterodimeric transcription factor by PKC to form the AP-1 complex results in a significant increase in protein expression(30). In the early stages of sepsis, PKC is activated and its subcellular localization is altered in the cardiomyocyte (31); however, the downstream proteins modified by PKC are activated and whether they play a potential role in cardiac dysfunction in sepsis have yet to be addressed.
Phosphoinositide 3-kinases
The biologic effects of sepsis-induced activation of many kinases that have been well studied in other organs remain elusive in the heart and the cardiomyocyte specifically. Recent studies have revealed that phosphoinositide 3-kinase (PI3K), formerly identified as an important inflammatory mediator in the lung (32) and pancreas (33), may specifically alter cardiac contractility (34). Phosphoinositide 3-kinases are a family of lipid kinases that phosphorylate the 3′OH group of the inositol ring. They are divided into two classes (class I and class II) based on the receptor that leads to their activation. PI3K α, a class I PI3K, is an important regulator of cardiomyocyte hypertrophy by increasing the rate of protein synthesis in the cell (34) PI3K γ, a class II PI3K, negatively modulates cardiac contractility (34). In models of ventricular pressure overload, PI3K γ is activated and is thought to act in multiple ways to modulate inotropic function (Figure 2) (35). PI3K γ activation may lower baseline levels of cAMP and suppress phospholamban phosphorylation with subsequent attenuation of SERCA2 activity(34). It has also been thought to act by direct interaction with β–adrenergic receptor kinase-1 (βARK 1) in facilitating endocytic desensitization of β-adrenergic receptors (36). Recent data has shown that PI3K γ also interacts with the phosphodiesterase inhibitor PDE3b leading to its activation and subsequent breakdown of camp (37). In cultured cardiomyocytes, PI3K γ has also been shown to be involved in calcium regulation. Inhibition of PI3K γ reduced the depletion of calcium stored in the sarcoplasmic reticulum and altered calcium cycling by modulating the L-type calcium channel (38). These data reflect an increasing body of data that is examining the role of PI3K γ in cardiac dysfunction. Whether these elucidated mechanisms are similar in the biology of sepsis-induced myocardial dysfunction remains an active area of investigation.
Figure 2. Proposed mechanisms of PI3K γ.

Changes in PI3K γ activity leads to altered myocardial function. Many mechanisms have been hypothesized for the negative inotropic effect of PI3K γ. Interactions between PI3K γ and both PDE3B and βARK1 have been reported.
To date, the inability to clearly define the role of PI3K in the biology of myocardial dysfunction has precluded it from being a specific therapeutic target. For instance, independent of sepsis studies, many investigations of the failing heart have concluded that PI3K γ acts as a negative inotrope. In contrast, other studies of myocardial injury have shown that the activation of all the PI3K isoforms, including PI3K γ, may provide a cardioprotective signal (39-41) Recent studies from our own lab have tried to gain further insight into this controversy. In a mouse model of endotoxemia, we have found that PI3K activity and the phosphorylation of downstream mediators are increased in the myocardium (Figure 3, unpublished data). Our data, which is consistent with other models advocating a cardioprotective role (39, 41), revealed that PI3Kγ deficient mice have a dramatic increase in mortality following induction of endotoxemia. Although the etiology of death may be multifactorial, echocardiographic indices of their systolic function reveal extremely poor function. Further results from our laboratory using a mouse model of sepsis have identified changes in many interrelated signaling cascades related to the PI3K pathway including changes in NF-κB activation, increased inducible nitric oxide synthase transcription, and AKT activation.
Figure 3. PI3K γ activity in murine endotoxemia.

Thin layer chromatography of myocardial tissue 6 and 12 hours following intraperitoneal LPS injection compared to control (saline injection).
Furthermore, these changes in intracellular signaling appear to lead to an increase in myocardial apoptosis. Although apoptosis is essential during development of the heart, it also may occur as a dysregulated response to injury and may explain the poor function observed in endotoxemic PI3K γ deficient mice. Further studies using models of sepsis, evaluated in the context of results from other models of altered myocardial function, should ultimately provide further clarification of the role of PI3K in mediating myocardial dysfunction.
Counter-regulation of kinases by phosphatases
As reviewed above, the activity of protein kinases are essential to the fine-tuned regulation of cardiac regulatory proteins and appear to play critical role in pathologic states of cardiac dysfunction. The cell physiologic process of kinase-dependent phosphorylation is counter-regulated by a family of de-phosphorylating proteins called phosphatases. Two general families of phosphatases are known, those that principally target serine or threonine residues (Ser/Thr phosphatases) and those that principally target tyrosine residues (Tyr phosphatases) (Reviewed in (42)).
Role of Phosphatases in the Heart
With regards to the pathophysiology of sepsis-induced myocardial depression, little is known about the role that phosphatases play in opposing the action of the PKA, PKC, and PI3 kinases. Two Ser/Thr phosphatases, known to play active roles in the normal physiology of the heart are protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A). These phosphatases are important regulators of many components of the excitation-coupling cascade. For example, PP2A seems to regulate myofilament function through the de-phosphorylation of troponin I in response to calcium (43). In transgenic mice, overexpression of PP2A in the myocardium significantly decreased the phosphorylation state of both phospholamban and troponin and led to increased necrosis, fibrosis, and poor cardiac contractility (44). Other studies have pointed to PP1 as a phosphatase targeting phospholamban which is responsible for regulating sarcoplasmic reticulum function by influencing uptake and release of calcium (45). The activity of PP1 is regulated by the two inhibitors, Inh-1 and Inh-2, which are activated by PKA-dependent phosphorylation. In heart failure models, increased PP1 activity is partially due to dephosphorylation and inactivation of its modifying inhibitor, Inh-1. As a result, the increase activity of PP1 promotes dephosphorylation of phospholamban and inhibition of the sarcoplasmic reticulum calcium-pump. Kranias et al reported that an increase in the activity of Inh-1 resulted in selective enhancement of phospholamban phosphorylation and augmentation of cardiac contractility (46). Thus, Inh-1 may represent an attractive novel therapeutic target for heart failure secondary to many insults; however, whether this same biology extends to sepsis-induced myocardial dysfunction remains a focus of on-going studies.
Although published data implies that disturbances in protein phosphatase expression and activity may cause or aggravate cardiac failure, there is no data on the role of phosphatases in the heart during sepsis. However, there is reason to believe this will be the case, as there is evidence of altered phosphatase activity in the lung in a murine model of sepsis (42). Thus, data derived from other organ systems affected by sepsis support our hypothesis that alteration in the balance between kinases and phosphatases may play a key role in the cardiac dysfunction induced by sepsis. For example, in preliminary studies we have observed a two-fold increase in PP2A activity in the heart as well as in isolated cardiomyocytes following lipopolysaccharide treatment. In an animal model of endotoxemia, PP2A co-localized to the myofilament and led to changes in the phosphorylation state of troponin I (unpublished data). Alterations in the degree of troponin I phosphorylation, known to disrupt the calcium sensitivity of the myofilament, are likely to result in poor contractility. Currently, studies to examine the effects on the heart of administering a selective PP2A inhibitor to the endotoxemic animal are being conducted.
Conclusion
Although the evolution, severity, and temporal manifestation of heart failure may vary considerably, the characteristics of abnormal ventricular performance seem comparable among many models that include sepsis. It has become clear that abnormal calcium sensitivity and alterations in cardiac contractile proteins result in ventricular dysfunction. As we now know these processes are regulated by protein phosphorylation in sepsis, it seems logical that among the multiple cellular pathways activated in the failing heart, specific phosphatases and/or kinases are going to play a crucial role in contractile dysfunction. Unfortunately, by the shear complexity of the systemic inflammatory response triggered by sepsis, numerous experimental obstacles stand in our way of achieving further understanding of this state of myocardial failure. Furthermore, it is important to recognize the dynamic nature of this pathobiology, in that distinct phases of sepsis, may require a different mechanistic understanding and therefore, a different therapeutic strategy. Nevertheless, a growing body of intriguing data continues to be derived from the use of various models of heart failure that are continuing to elucidate those key components, including specific kinases and phosphatases, that are regulating alterations in myocardial function. The ultimate hope is that in gaining a greater understanding of the myocardial response to septic shock, novel therapeutic targets will be identified and enable clinicians at the bedside to more effectively manage the patient with sepsis.
Acknowledgments
This work was supported by RO1 GM66839-01 to TPS and American Academy of Pediatrics Section on Cardiology and Cardiac Surgery Research Fellowship Award and American Heart Association Post-doctoral Fellowship Award (0425371B) to AL.
References
- 1.DuPont HL, Spink WW. Infections due to gram-negative organisms: an analysis of 860 patients with bacteremia at the University of Minnesota Medical Center, 1958-1966. Medicine (Baltimore) 1969;48:307–32. doi: 10.1097/00005792-196907000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Carcillo JA, Fields AI. Clinical practice parameters for hemodynamic support of pediatric and neonatal patients in septic shock. J Pediatr (Rio J) 2002;78:449–66. [PubMed] [Google Scholar]
- 3.Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167:695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 4.Parker MM, Shelhamer JH, Natanson C, Alling DW, Parrillo JE. Serial cardiovascular variables in survivors and nonsurvivors of human septic shock: heart rate as an early predictor of prognosis. Crit Care Med. 1987;15:923–9. doi: 10.1097/00003246-198710000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Pollack MM, Fields AI, Ruttimann UE. Sequential cardiopulmonary variables of infants and children in septic shock. Crit Care Med. 1984;12:554–9. doi: 10.1097/00003246-198407000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Pollack MM, Fields AI, Ruttimann UE. Distributions of cardiopulmonary variables in pediatric survivors and nonsurvivors of septic shock. Crit Care Med. 1985;13:454–9. doi: 10.1097/00003246-198506000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Balk RA. Severe sepsis and septic shock. Definitions, epidemiology, and clinical manifestations. Crit Care Clin. 2000;16:179–92. doi: 10.1016/s0749-0704(05)70106-8. [DOI] [PubMed] [Google Scholar]
- 8.Parrillo JE, Burch C, Shelhamer JH, Parker MM, Natanson C, Schuette W. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. J Clin Invest. 1985;76:1539–53. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, Michael P, Brabant D, Parissenti AM, Ramana CV, Xu X, Parrillo JE. Human serum from patients with septic shock activates transcription factors STAT1, IRF1, and NF-kappaB and induces apoptosis in human cardiac myocytes. J Biol Chem. 2005;280:42619–26. doi: 10.1074/jbc.M508416200. [DOI] [PubMed] [Google Scholar]
- 10.Celis R, Torre-Martinez G, Torre-Amione G. Evidence for activation of immune system in heart failure: is there a role for anti-inflammatory therapy? Curr Opin Cardiol. 2008;23:254–60. doi: 10.1097/HCO.0b013e3282fbfbc7. [DOI] [PubMed] [Google Scholar]
- 11.Levy RJ, Deutschman CS. Evaluating myocardial depression in sepsis. Shock. 2004;22:1–10. doi: 10.1097/01.shk.0000129198.53836.15. [DOI] [PubMed] [Google Scholar]
- 12.Wu LL, Ji Y, Dong LW, Liu MS. Calcium uptake by sarcoplasmic reticulum is impaired during the hypodynamic phase of sepsis in the rat heart. Shock. 2001;15:49–55. doi: 10.1097/00024382-200115010-00008. [DOI] [PubMed] [Google Scholar]
- 13.Ruan Q, Nagueh SF. Usefulness of isovolumic and systolic ejection signals by tissue Doppler for the assessment of left ventricular systolic function in ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2006;97:872–5. doi: 10.1016/j.amjcard.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 14.Vogel M, Cheung MM, Li J, Kristiansen SB, Schmidt MR, White PA, Sorensen K, Redington AN. Noninvasive assessment of left ventricular force-frequency relationships using tissue Doppler-derived isovolumic acceleration: validation in an animal model. Circulation. 2003;107:1647–52. doi: 10.1161/01.CIR.0000058171.62847.90. [DOI] [PubMed] [Google Scholar]
- 15.Vlahos CJ, McDowell SA, Clerk A. Kinases as therapeutic targets for heart failure. Nat Rev Drug Discov. 2003;2:99–113. doi: 10.1038/nrd1009. [DOI] [PubMed] [Google Scholar]
- 16.Neumann J. Altered phosphatase activity in heart failure, influence on Ca2+ movement. Basic Res Cardiol. 2002;97 1:I91–5. doi: 10.1007/s003950200036. [DOI] [PubMed] [Google Scholar]
- 17.Jones WK, Brown M, Ren X, He S, McGuinness M. NF-kappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc Toxicol. 2003;3:229–54. doi: 10.1385/ct:3:3:229. [DOI] [PubMed] [Google Scholar]
- 18.Sulakhe PV, Vo XT. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol Cell Biochem. 1995;149-150:103–26. doi: 10.1007/BF01076569. [DOI] [PubMed] [Google Scholar]
- 19.Yang SL, Hsu C, Lue SI, Hsu HK, Liu MS. Protein kinase a activity is increased in rat heart during late hypodynamic phase of sepsis. Shock. 1997;8:68–72. doi: 10.1097/00024382-199707000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 21.Kiriazis H, Kranias EG. Genetically engineered models with alterations in cardiac membrane calcium-handling proteins. Annu Rev Physiol. 2000;62:321–51. doi: 10.1146/annurev.physiol.62.1.321. [DOI] [PubMed] [Google Scholar]
- 22.Liu MS, Wu LL. Reduction in the Ca2(+)-induced Ca2+ release from canine cardiac sarcoplasmic reticulum following endotoxin administration. Biochem Biophys Res Commun. 1991;174:1248–54. doi: 10.1016/0006-291x(91)91555-q. [DOI] [PubMed] [Google Scholar]
- 23.Wu LL, Tang C, Dong LW, Liu MS. Altered phospholamban-calcium ATPase interaction in cardiac sarcoplasmic reticulum during the progression of sepsis. Shock. 2002;17:389–93. doi: 10.1097/00024382-200205000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–65. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 25.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 26.Tavernier B, Mebazaa A, Mateo P, Sys S, Ventura-Clapier R, Veksler V. Phosphorylation-dependent alteration in myofilament ca2+ sensitivity but normal mitochondrial function in septic heart. Am J Respir Crit Care Med. 2001;163:362–7. doi: 10.1164/ajrccm.163.2.2002128. [DOI] [PubMed] [Google Scholar]
- 27.Tavernier B, Li JM, El-Omar MM, Lanone S, Yang ZK, Trayer IP, Mebazaa A, Shah AM. Cardiac contractile impairment associated with increased phosphorylation of troponin I in endotoxemic rats. Faseb J. 2001;15:294–6. doi: 10.1096/fj.00-0433fje. [DOI] [PubMed] [Google Scholar]
- 28.Powers FM, Farias S, Minami H, Martin AF, Solaro RJ, Law WR. Cardiac myofilament protein function is altered during sepsis. J Mol Cell Cardiol. 1998;30:967–78. doi: 10.1006/jmcc.1998.0661. [DOI] [PubMed] [Google Scholar]
- 29.Layland J, Cave AC, Warren C, Grieve DJ, Sparks E, Kentish JC, Solaro RJ, Shah AM. Protection against endotoxemia-induced contractile dysfunction in mice with cardiac-specific expression of slow skeletal troponin I. Faseb J. 2005;19:1137–9. doi: 10.1096/fj.04-2519fje. [DOI] [PubMed] [Google Scholar]
- 30.Shubeita HE, Martinson EA, Van Bilsen M, Chien KR, Brown JH. Transcriptional activation of the cardiac myosin light chain 2 and atrial natriuretic factor genes by protein kinase C in neonatal rat ventricular myocytes. Proc Natl Acad Sci U S A. 1992;89:1305–9. doi: 10.1073/pnas.89.4.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang SL, Hsu C, Lue SI, Hsu HK, Yang J, Liu MS. Protein kinase C activity is increased in rat heart during the early hyperdynamic phase of sepsis. Shock. 1998;9:199–203. doi: 10.1097/00024382-199803000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Lionetti V, Lisi A, Patrucco E, De Giuli P, Milazzo MG, Ceci S, Wymann M, Lena A, Gremigni V, Fanelli V, Hirsch E, Ranieri VM. Lack of phosphoinositide 3-kinase-gamma attenuates ventilator-induced lung injury. Crit Care Med. 2006;34:134–41. doi: 10.1097/01.ccm.0000190909.70601.2c. [DOI] [PubMed] [Google Scholar]
- 33.Lupia E, Goffi A, De Giuli P, Azzolino O, Bosco O, Patrucco E, Vivaldo MC, Ricca M, Wymann MP, Hirsch E, Montrucchio G, Emanuelli G. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Pathol. 2004;165:2003–11. doi: 10.1016/s0002-9440(10)63251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–49. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 35.Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–87. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 36.Naga Prasad SV, Jayatilleke A, Madamanchi A, Rockman HA. Protein kinase activity of phosphoinositide 3-kinase regulates beta-adrenergic receptor endocytosis. Nat Cell Biol. 2005;7:785–96. doi: 10.1038/ncb1278. [DOI] [PubMed] [Google Scholar]
- 37.Alloatti G, Montrucchio G, Lembo G, Hirsch E. Phosphoinositide 3-kinase gamma: kinase-dependent and -independent activities in cardiovascular function and disease. Biochem Soc Trans. 2004;32:383–6. doi: 10.1042/bst0320383. [DOI] [PubMed] [Google Scholar]
- 38.Marcantoni A, Levi RC, Gallo MP, Hirsch E, Alloatti G. Phosphoinositide 3-kinasegamma (PI3Kgamma) controls L-type calcium current (ICa,L) through its positive modulation of type-3 phosphodiesterase (PDE3) J Cell Physiol. 2006;206:329–36. doi: 10.1002/jcp.20467. [DOI] [PubMed] [Google Scholar]
- 39.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–38. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102:102–9. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 41.Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–3. [PubMed] [Google Scholar]
- 42.Shanley TP, Vasi N, Denenberg A, Wong HR. The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol. 2001;166:966–72. doi: 10.4049/jimmunol.166.2.966. [DOI] [PubMed] [Google Scholar]
- 43.Jaquet K, Thieleczek R, Heilmeyer LM., Jr Pattern formation on cardiac troponin I by consecutive phosphorylation and dephosphorylation. Eur J Biochem. 1995;231:486–90. doi: 10.1111/j.1432-1033.1995.tb20722.x. [DOI] [PubMed] [Google Scholar]
- 44.Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279:40827–34. doi: 10.1074/jbc.M405770200. [DOI] [PubMed] [Google Scholar]
- 45.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–35. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]