

Abstract
Gene delivery biomaterials need to be designed to efficiently achieve nuclear delivery of plasmid DNA. Polycations have been used to package DNA and other nucleic acids within sub-micron sized particles, offering protection from shear-induced or enzymatic degradation. However, cytotoxicity issues coupled with limited in vivo transfection efficiencies minimize the effectiveness of this approach. In an effort to improve upon existing technologies aimed at delivering nucleic acids, an alternative approach to DNA packaging was explored. Peptide nucleic acids (PNAs) were used to directly functionalize DNA with poly(ethylene glycol) (PEG) chains that provide a steric layer and inhibit multimolecular aggregation during complexation. DNA prePEGylation by this strategy was predicted to enable the formation of more homogeneous and efficiently packaged polyplexes.
In this work, DNA-PNA-peptide-PEG (DP3) conjugates were synthesized and self-assembled with 25 kDa poly(ethylenimine) (PEI). Complexes with small standard deviations and average diameters ranging from 30 – 50 nm were created, with minimal dependence of complex size on N:P ratio (PEI amines to DNA phosphates). Furthermore, PEI-DNA interactions were altered by the derivitization strategy, resulting in tighter compaction of the PEI-DP3 complexes in comparison with PEI-DNA complexes. Transfection experiments in Chinese Hamster Ovary (CHO) cells revealed comparable transfection efficiencies but reduced cytotoxicities of the PEI-DP3 complexes relative to PEI-DNA complexes. The enhanced cellular activities of the PEI-DP3 complexes were maintained following the removal of free PEI from the PEI-DP3 formulations, whereas the cellular activity of the conventional PEI-DNA formulations was reduced by free PEI removal. These findings suggest that DNA prePEGylation by the PNA-based strategy might provide a way to circumvent cytotoxicity and formulation issues related to the use of PEI for in vivo gene delivery.
Introduction
Non-viral, biomimetic nucleic acid delivery systems have been developed with the aim of designing modular, targeted, and safe constructs for gene therapy. Such an approach has the potential to bring about practical pharmaceutical products with great improvement in combating disease. While viral vectors have achieved efficient delivery of exogenous DNA, their innate immunogenicity and toxicity have raised significant concerns over their clinical development. Despite these limitations, the multiple functionalities and responsiveness of these viral systems provide key insight into how biomimetic vehicles can be more effectively designed.
One widely explored method of non-viral vehicle formulation involves the use of polycations to condense DNA into nano- to submicron-scale particles.(1-5) For example, poly(ethylenimine) (PEI) is a cationic polymer that contains a high density of primary, secondary, and tertiary amine groups, and has been used to package DNA and RNA within nanosized complexes.(2, 5-7) Polyplexes made from PEI constitute a highly effective delivery system in vitro, an effect which has been attributed to the capacity of PEI to promote endosomal escape via pH-buffering. (8-10) Unfortunately, concerns about the toxicity of PEI (11) and challenges in formulating PEI-based vehicles have limited its applicability in vivo. Cationic polyplexes tend to aggregate and interact with serum proteins after systemic administration, leading to their rapid phagocytic elimination. (12, 13) They are also non-specifically internalized by liver cells. (14-16)
One way to overcome the serum instability and non-specificity of polyplexes is through the steric stabilization of polyplexes with hydrophilic, uncharged polymers. Poly(ethylene glycol) (PEG) has been conjugated to PEI and other DNA-condensing polymers via reaction with primary amine groups along the backbone of the polycation, resulting in enhanced polyplex stability.(6, 17-20) To enhance cell specific uptake, cell-targeting and other functional peptides have been added directly to the polycation or to the conjugated PEG.(2, 6, 21-24) The PEGylation and functionalization of polycationic gene delivery carriers has been explored both pre- and postcomplexation with DNA. Precomplexation PEGylation, involving the complexation of DNA with PEGylated complexation agents, was developed with the intention of minimizing polyplex aggregation during formulation. Polyplexes are inherently heterogeneous due to uncontrolled self-aggregation during the formulation process. Consequently, PEGylated complexation agents have the potential to produce more monodisperse particles. (22, 25, 26) However, the charge density of the polycation is reduced by PEGylation, making precomplexation PEGylation ineffective for low molecular weight polycations, (3, 27) and in general, lowering the condensation efficiency. (4, 25, 26, 28-30)
The addition of PEG postcomplexation can be accomplished by PEG grafting onto surface-exposed amine groups on the polyplex, and has been shown to effectively stabilize PEI-based polyplexes containing high molecular weight PEI; (6) in contrast, polyplexes formed from lower molecular weight polycations can be destabilized by this process. (31) Surface PEGylation has also been shown to alter the intracellular trafficking of particles and to reduce their gene transfer capability.(20) In an effort to circumvent these limitations, non-disruptive PEGylation strategies have been pursued. For example, cyclodextrin-based polycations have been developed that allow DNA complexation and PEGylation in a single-step reaction. PEGylation occurs by inclusion complex formation between adamantine-PEGs and cyclodextrins, and has no adverse effects on DNA-cation interactions.(22, 31, 32)
To enable the formulation of polyplexes appropriate for systemic administration and efficient cellular utilization, we have explored a novel formulation strategy that employs peptide nucleic acid (PNA)-peptide conjugates as linkers for the direct and reversible PEGylation of DNA. PNAs are nucleic acid analogs that contain a peptide backbone and can hydrogen bond to complementary DNA (or RNA) via either conventional Watson-Crick base pairing in the anti-parallel direction or Hoogsteen base pairing in the parallel direction. Because PNAs contain a neutral backbone, the PNA-DNA duplex is more stable than the corresponding DNA-DNA duplex, and PNAs can interact with one strand of double-stranded DNA by strand invasion.(33-35) Symmetric PNA oligomers linked by a flexible spacer (PNA “clamps”; Figure 1) form stable PNA-DNA-PNA triplexes.(35-38) PNA clamp binding to plasmid DNA outside of the coding region does not interfere with transcriptional activity, and has been previously used to label plasmids with fluorophores and other tags that enable DNA tracking or targeting during gene delivery.(39, 40)
Figure 1.
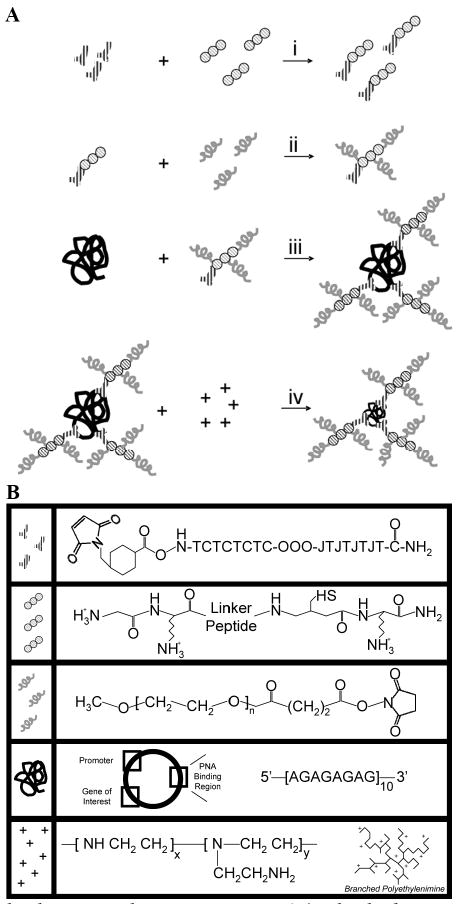
PNA-Based Vehicle Formulation Design. (A) The linker peptide (circles; see detailed expansion in [B]) was bound to the PNA (“Y”s) by the reaction of the cysteine thiol with the terminal PNA maleimide (i). Then, succinimidyl-PEG chains (grey squiggles) were bound to the amines on the peptide N-terminus and lysine residues (ii). The PNA-peptide-PEG conjugates were purified and then bound to GWiz plasmid DNA (black scribble) via sequence-specific hydrogen bonding interactions (iii). The final DNA-PNA-peptide-PEG (DP3) conjugate was purified, and 25 kDa branched PEI (+) was added to complex the DNA (iv). (B) Detailed structures of the components of the DP3 conjugates. Figure not to scale.
Our formulation design scheme (Figure 1) employs PNA clamps as linkage points for the direct functionalization of plasmid DNA with protease-sensitive peptides and PEG. The peptide linker serves as a cleavable bridge that will allow cell-triggered dePEGylation (by protease secretion), which we expect should promote improved DNA internalization. Furthermore, because PEG prevents polyplex aggregation, we expect that prePEGylation of the DNA followed by polyplex formation should enable the creation of single plasmid complexes with small standard deviations and size, without compromising the efficiency of polycation-DNA compaction. Precise particle size control during formulation can be beneficial in overcoming a variety of physiological barriers such as vascular extravasation (41-45), interstitial transport (46, 47), and cellular internalization (48-53). For example, studies on the design requirements for effective transcapillary flux of antibody therapies, have demonstrated that smaller, positively charged molecules are more efficient at vascular extravasation. (54-56) With respect to interstitial transport, smaller adenoviral particles (∼25 nm) move more efficiently than larger particles (∼100 nm) through multicellular extracellular matrix spheroids.(47) Furthermore, previous studies have suggested that particles 50 nm or less are optimal for receptor-mediated endocytosis.(57, 58) Coincidentally, DNA polyplexes containing a single plasmid are believed to be approximately 30 nm in diameter.(59) Hence, methods to form homogeneous, small particles have clear advantages with respect to in vivo efficacy as well as from a manufacturing/patient-dosing perspective.
In this work, we demonstrate the development of DNA-PNA-peptide-PEG (DP3) conjugates, and observe the corresponding effect of DNA prePEGylation on polyplex formation and in vitro bioactivity. The starting components for these polyplexes (DP3 conjugates and PEI) represent stable compounds, and the methods for polyplex formation are amenable to bedside formulation. Furthermore, with the first demonstration of direct DNA PEGylation for gene therapy, we show that we can form 30-50 nm particles of a homogeneous size distribution that have improved nucleic acid packaging efficiency, low cytotoxicity, and activity that is independent of free PEI in solution. Proof-of-principle of our PNA-based design will lay the foundation for its exploration in vivo.
Materials and Methods
Reagents and Materials
PNA clamps (AlexaFluor488/AlexaFluor555/maleimide-TCTCTCTC-OOO-JTJTJTJT-CONH2, (34) where O = 8-amino-3,6-dioxaoctanoic acid and J = pseudoisocytosine) with either an AlexaFluor488, AlexaFluor555, or maleimide terminus were synthesized by solid-phase synthesis and HPLC-purified to >95% purity by Panagene (Daejeon, Korea). The GWiz green fluorescent protein (GFP)-expressing plasmid containing 10 sequential copies of the repeated PNA binding sequence 5′-AGAGAGAG-3′ was purchased from Genlantis (San Diego, CA) (see Supplementary Figure 1D for the sequence of the PNA binding region), and the non-PNA binding plasmid (SV40-Renilla) was purchased from Promega (Madison, WI). Both plasmids were amplified in E. coli and purified with a Qiagen MegaPrep kit. Briefly, competent DH-5α cells were transformed with the appropriate plasmid and cultured overnight in Lysogeny Broth. Cells were lysed and the amplified plasmid was purified and recovered in accordance with the manufacturer's protocols. A 22 amino acid linker peptide (NH2-GKGGPQG/IWGQGRGDSPGDRCK-CONH2) was synthesized by solid-phase synthesis and HPLC-purified to >95% by Global Peptide Services (Huntsville, AL). This peptide contains an MMP-1-sensitive region (boldfaced with the exact cleavage site indicated by a backslash).(60) α–methoxy, ω-succinimide poly(ethylene glycol) (MW=5,000 g/mol) was synthesized, purified, and analyzed by gel permeation chromatography (GPC) and Fourier transform infrared spectroscopy (FTIR) by Polymer Source, Inc. (Montreal, Canada). 25 kDa branched poly(ethylenimine) (PEI) was purchased from Sigma (St. Louis, MO). Polymer molecular weight was validated by light scattering and GPC by the manufacturer.
Conjugate Development and Particle Formation
PNA-Peptide-PEG Conjugate Formation
To form PNA-peptide conjugates, 2 μM of the maleimide-terminated PNA and 0.2 μM of peptide were incubated in 100 μL of 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4) overnight at 37°C. A 10-fold excess of PNA was used in this reaction to minimize the presence of unreacted peptide at the end of the reaction. To derivatize the PNA-peptides with succinimide-terminated PEG, 1.25 μL of 4 mM PEG in 20 mM HEPES buffer (pH 7.4) was added to the PNA-peptide reaction mixture (corresponding to a 250-fold molar excess), and the resulting solution was incubated overnight at 37°C. Following this reaction, unreacted components were removed via centrifugal filtration with a YM-10 centrifugal filtration column (10,000 Da molecular weight cut-off [MWCO], Millipore, Billerica, MA). For this process, the membrane was first primed with a 500 μL distilled water rinse at 12,000g for 30 minutes in a table-top centrifuge. Following this rinse, sample was added to the column and centrifuged for 30 minutes at 12,000g. Three separate wash steps followed; in each wash, 500 μL of distilled water was added to the column and subsequently centrifuged for 30 minutes at 12,000g. To collect the retentate at the end of the washes, the membrane was inverted into a clean tube, and 20 μL of distilled water was added to the membrane. The column was then centrifuged for 2 minutes at 12,000g. The final recovered volumes were raised to 100 μL by the addition of 20 mM HEPES buffer (pH 7.4).
Estimation of PNA-DNA Hybridization Efficiency
0.6 pM of the GWiz PNA-binding plasmid or of the SV40-Renilla non-PNA-binding plasmid was incubated with 0 – 200 pM of AF-PNA in a 10 mM HEPES/50 mM NaCl Buffer (pH 7.4) overnight at 37°C. After incubation, the samples were analyzed by gel electrophoresis on a 1% agarose gel, and the gel was stained with a 1% ethidium bromide solution and visualized on a Bio-Rad Gel Doc XR (Hercules, CA).
DP3 Formation by Hybridization of DNA with PNA-Peptide-PEG Conjugates
6 pM of DNA was added to the purified PNA-peptide-PEG conjugates in a total volume of 100 μL of 20 mM HEPES buffer (pH 7.4), and the mixture was allowed to incubate overnight at 37°C.(39) Unbound PNA-peptide-PEG was removed via centrifugal filtration by a similar procedure as that described for the PNA-peptide-PEG conjugates; a YM-100 column was used (100,000 MWCO, Millipore) and spin times were reduced to 10 minutes per spin to prevent membrane clogging.
Assessment of DP3 Conjugate Stoichiometry
Tryptophan fluorescence analyses were performed on a Jasco FP-6500 spectrofluorimeter (excitation λ = 280 nm; emission λ = 350 nm). An ETC-273T temperature controller and a Julabo AWC-100 water chiller enabled sample analysis to be done at 25°C. For analysis of peptide standards, peptide was dissolved in a 20 mM HEPES buffer (pH 7.4) at the indicated concentrations and assessed fluorimetrically.
UV/Vis spectroscopy measurements were performed on a Nanodrop ND 1000 spectrophotometer (ND-1000) (Thermo Scientific – Nanodrop Products, Wilmington, DE). DNA concentrations were determined as per the manufacturer's protocol. Briefly, 2 μL of sample were placed onto the sample platform. The absorbances at 230 nm, 260 nm, and 280 nm were analyzed, and a corresponding concentration and measure of DNA purity were assessed.
Attenuated total reflectance FTIR (ATR-FTIR) analyses were performed on a Spectrum One FTIR Spectrometer (Perkin Elmer, Waltham, MA). 10 μL of each sample were dried with air onto a diamond ATR crystal, and 200 runs per sample were subsequently conducted under ambient conditions. Spectra were acquired between 4000 cm-1 and 1000 cm-1.
DNA Complexation
25 kDa PEI solutions were added to DP3 conjugate solutions or free plasmid solutions at varying N:P (PEI nitrogen/DNA phosphate) ratios, following existing protocols.(5) Briefly, a 500 μL PEI solution was added to a 500 μL DP3 (or free plasmid) solution containing 20 μg of plasmid. The PEI solution was added to the DNA solution in a drop-wise fashion while vortexing, and the combined solutions were allowed to form complexes at room temperature for 10 minutes prior to sample analysis.
DNA Complex Purification
Free PEI was removed from the suspensions of PEI complexes by centrifugal filtration of the complex suspensions with a YM-100 filter. First, the filters were primed with 500 μL of double distilled water (ddH2O) for 5 minutes at 12,000g. Then, 500 μL of complex suspension were added and centrifuged at 100g for 30 minutes. One 500 μL wash was performed with ddH20. Complex sizes were assessed via dynamic light scattering (DLS) prior to and following purification.
Polyplex Characterization
Dynamic Light Scattering (DLS) and Zeta Potential Analyses
DLS and zeta potential analyses were performed at a 90° angle with a Brookhaven (Holtsville, NY) BI9000AT correlator and a Lexel (Fremont, CA) model 95 argon ion laser operating at 488 nm. All measurements were performed at 20 ± 0.2°C, with sample temperature controlled by an external circulating bath. For DLS, the CONTIN algorithm was used to analyze the autocorrelation functions and estimate the mean diameters and size distributions of the complexes formed. The measured effective hydrodynamic diameters for each sample run were the average values from a total of 5 runs of 2 minutes each, while the measured zeta potentials for each sample run were the average of 10 runs of 2 minutes each.
Atomic Force Microscopy
Atomic force microscopy (AFM) was performed on a Nanoscope IIIa atomic force microscope (Digital Instruments, Santa Barbara, CA) used in conjunction with an MLCT cantilever tip (60 nm backside gold coating, 3 μm tip, Veeco, Santa Barbara, CA). Contact mode AFM images were collected under ambient conditions at approximately 25°C. Samples were prepared by the complex formulation procedure described above. 50 μL of complex suspension containing 1 μg of DNA was dried for 24 hours onto a 77 mm2 mica substrate. Height and deflection error modes were used for imaging, with scan rates of ∼1 Hz. Image processing was performed using the Nanoscope IIIa software package.
Scanning Electron Microscopy
Scanning electron microscopy (SEM) was performed on a Hitachi S-4700 field emission scanning electron microscope (Pleasanton, CA). 50 μL of complex suspension containing 1 μg of DNA was prepared as described and dried overnight onto a 127 mm2 glass coverslip, fixed with glutaraldehyde, and treated with osmium tetroxide via standard protocols. The complexes were then sputtered with gold, and the sputtered samples were fixed in the sample holder and placed in the vacuum chamber of the microscope under low vacuum (10-3 torr) for imaging.
Ethidium Bromide Exclusion Assays and Nuclease Protection Assays
Ethidium bromide exclusion assays were performed by agarose gel electrophoresis. Complexes were formed as described, over a range of N:P ratios from 0 to 10. 10 μL of each sample were analyzed on 1% agarose gels formed in the presence of 1% ethidium bromide, and the resulting gels were imaged as described.
Nuclease protection assays were conducted by a modification of existing protocols. (22) Briefly, 10 μL of 100% mouse serum (Sigma) were added to 10 μL of complex suspension (200 ng of DNA) and the mixture was incubated for 4 hours at 37°C. Subsequently, termination buffer (labeled STOP) containing 38 mM EDTA, 154 mM NaOH, and 38 mM NaCl was added to stop the serum degradation process, and the resulting mixture was incubated for 15 minutes at 4°C. DNA was then displaced from the complexes by incubation with 1 mg/mL heparin (stock solution of 10 mg/mL) (Sigma). Samples were then analyzed by agarose gel electrophoresis as described.
In Vitro Transfection and Viability Assays
Cell Culture
Chinese hamster ovary (CHO) cells (ATCC, Manassas, VA) were maintained in a humidified, 37°C atmosphere in Dulbecco's modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 100 μg/mL penicillin, and 100 μg/mL streptomycin (Invitrogen).
Transfection
For transfection, 200,000 cells were seeded into each well of a 6 well dish and the cells were grown overnight. Immediately prior to transfection, the growth medium was removed and replaced with 2 mL of serum free Opti-Mem medium (Invitrogen). 500 μL of complex suspension (containing 10 μg DNA) was formed and added to the cells in a drop-wise manner. The complexes were then allowed to incubate with the cells in serum free medium for 3 hours. At that time, the serum free medium was removed, the cells were washed in PBS, and growth medium was added. The cells were analyzed by flow cytometry and fluorescence microscopy 24 hours after transfection. For flow cytometry, the cells were trypsinized, washed in 0.1% BSA, and reconstituted in PBS for analysis with a BD Biosciences FACSCalibur cytometer (San Jose, CA). For fluorescence microscopy, live cells were imaged with a Leica microscope according to established protocols.
Viability
For cell viability assays, an ethidium homodimer dye (Invitrogen) was used to stain for dead cell nuclei. 4 μL of a 2 mM dye solution was added to the transfected cell cultures, and the cells and dye were incubated for 10 minutes at room temperature. The cells were subsequently imaged by fluorescence microscopy.
Results
DP3 Conjugate Formation
PNA-Peptide-PEG Conjugate Formation
The PNA-peptide-PEG conjugates were formed by the step-wise reaction of the individual components, as summarized in Figure 1. In the first step, PNA-peptide conjugates were formed by the reaction of the N-terminal PNA maleimide groups on the maleimide-terminated PNAs with the cysteine thiol residues near the C-terminus of the peptides. Next, the N-terminus and the two lysine side chain amine groups on the peptides were reacted with succinimide-terminated PEG. The PNA-peptide-PEG conjugates were purified by centrifugal microconcentration, and the stoichiometry of these conjugates was assessed by fluorescence spectroscopy and ATR-FTIR, as discussed below.
PNA-Peptide-PEG Hybridization with Plasmid DNA
To estimate the PNA-peptide-PEG:DNA incubation ratio that would enable saturation of the DNA, the GWiz PNA-binding plasmid and the SV40-Renilla non-PNA-binding plasmid were incubated with AlexaFluor488-labeled PNA (AF-PNA) over a range of ratios to enable PNA strand invasion and PNA-DNA hydrogen bonding. The resulting mixtures were analyzed by gel electrophoresis (Supplementary Figure 1). An approximate 3-fold molar site excess of AF-PNA:PNA binding sites (equal to a 30-fold molar excess of AF-PNA:GWiz) was effective at achieving saturation of GWiz DNA with PNA, consistent with previous work (39), whereas no binding of AF-PNA to the SV40-Renilla DNA was observed.
To form the hybridized DP3 conjugates, the GWiz PNA-binding plasmid was incubated with the PNA-peptide-PEG conjugates, and the DP3 conjugates were purified by centrifugal microconcentration. Intrinsic fluorescence spectroscopy and ATR-FTIR spectroscopy were used to analyze the stoichiometry of the DP3 conjugates. The peptide incorporated into the conjugates included a tryptophan residue whose fluorescence enabled analysis of peptide concentration. Figure 2 illustrates the fluorescence emission spectrum of the DP3 conjugate, as well as the spectra for various control samples that were prepared identically but in the absence of one of the conjugate components. The observed fluorescence of the DP3 conjugate corresponded to a peptide concentration that was a 7-fold molar excess over the concentration of the plasmid (assessed by UV/Vis spectroscopy; data not shown), suggesting that PNA-peptide-PEG conjugates were bound to approximately 70% of the available PNA binding sites on the plasmid. Samples prepared identically, but in the absence of peptide, exhibited minimal fluorescence, as expected; samples prepared in the absence of PNA also exhibited minimal fluorescence, suggesting that PNA-peptide-PEG hybridization to the plasmid was mediated by PNA-DNA interactions. Samples prepared in the absence of DNA exhibited minimal fluorescence, and provided validation of the efficiency of the purification process.
Figure 2.
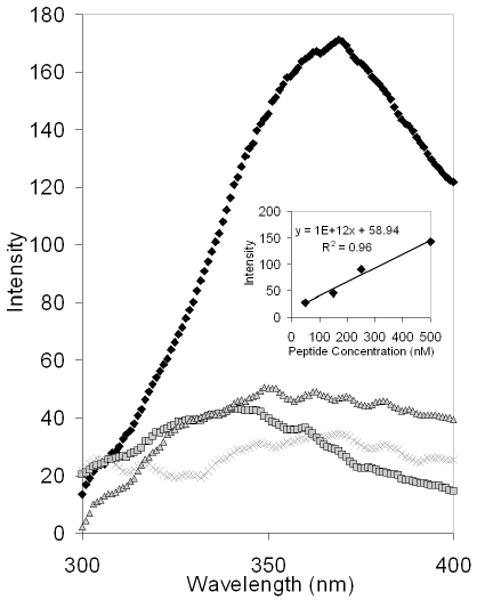
Intrinsic Fluorescence of the DNA-PNA-peptide-PEG Conjugate. The purified DP3 conjugates and conjugates prepared identically but in the absence of one component were analyzed via intrinsic fluorescence spectroscopy to detect tryptophan residues present in the linker peptide (excitation 280 nm; emission 350 nm). The DP3 conjugate (black diamonds) exhibited significant fluorescence at 350 nm, indicative of the presence of the linker peptide. In contrast, samples prepared in the absence of DNA (grey squares), PNA (grey “X”s), or peptide (grey triangles) had minimal fluorescence at the same wavelength. Inset: the fluorescence emission peak intensities of various linker peptide solutions were analyzed (excitation 280 nm; emission 350 nm).
ATR-FTIR spectroscopy was used as a secondary means of conjugate validation (Figure 3). The DP3 sample spectrum exhibited characteristic peaks indicative of DNA, PNA, peptide, and PEG (Figure 3A, bottom). The DNA spectrum (Figure 3A, top) had characteristic vibrational signals in the 1000-1500 cm-1 region. The PNA and the peptide spectra both exhibited characteristic amide bond signals (1630 cm-1), as expected (Figure 3, B and C), whereas the PEG spectrum had a signal at around 2800 cm-1, indicative of the carbon-carbon vibrational stretching along the backbone of the polymer (Figure 3D). In contrast, conjugate samples that were missing one or more of the conjugate components exhibited spectra missing the corresponding signals (Figure 3A, top and middle). Notably, the presence of the characteristic PEG signal only in the DP3 conjugate sample suggests direct DNA prePEGylation.
Figure 3.
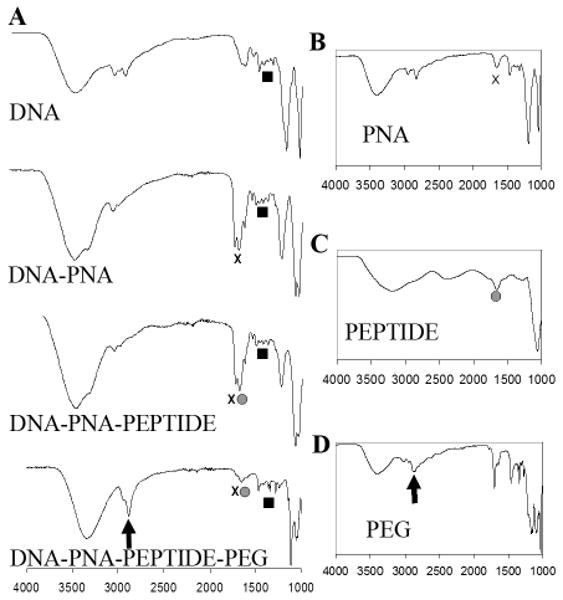
ATR-FTIR Characterization of the DP3 Conjugates. (A) ATR-FTIR spectra of the GWiz plasmid (top), the DP3 conjugate (bottom), and conjugates lacking one or more conjugate components (middle). The main absorption bands of DNA (black squares) are: 1063 cm-1 (symmetric stretching vibration of the phosphate groups), 1222 cm-1 (antisymmetric stretching vibration of the phosphate group), 1017 cm-1 (vibration of C=N of ribose), and 1693 cm-1 (vibrations of C6=O of guanine and C4=O of thymine). Amide bonds, found in both PNA (B) and peptide (C), have an expected peak at approximately 1630 cm-1 (black “x”s in [A] and [B] and grey circles in [A] and [C]). Peaks attributable to PEG (D) are observed at 2890 cm-1 (black arrows in [A] and [D]), indicative of the –CH2- stretching vibrations.
Physical Characterization of DNA Complexes
Complex Size and Charge
The DP3 conjugates and unmodified plasmids were self-assembled with 25 kDa PEI and the resulting complexes were analyzed by DLS. As shown in Figure 4, the PEI-DP3 complexes were smaller and less heterogeneous than the PEI-DNA complexes at an N:P ratio of 10. By a lognormal statistical fit, the mean of the PEI-DP3 size distribution was reduced in comparison with that of the PEI-DNA size distribution from approximately 100 nm to 40 nm; the standard deviation of the PEI-DP3 size distribution was reduced by half (from 28 nm to 14 nm) in comparison with that of the PEI-DNA size distribution. As previously mentioned, the wide standard deviation of PEI-DNA complex diameters is typical for these systems, and is a direct consequence of the multimolecular aggregation that occurs during the formulation process. To further examine this observation, AFM and SEM were utilized to characterize the PEI-DP3 and PEI-DNA complexes (Figure 5, A and B). Complex suspensions were dried onto AFM and SEM substrates and the substrates were subsequently analyzed. AFM and SEM images of the complexes supported the finding that smaller and less heterogeneous complexes formed when the DNA was directly PEGylated prior to complexation.
Figure 4.
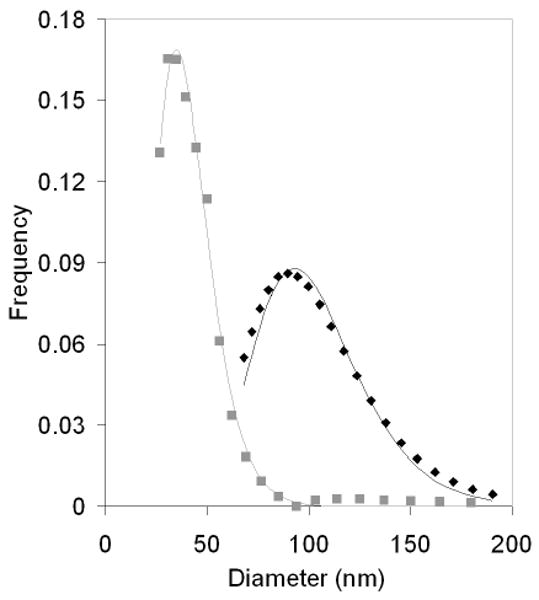
PEI-DP3 and PEI-DNA Polyplex Size Distributions. PEI-DP3 (grey squares) and PEI-DNA (black diamonds) polyplexes were formed at an N:P of 10 and analyzed via DLS. The resulting autocorrelation functions were transformed into the relevant size distributions by the CONTIN algorithm.
Figure 5.
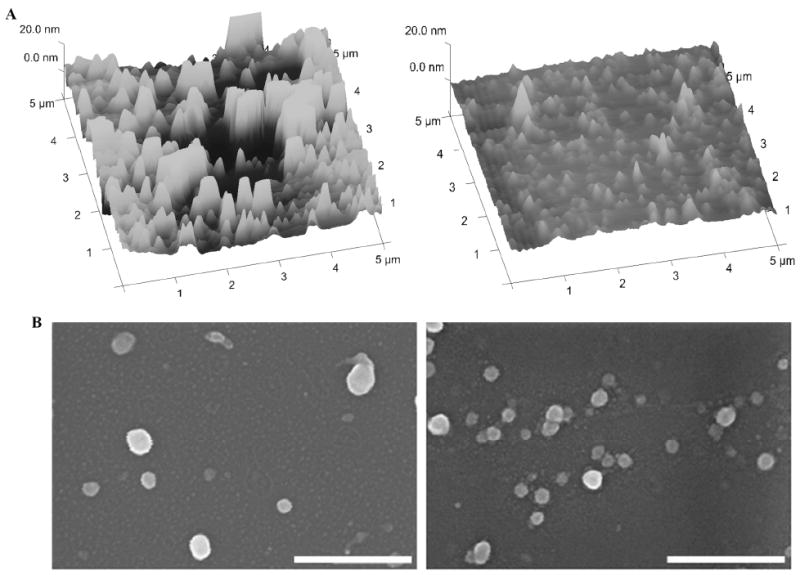
PEI-DP3 and PEI-DNA Polyplex Shape and Size. (A) AFM images of PEI-DNA (left) and PEI-DP3 (right) complexes dried on a mica strip and imaged in contact mode (N:P = 10). (B) SEM images of PEI-DNA (left) and PEI-DP3 (right) complexes dried onto glass substrates (N:P = 10). Scale bar = 500 nm.
The size of PEI-DNA complexes depends upon the N:P ratio at which they are formed because of the dependence of colloidal stability on surface charge. (61) In contrast, it was expected that DNA prePEGylation would reduce this N:P dependence by steric stabilization of the PEI-DP3 complexes. DLS and zeta potential analyses were therefore used to assess the mean complex size and zeta potential as a function of the N:P ratio (Figure 6). As predicted, the PEI-DP3 complexes had similar average diameters, regardless of the amount of polycation introduced, whereas the PEI-DNA complexes were larger at lower N:P ratios (Figure 6A). Zeta potential analyses (Figure 6B) revealed no significant differences between the trends for the PEI-DP3 and PEI-DNA samples.
Figure 6.
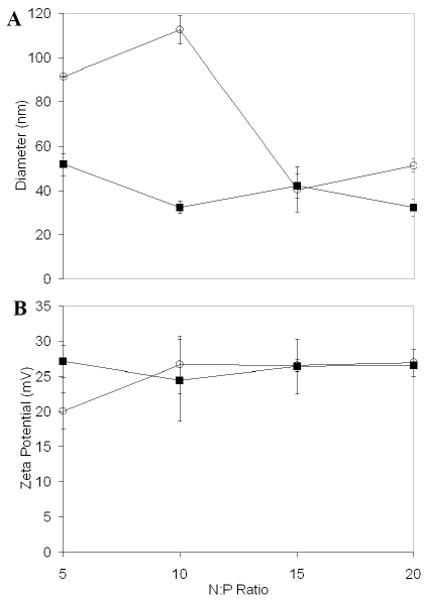
PEI-DP3 and PEI-DNA Polyplex Characterization by Light Scattering. (A) DLS was used to assess PEI-DP3 (black squares) and PEI-DNA (open circles) polyplex diameters at N:P = 5 - 20. Number averaged distributions obtained with the CONTIN algorithm were used to calculate average polyplex diameters. Statistically significant differences are noted at N:P ratios of 5 (p=0.006) and 10 (p=0.021). (B) Phase analysis light scattering was employed to determine the zeta potentials of polyplexes prepared at the same N:P ratios. All data represent the mean values obtained from three separately prepared and analyzed samples per data point. Error bars represent one standard deviation from the mean.
Complexation Efficiency and Nuclease Protection
Because polymer PEGylation has been shown to destabilize polyplexes (4, 25, 26, 28-30), the complexation efficiency and nuclease protection of the PEI-DP3 complexes were explored. Ethidium bromide exclusion assays revealed that complexation efficiency increased as a consequence of the DNA prePEGylation strategy employed with the DP3 conjugate (Figure 7). In this assay, any reduction in DNA band intensity is a consequence of the complexation process excluding ethidium bromide from the nucleic acid, inhibiting its ability to intercalate and fluoresce. At low N:P ratios, the band intensity (as quantified via optical density measurements) for supercoiled DP3 was lower than that for DNA (Figure 7, A and B), suggesting that PEI was able to more effectively condense the PEGylated nucleic acid.
Figure 7.
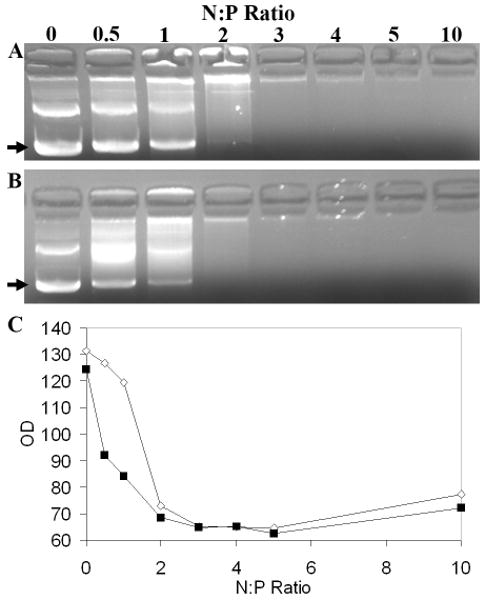
Assessment of Complexation Efficiency by Ethidium Bromide Exclusion Assays. PEI-DNA (A) and PEI-DP3 (B) polyplexes were prepared and analyzed by agarose gel electrophoresis at N:P ratios ranging from 0 to 10. The bands corresponding to supercoiled plasmid DNA are indicated (arrows). (C) Optical density measurements of the supercoiled DNA band for PEI-DP3 (black boxes) and PEI-DNA (white diamonds) polyplexes for the gel shown in A. The trends in optical density were consistent in replicates of the same experiment. Other bands on the gel include open circular and nicked plasmid DNA, as well as DNA aggregates found in the wells of the gel.
Nuclease protection assays were used to examine the resistance of DNA within the PEI-DP3 complexes to serum nucleases. Consistent with the finding that the PEI-DP3 complexes were packaged efficiently at N:P ratios greater than 3, these complexes were also able to protect DNA from enzymatic degradation (Supplementary Figure 2). Previous studies have shown the effectiveness of PEI at achieving this goal. (62, 63)
Complex Bioactivity
Transfection Efficiency and Cell Viability
To determine if DNA prePEGylation and the resulting alterations in complex size influenced cellular transfection, the PEI-DP3 and PEI-DNA complexes were used to transfect CHO cells. After 24 hours, a reduction in the transfection efficiency was observed when the PEI-DP3 complexes were used for transfection in comparison with when the PEI-DNA complexes were used (PEI-Mock complexes containing a non-GFP-expressing plasmid were also examined to account for any non-specific autofluorescence associated with PEI) (Figure 8, A and C). Similar trends were observed when transfection was assessed by flow cytometry and image analysis. However, while the transfection efficiency was somewhat lower with PEI-DP3 than with PEI-DNA, the cytotoxicity of the PEI-DP3 complexes was significantly reduced relative to those of both the PEI-Mock and the PEI-DNA complexes (Figure 8, B and D).
Figure 8.
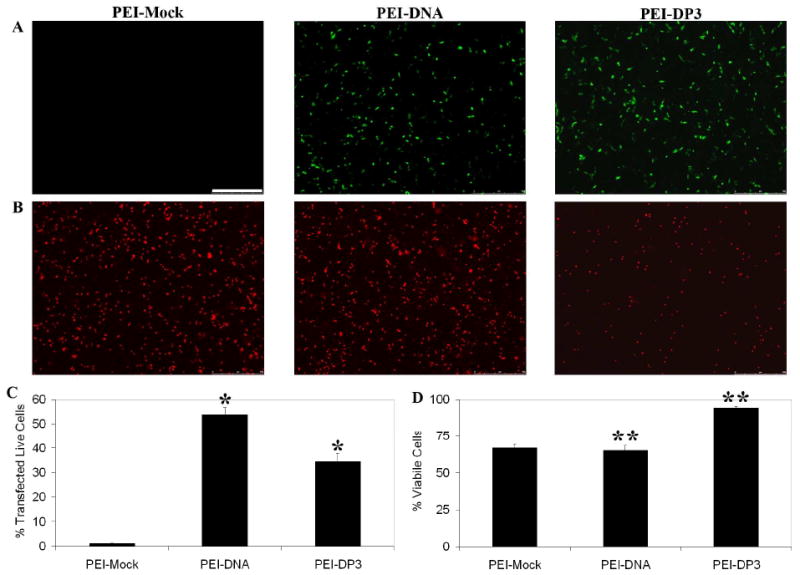
Transfection Efficiency and Cytotoxicity Assays. The transfection efficiencies of PEI-Mock (complexes made with PEI and a non-GFP-expressing plasmid), PEI-DNA, and PEI-DP3 complexes formulated at N:P = 10 were assessed in CHO cells 24 h post-transfection by fluorescence microscopy (A) and flow cytometry (C) to detect GFP fluorescence. The scale bar (shown in [A]) = 500 μm. The data in (C) represent the mean values obtained from three separate transfection experiments, and the error bars represent one standard deviation from the mean. Statistically significant differences were observed between the indicated samples (p [*] = 0.038). (B) The cytotoxicities of the same complexes were evaluated with a ethidium homodimer stain and fluorescence microscopy (B). Quantification was performed by image analysis (D), and [**] indicate a statistically significant difference between the indicated samples (p [**] = 0.0001).
Complex Purification Effects
Previous work has shown that the in vitro cytotoxicity of PEI-DNA formulations depends largely upon the excess PEI that remains in solution following complexation, and thus can be reduced through complex purification via size exclusion chromatography. (64) To examine the effects on PEI-DP3 transfection efficiency and cytotoxicity following free PEI removal, centrifugal filtration was used to purify the complexes. As expected, a reduction in cytotoxicity was observed for the PEI-Mock and PEI-DNA complexes following purification (Figure 9). However, the PEI-DP3 complexes retained their low level of cytotoxicity following purification, suggesting that the amount of free PEI in solution following complexation was significantly reduced in these formulations in comparison with conventional PEI-DNA formulations.
Figure 9.
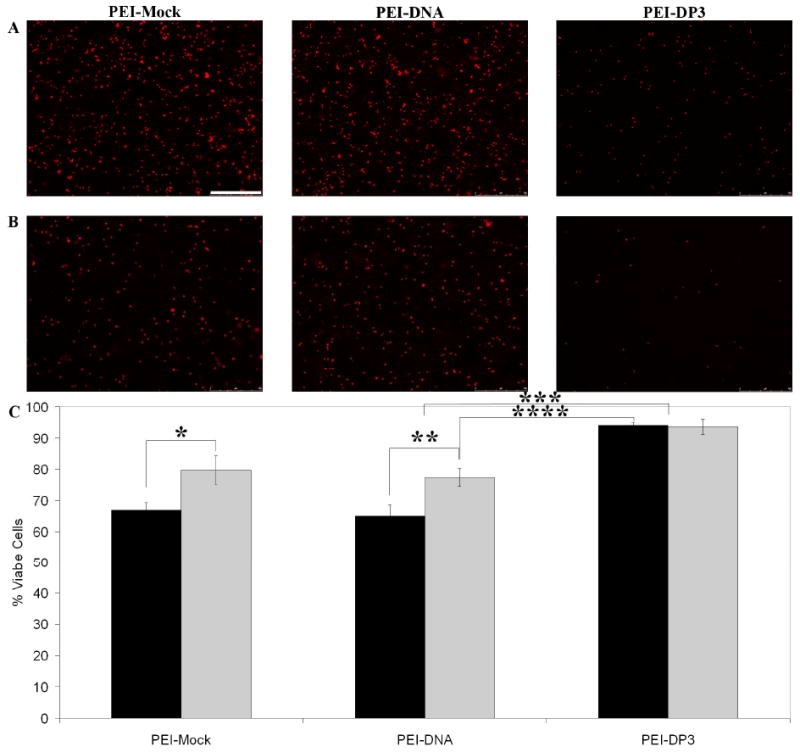
Effect of Complex Purification on Cytotoxicity. Unpurified (A) and purified (B) PEI-Mock, PEI-DNA, and PEI-DP3 complexes were used at N:P = 10 to transfect CHO cells, and the cytotoxicity of each formulation was analyzed by ethidium homodimer staining and fluorescence microscopy. Quantification for the unpurified (black bars) and purified (grey bars) formulations was performed by image analysis (C), and indicated statistically significant differences between the indicated samples (p [*] = 0.002; p [**] = 0.02; p [***] = 0.00005; p [****] = 0.00005). All data represent the mean values obtained from three separate transfection experiments, and the error bars represent one standard deviation from the mean. The scale bar (shown in [A] = 500 μm).
While purification of PEI-DNA complexes lowers their cytotoxicity, previous work has shown that the removal of the excess PEI in solution reduces transfection efficiency. (64) This observation was previously attributed to the role of free PEI in solution in assisting endosomal buffering following cellular internalization.(64) This particular point is of concern when considering the potential in vivo applicability of PEI-DNA complexes, since the in vitro concentration of free PEI is not an appropriate reflection of the physiological situation. Figure 10 illustrates that the transfection efficiency of the PEI-DP3 complexes increased following purification, whereas that of the PEI-DNA complexes decreased. These changes resulted in transfection levels that were approximately 12% higher when PEI-DP3 was used for transfection as opposed to PEI-DNA (as confirmed by both image analysis [data not shown] and flow cytometry).
Figure 10.
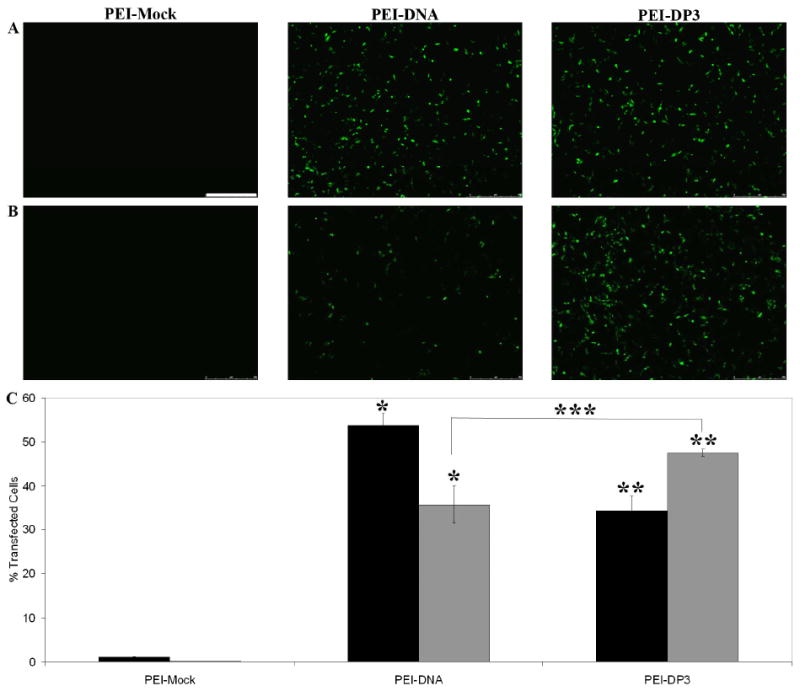
Effect of Complex Purification on Transfection Efficiency. Unpurified (A) and purified (B) PEI-Mock, PEI-DNA, and PEI-DP3 complexes were used at N:P = 10 to transfect CHO cells, and the transfection efficiency obtained by each formulation was analyzed by ethidium homodimer staining and fluorescence microscopy. Quantification was performed by flow cytometry for both the unpurified (black bars) and purified (grey bars) formulations (C). All data represent the mean values obtained from three separate transfection experiments, and the error bars represent one standard deviation from the mean. Statistically significant differences were observed between the indicated samples (p [*] = 0.045; p [**] = 0.045; p [***] = 0.046). The scale bar (shown in [A] = 500 μm).
Discussion
PEI-based formulations are highly efficient in vitro gene delivery vehicles, in large part because of their capacity to induce endosomal release by pH-buffering. (8) Unfortunately, the in vivo applicability of these formulations is questionable, given the inherent cytotoxicity of PEI (64) and challenges in formulation. In particular, the process of multi-molecular aggregation during polyplex formation is a significant issue that results in highly heterogeneous complexes at sizes larger than the optimum for either receptor-mediated internalization or for effective in vivo administration. Furthermore, the in vitro activity of PEI-based formulations depends upon the presence of free PEI at levels higher than would be feasible in vivo. (64) To address these issues in PEI polyplex formulation, a novel strategy for DNA prePEGylation has been explored in this work with the goals of reducing aggregation during formulation and producing less heterogeneous and more efficiently-packaged polyplexes.
With this aim in mind, PNA-peptide-PEG conjugates were synthesized as the basis for a new class of gene delivery materials. When these PNA-peptide-PEGs were hybridized with DNA, the resulting DP3 conjugates behaved differently than unmodified DNA when they were self-assembled with PEI. The PEI-DP3 complexes were smaller and less heterogeneous than PEI-DNA complexes formulated at the same N:P ratio (Figures 4-6). This observation was confirmed via DLS analysis of particle scattering, as well as by AFM and SEM imaging, which enabled visualization of individual particles. Furthermore, when the N:P ratio was varied, the size of the PEI-DP3 complexes remained constant at ∼30 – 50 nm, whereas the size of the PEI-DNA complexes was larger at low N:P ratios. Given that DNA polyplexes containing a single plasmid are believed to be approximately 30 nm in diameter,(59) this result suggested that single plasmid-containing complexes were formed with the DP3 conjugate. This effect was attributed to steric stabilization of the PEI-DP3 complexes by DNA prePEGylation. However, unlike typical surface PEGylation strategies which introduce a brush layer of PEG, the observed shifts in the complex size distribution were the result of a small number of PEG molecules bound to each plasmid through the PNA-peptide linker (∼10-20 PEG molecules, assuming the PNA-peptides were saturated with PEG). As a result, PNA-based DNA prePEGylation did not produce measurable effects on polyplex zeta potential (Figure 6B). Furthermore, the PEI-DP3 complexes did not exhibit significant improvements in salt stability in comparison with PEI-DNA complexes, presumably because a large amount of their surface area was not covered by PEG (data not shown).
Although polymer PEGylation has been shown to destabilize polyplexes (4, 25, 26, 28-30), the complexation efficiency and nuclease protection of the PEI-DP3 complexes were not adversely affected by DNA prePEGylation (Figure 7 and Supplementary Figure 2). To the contrary, ethidium bromide exclusion assays suggested that PEI formed tighter complexes at low charge ratios with the DP3 conjugates than with DNA, suggesting that more polycation was able to bind per DP3-contained plasmid. With more polycation per plasmid, less free PEI would remain in solution. This result would be highly desirable from a cytotoxicity perspective, since the presence of free polycation in solution has been shown to contribute to cell death. (64) Furthermore, it was consistent with our findings on the cellular activity of the PEI-DP3 complexes (discussed subsequently).
Recognizing the physical differences associated with this proposed formulation, PEI-DP3 complex bioactivity was explored in CHO cells. When transfections were performed with unpurified PEI-DP3 and PEI-DNA complexes (i.e., formulations containing free PEI, as are typically used (7-9, 21)), the transfection efficiencies of the PEI-DP3 complexes were slightly lower than those of the PEI-DNA complexes (Figure 8, A and C). However, a 30% reduction in cytotoxicity was observed for the PEI-DP3 complexes, most likely as a consequence of the improved complexation brought on by DNA prePEGylation (and the resulting reductions in the levels of free PEI in solution) (Figure 8, B and D).1 In an effort to further investigate this effect, free PEI was removed from the formulations and cellular transfections were performed with the purified complexes. The cytotoxicity of the purified PEI-DP3 complexes remained significantly lower than that for the conventional PEI formulations, whereas the transfection efficiency of these complexes increased following purification (Figures 9 and 10). In contrast, the cytotoxicity of the purified PEI-DNA complexes was lower after purification, as expected, and the transfection efficiency of these complexes decreased.
The effects of free PEI removal on the transfection efficiencies achieved by both the PEI-DP3 and the PEI-DNA complexes were attributed in part to the known in vitro activity of PEI. The findings with the PEI-DNA complexes were consistent with previous work suggesting that free PEI in solution significantly adds to the activity of PEI formulations by enhancing endosomal buffering. (64) Thus, the removal of free PEI would be expected to adversely affect transfection efficiency, as was observed. Clearly, a dependence on free polycation to promote efficient gene delivery is not desirable for in vivo applications. The PEI-DP3 complexes appeared to overcome this issue through the inclusion of more PEI per plasmid within the complex, which would enhance the innate endosomal buffering capacity of the complexes. The reasons for the increase in transfection efficiency following the removal of free PEI from the PEI-DP3 formulations were less clear. One possibility is that with the unpurified PEI-DP3 formulations, the residual PEI in solution blocks polyplex internalization by saturating the available binding sites on the cells.
With a subtle modification of plasmid DNA (the conjugation of ∼10-20 PEG molecules), the complexation and delivery efficiency of a well known polymer in the gene delivery field has been significantly altered. The effects of DNA prePEGylation on the size distribution, cytotoxicity, and gene delivery efficiency of PEI-based polyplexes are expected to have important impacts on in vivo activity. As discussed, particles of ∼10 – 100 nm are capable of both avoiding renal clearance (65), efficiently exiting the bloodstream, and travelling through interstitial tissues. (46, 47) Furthermore, the low cytotoxicity and innate activity of the PEI-DP3 complexes in the absence of free PEI should significantly improve in vivo effectiveness. Future work will explore both the dose response of the PEI-DP3 complexes at various N:P ratios in multiple cell lines, and the potential benefits of this modified nucleic acid formulation in vivo. The described studies suggest the importance of future efforts focused not only on developing new biomaterials, but also on improving existing systems in the field.
Supplementary Material
Acknowledgments
M.O.S. thanks the NSF (CBET-0707583), the University of Delaware Research Foundation, the USARO (W911NF-08-1-0241), and the NIH/NCRR INBRE (2 P20 RR016472-07) for financial support for this project. P.G.M. acknowledges fellowship funding from the NSF IGERT Program (DGE-0221651) and from Merck Research Laboratories.
Footnotes
We do not believe that the differences in bioactivity between the two formulations were related to the MMP-1-cleavable peptide incorporated into the PEI-DP3 complexes, given that CHO cells do not express high levels of MMP-1 under the conditions studied.
Supporting Information Available: Validation of DNA-PNA conjugation. Nuclease stability of modified and unmodified complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Demeneix B, Behr J, Boussif O, Zanta MA, Abdallah B, Remy J. Gene transfer with lipospermines and polyethylenimines. Adv Drug Deliv Rev. 1998;30:85–95. doi: 10.1016/s0169-409x(97)00109-9. [DOI] [PubMed] [Google Scholar]
- 2.Ogris M, Steinlein P, Carotta S, Brunner S, Wagner E. DNA/polyethylenimine transfection particles: influence of ligands, polymer size, and PEGylation on internalization and gene expression. AAPS PharmSci. 2001;3:E21. doi: 10.1208/ps030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrett SW, Davies OR, Milroy DA, Wood PJ, Pouton CW, Threadgill MD. Synthesis and characterisation of polyamine-poly(ethylene glycol) constructs for DNA binding and gene delivery. Bioorg Med Chem. 2000;8:1779–1797. doi: 10.1016/s0968-0896(00)00113-9. [DOI] [PubMed] [Google Scholar]
- 4.Wolfert MAS, L W. Atomic force microscopic analysis of the influence of the molecular weight of poly(L)lysine on the size of polyelectrolyte complexes formed with DNA. Gene Therapy. 1996;3:269–273. [PubMed] [Google Scholar]
- 5.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92:7297–301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999;6:595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 7.Godbey WT WK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60:149–60. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 8.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. PNAS. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 10.Brunner S, Sauer T, Carotta S, Cotten M, Saltik M, Wagner E. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 2000;7:401–407. doi: 10.1038/sj.gt.3301102. [DOI] [PubMed] [Google Scholar]
- 11.Chollet P, Favrot M, Hurbin A, Coll JL. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J Gene Med. 2001;4:84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- 12.Dash P, Read M, Barrett L, Wolfert M, Seymour L. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6:643–650. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa M, Takemura S, Takakura Y, Hashida M. Targeted delivery of plasmid DNA to hepatocytes in vivo: optimization of the pharmacokinetics of plasmid DNA/galactosylated poly(L-lysine) complexes by controlling their physicochemical properties. J Pharm Exp Ther. 1998;287:408–415. [PubMed] [Google Scholar]
- 14.Burke RS PS. Extracellular barriers to in Vivo PEI and PEGylated PEI polyplex-mediated gene delivery to the liver. Bioconjug Chem. 2008;19:693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- 15.Collard WT, Yang Y, Kwok KY, Park Y, Rice KG. Biodistribution, metabolism, and in vivo gene expression of low molecular weight glycopeptide polyethylene glycol peptide DNA co-condensates. J Pharm Sci. 2000;89:499–512. doi: 10.1002/(SICI)1520-6017(200004)89:4<499::AID-JPS7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Ward CM, Read ML, Seymour LW. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 2001;97:2221–2229. doi: 10.1182/blood.v97.8.2221. [DOI] [PubMed] [Google Scholar]
- 17.Kircheis R, Blessing T, Brunner S, Wightman L, Wagner E. Tumor targeting with surface-shielded ligand--polycation DNA complexes. J Control Release. 2001;72:165–70. doi: 10.1016/s0168-3659(01)00272-3. [DOI] [PubMed] [Google Scholar]
- 18.Brus C PH, Aigner A Czubayko F, Kissel T. Physicochemical and biological characterization of polyethylenimine-graft-poly(ethylene glycol) block copolymers as a delivery system for oligonucleotides and ribozymes. Bioconjug Chem. 2004;15:677–84. doi: 10.1021/bc034160m. [DOI] [PubMed] [Google Scholar]
- 19.Merdan T KK, Petersen H Bakowsky U, Voigt KH Kopecek J, Kissel T. PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconjug Chem. 2005;16:785–92. doi: 10.1021/bc049743q. [DOI] [PubMed] [Google Scholar]
- 20.Mishra S, Webster P, Davis ME. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur J Cell Biol. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 21.Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Different strategies for formation of pegylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem. 2001;12:529–37. doi: 10.1021/bc0001488. [DOI] [PubMed] [Google Scholar]
- 22.Bartlett DW DM. Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug Chem. 2007;18:456–468. doi: 10.1021/bc0603539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner E, Zenke M, Cotten M, Beug H, Birnstiel ML. Transferrin-polycation conjugates as carriers for DNA uptake into cells. Proc Natl Acad Sci U S A. 1990;87:3410–4. doi: 10.1073/pnas.87.9.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varga CM, Wickham TJ, Lauffenburger DA. Receptor-mediated targeting of gene delivery vectors: insights from molecular mechanisms for improved vehicle design. Biotechnol Bioeng. 2000;70:593–605. doi: 10.1002/1097-0290(20001220)70:6<593::aid-bit1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 25.Rimann M, Luhmann T, Textor M, Guerino B, Ogier J, Hall H. Characterization of PLL-g-PEG-DNA Nanoparticles for the Delivery of Therapeutic DNA. Bioconjug Chem. 2008;19:548–57. doi: 10.1021/bc7003439. [DOI] [PubMed] [Google Scholar]
- 26.Choi YH, Liu F, Kim JS, Choi YK, Park JS, Kim SW. Polyethylene glycol-grafted poly-L-lysine as polymeric gene carrier. J Control Release. 1998;54:39–48. doi: 10.1016/s0168-3659(97)00174-0. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen HK, Lemieux P, Vinogradov SV, Gebhart CL, Guerin N, Paradis G, Bronich TK, Alakhov VY, Kabanov AV. Evaluation of polyether-polyethylenimine graft copolymers as gene transfer agents. Gene Ther. 2000;7:126–138. doi: 10.1038/sj.gt.3301052. [DOI] [PubMed] [Google Scholar]
- 28.Kaminskas LM BB, Karellas P Krippner GY, Lessene R Kelly B, Porter CJ. The impact of molecular weight and PEG chain length on the systemic pharmacokinetics of PEGylated poly l-lysine dendrimers. Mol Pharm. 2008;5:449–463. doi: 10.1021/mp7001208. [DOI] [PubMed] [Google Scholar]
- 29.Walsh M, Tangney M, O'Neill MJ, Larkin JO, Soden DM, McKenna SL, Darcy R, O'Sullivan GC, O'Driscoll CM. Evaluation of cellular uptake and gene transfer efficiency of pegylated poly-L-lysine compacted DNA: implications for cancer gene therapy. Mol Pharm. 2006;3:644–653. doi: 10.1021/mp0600034. [DOI] [PubMed] [Google Scholar]
- 30.Pun SH, Davis ME. Development of a nonviral gene delivery vehicle for systemic application. Bioconjug Chem. 2002;13:630–9. doi: 10.1021/bc0155768. [DOI] [PubMed] [Google Scholar]
- 31.Pun SH, Davis ME. Development of a nonviral gene delivery vehicle for systemic application. Bioconjug Chem. 2002;13:630–639. doi: 10.1021/bc0155768. [DOI] [PubMed] [Google Scholar]
- 32.Pun SH BN, Liu A Jensen G, Machemer T Quijano E, Schluep T Wen S, Engler H Heidel J, Davis ME. Cyclodextrin-modified polyethylenimine polymers for gene delivery. Bioconjug Chem. 2004;15:831–840. doi: 10.1021/bc049891g. [DOI] [PubMed] [Google Scholar]
- 33.Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995;23:217–22. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zelphati O, Liang X, Hobart P, Felgner PL. Gene chemistry: functionally and conformationally intact fluorescent plasmid DNA. Hum Gene Ther. 1999;10:15–24. doi: 10.1089/10430349950019156. [DOI] [PubMed] [Google Scholar]
- 35.Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993;365:566–8. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- 36.Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, Nielsen PE. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol. 1994;48:1310–3. doi: 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 37.Zelphati O, Liang X, Nguyen C, Barlow S, Sheng S, Shao Z, Felgner PL. PNA-dependent gene chemistry: stable coupling of peptides and oligonucleotides to plasmid DNA. Biotechniques. 2000;28:304–10. 312–4, 316. doi: 10.2144/00282rr01. [DOI] [PubMed] [Google Scholar]
- 38.Dean DA. Peptide nucleic acids: versatile tools for gene therapy strategies. Adv Drug Deliv Rev. 2000;44:81–95. doi: 10.1016/s0169-409x(00)00087-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang KW, Hoffman EP, Huang L. Targeted delivery of plasmid DNA to myogenic cells via transferrin-conjugated peptide nucleic acid. Mol Ther. 2000;1:236–43. doi: 10.1006/mthe.2000.0043. [DOI] [PubMed] [Google Scholar]
- 40.Koppelhus U, Nielsen PE. Cellular delivery of peptide nucleic acid (PNA) Adv Drug Deliv Rev. 2003;55:267–80. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 41.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–6. [PubMed] [Google Scholar]
- 42.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–39. [PMC free article] [PubMed] [Google Scholar]
- 43.Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 44.Sevick EM, Jain RK. Measurement of capillary filtration coefficient in a solid tumor. Cancer Res. 1991;51:1352–5. [PubMed] [Google Scholar]
- 45.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 46.Goodman TT, Ng CP, Pun SH. 3-D tissue culture systems for the evaluation and optimization of nanoparticle-based drug carriers. Bioconjug Chem. 2008;19:1951–9. doi: 10.1021/bc800233a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enger PO, Thorsen F, Lonning PE, Bjerkvig R, Hoover F. Adeno-associated viral vectors penetrate human solid tumor tissue in vivo more effectively than adenoviral vectors. Hum Gene Ther. 2002;13:1115–25. doi: 10.1089/104303402753812511. [DOI] [PubMed] [Google Scholar]
- 48.Aoyama Y, Kanamori T, Nakai T, Sasaki T, Horiuchi S, Sando S, Niidome T. Artificial viruses and their application to gene delivery. Size-controlled gene coating with glycocluster nanoparticles. J Am Chem Soc. 2003;125:3455–7. doi: 10.1021/ja029608t. [DOI] [PubMed] [Google Scholar]
- 49.Nakai T, Kanamori T, Sando S, Aoyama Y. Remarkably size-regulated cell invasion by artificial viruses. Saccharide-dependent self-aggregation of glycoviruses and its consequences in glycoviral gene delivery. J Am Chem Soc. 2003;125:8465–75. doi: 10.1021/ja035636f. [DOI] [PubMed] [Google Scholar]
- 50.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J Am Chem Soc. 2004;126:6520–1. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 51.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–8. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 52.Wagner E, Cotten M, Foisner R, Birnstiel ML. Transferrin-polycation-DNA complexes: the effect of polycations on the structure of the complex and DNA delivery to cells. Proc Natl Acad Sci U S A. 1991;88:4255–9. doi: 10.1073/pnas.88.10.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perales JC, Ferkol T, Beegen H, Ratnoff OD, Hanson RW. Gene transfer in vivo: sustained expression and regulation of genes introduced into the liver by receptor-targeted uptake. Proc Natl Acad Sci U S A. 1994;91:4086–90. doi: 10.1073/pnas.91.9.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rippe B, Haraldsson B. Fluid and protein fluxes across small and large pores in the microvasculature. Application of two-pore equations. Acta Physiol Scand. 1987;131:411–28. doi: 10.1111/j.1748-1716.1987.tb08257.x. [DOI] [PubMed] [Google Scholar]
- 55.Melkko S, Halin C, Borsi L, Zardi L, Neri D. An antibody-calmodulin fusion protein reveals a functional dependence between macromolecular isoelectric point and tumor targeting performance. Int J Radiat Oncol Biol Phys. 2002;54:1485–90. doi: 10.1016/s0360-3016(02)03927-5. [DOI] [PubMed] [Google Scholar]
- 56.Dellian M, Yuan F, Trubetskoy VS, Torchilin VP, Jain RK. Vascular permeability in a human tumour xenograft: molecular charge dependence. Br J Cancer. 2000;82:1513–8. doi: 10.1054/bjoc.1999.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao H, Shi W, Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. 2005;102:9469–74. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244:105–15. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 59.Dauty E, Remy JS, Zuber G, Behr JP. Intracellular delivery of nanometric DNA particles via the folate receptor. Bioconjug Chem. 2002;13:831–9. doi: 10.1021/bc0255182. [DOI] [PubMed] [Google Scholar]
- 60.Lutolf MP LFJ, Schmoekel HG Metters AT, Weber FE Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiemenez P, Rajagopalan R. Principles of Colloid and Surface Chemistry. 3rd. CRC; 1997. [Google Scholar]
- 62.Vinogradov SV, Bronich TK, Kabanov AV. Self-assembly of polyamine-poly(ethylene glycol) copolymers with phosphorothioate oligonucleotides. Bioconjug Chem. 1998;9:805–12. doi: 10.1021/bc980048q. [DOI] [PubMed] [Google Scholar]
- 63.Ferrari S, Pettenazzo A, Garbati N, Zacchello F, Behr JP, Scarpa M. Polyethylenimine shows properties of interest for cystic fibrosis gene therapy. Biochim Biophys Acta. 1999;1447:219–25. doi: 10.1016/s0167-4781(99)00153-0. [DOI] [PubMed] [Google Scholar]
- 64.Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J Gene Med. 2004;6:1102–11. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- 65.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165–70. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.