

Abstract
In the present study, we characterized the generation of prostaglandin (PG)E2 in human neutrophils. We found that the Ca2+-dependent type IV cytosolic phospholipase A2 (cPLA2) was pivotally involved in the COX-2-mediated generation of PGE2 in response to a calcium ionophore, as determined by the use of selected PLA2 inhibitors. PGE2 biosynthesis elicited by bacterial-derived peptides or by phagocytic stimuli acting on cell surface receptors also showed to be dependent on cPLA2 activity. We then assessed metabolism of unesterified arachidonic acid (AA), and observed that PGE2 production becomes favored over that of LTB4 with higher AA concentrations. Withdrawal of calcium prevented the generation of PGE2 in response to a calcium ionophore but did not affect the up-regulation of COX-2 or its capacity to convert AA, thus limiting its implication at the level of cPLA2 activation. Of the main eicosanoids produced by neutrophils, only LTB4 was able to up-regulate COX-2 expression. Finally, the only PGE synthase isoform found in neutrophils is microsomal PGE synthase-1; it co-localized with COX-2 and its expression appeared mainly constitutive. These results highlight key differences in regulatory processes of the 5-LO and COX pathways, and enhance our knowledge at several levels in the PGE2 biosynthesis in neutrophils.
Keywords: Inflammation, Polymorphonuclear leukocyte, Cyclooxygenase, Fatty acid, Eicosanoids, Prostaglandins
1. Introduction
Polymorphonuclear neutrophils are often the first leukocytes to migrate toward inflammatory lesions where they perform host defense functions such as phagocytosis, release of proteolytic enzymes, generation of oxygen-derived reactive agents, and synthesis of an array of cytokines and chemokines [1–4]. These cells also biosynthesize a selection of arachidonic acid (AA)-derived lipid mediators of inflammation, termed eicosanoids. Indeed, neutrophils are recognized as a major source of leukotriene (LT)B4. More recently, they have been shown to generate prostaglandin (PG)E2 and thromboxane (TX) A2 via the inducible cyclooxygenase (COX-2) pathway [5–8].
The eicosanoids produced by neutrophils may have distinct –and sometimes divergent – biological activities. For instance, LTB4 is a potent chemoattractant and agonist for leukocytes, and is important in asthma, allergies and inflammation [9]. TXA2 is recognized for stimulating platelet aggregation and activation. PGE2 has both pro- and anti-inflammatory properties: it participates in regulating blood flow and vascular permeability, bronchial airway contraction, nociceptor activation and hyper responsiveness [10]. On the other hand, PGE2 inhibits important leukocyte effector functions, including chemotaxis, aggregation, superoxide production, lysosomal enzyme release and LTB4 generation [11–16]. Thus, the overall profile of eicosanoids generated by neutrophils may clearly influence the way these cells participate in orchestrating the inflammatory response and, as such, a better understanding of the mechanisms controlling their biosynthesis is of fundamental and clinical interest. The generation of LTs has received considerable attention. Much less is known, however, regarding the regulation of PGE2 biosynthesis in neutrophils.
The first step in eicosanoid generation is the release of AA from membrane phospholipids by a phospholipase (PL)A2 enzyme [17], in a Ca2+-dependent fashion. In neutrophils, type IV cytosolic (c)PLA2 has been identified as the preeminent isoform responsible for the mobilization of AA committed for the generation of LTs [18,19], which starts when 5-LO inserts an oxygen molecule at the fifth carbon of unesterified AA forming the hydroperoxyl intermediate, 5-hydroperoxyeicosatetraenoic acid (5-HPETE). The same enzyme also catalyzes a dehydration reaction, forming the unstable epoxide intermediate, LTA4, which can be further metabolized to LTB4 by LTA4 hydrolase. The COX enzyme, on the other hand, catalyzes two reactions by which AA is transformed to PGH2, the common precursor of all prostanoids. In humans, two distinct genes are known to encode COX isoforms [20], both contributing to the inflammatory process. COX-2 has attracted special attention as it is specifically induced during acute and chronic inflammation. Finally, PGH2 can be isomerized to PGE2, either non-enzymatically or by one or more of several PGE2 synthase isoforms [21], whereas TXA2 formation results from the activity of TXA2 synthase. In neutrophils, COX-2 largely prevails over COX-1 for the generation of prostanoids [8,22,23], while the identity of the PGE2 synthase isoform(s) involved with COX-2-derived PGE2 production remains to be determined.
We characterized in the present study the main metabolic steps which are governing biosynthesis of PGE2 in neutrophils: AA liberation and utilization, COX activation, and PGE2 synthase expression. Results indicate a key implication for Ca2+-dependent type IV cPLA2 and the constitutive presence of microsomal PGE2 synthase in these cells.
2. Experimental procedures
2.1. Materials
Adenosine deaminase (ADA) was purchased from Roche Applied Science (Indianapolis, Indiana, USA). CGS 21680 was from Research Biochemicals International (Natik, Maine, USA). Lipopolysaccharide (LPS; from E. coli 0111; B4), was obtained from Calbiochem-Novalbiochem Corp. (San Diego, CA, USA). DFP (diisopropylfluorophosphate) was from Serva Electrophoresis (Carl-Benz-Str7, Heidelberg). Leupeptin and aprotinin were obtained from ICN Biomedicals Inc. (Irvin, California, USA). LTB4, PGE2 and cTXA2 were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Pyrrophenone was a generous gift from Dr. K. Seno, Shionogi Research Laboratories (Osaka, Japan). fMLP and arachidonic acid, were obtained from Sigma (Oakville, ON, Canada). Recombinant human GM-CSF and TNF-α were purchased from Cedarlane (Hornby, ON, Canada). HPLC solvents (acetonitrile and methanol) were from Fisher (Ville St. Laurent, QC, Canada) and from VWR (Ville Mont-Royal, QC, Canada) respectively.
2.2. Neutrophil isolation
Neutrophils were isolated as originally described [24] with modifications [13]. Briefly, venous blood collected on isocitrate anticoagulant solution from healthy volunteers was centrifuged (250×g, 10 min), and the resulting platelet-rich plasma was discarded. Leukocytes were obtained following erythrocyte sedimentation in 2% Dextran T-500. Neutrophils were then separated from other leukocytes by centrifugation on a 10-ml Ficoll-Paque cushion. Contaminating erythrocytes were removed by a 15-s hypotonic lysis; purified granulocytes (>95 neutrophils, <5% eosinophils) contained fewer than 0.2% monocytes, as determined by esterase staining. Viability was greater than 98%, as determined by trypan blue dye exclusion. The whole cell isolation procedure was carried out sterilely at room temperature (RT).
2.3. Monocyte isolation
Monocytes were purified by elutriation, as previously described [25]. Purity of the obtained monocyte fraction (>85%) was assessed either by Giemsa staining of cytocentrifuged smears or by FACS analysis using an anti-CD14 monoclonal antibody (clone FMC 32, Serotec, Adelaide, South Australia). Contaminant cells were essentially all lymphocytes; platelets were rarely detected.
2.4. Cell incubations
2.4.1. Neutrophils
Neutrophils were resuspended at a concentration of 5×106 cells/ml in Hank’s balanced salt solution (HBSS; 37 °C) containing 10 mM HEPES pH 7.4, 1,6 mM Ca2+ and no Mg2+. Where mentioned, adenosine deaminase (ADA; 0.1 U/ml) was added to cell suspensions 20 min prior to stimulation with agonist(s). CGS 21680 (1 μM final concentration) was dissolved in DMSO and added to cell suspensions 10 min prior to stimulation. The final organic solvent concentration never exceeded 0.1% (v/v).
2.4.2. Monocytes
Elutriated monocytes were resuspended (2×106 cells/ml) in RPMI 1640 supplemented with 10% fetal calf serum and penicillin/streptomycin. Cells were distributed in 1 ml aliquots, in Minisorp tubes to minimize adhesion, and incubated at 37 °C. Unless stated otherwise, monocytes were treated with LPS at a final concentration of 2 μg/ml for 16 h. Cells were then stimulated for 60 min with 10 μM AA. For PGE2 measurements, cell suspensions were centrifuged and cell-free supernatants were stored at −20 °C.
2.5. Measurement of free arachidonic acid by liquid chromatography-mass spectrometry
Reactions were stopped by adding 2 volumes of ice-cold methanol containing 20 ng of D8-AA as an internal standard. Samples were processed as described above for HPLC analysis and the HPLC fractions containing AA (determined by using a 3H-AA standard) were collected. Samples were evaporated under reduced pressure (using a Speed Vac model SVC 100D; Savant Instruments Inc., Farmingdale, New York, USA) and redissolved into 100 μl of acetonitrile. AA was assayed by liquid chromatography-mass spectrometry (LC-MS), using a nebulizer-assisted electrospray (ion spray) interface coupled to a triple-quadrupole MS (API-III; PE Sciex, Thornhill, Ontario, Canada). Six μl aliquots of the samples were injected into the electrospray interface via the 20-μl loop of a Rheodyne injector (model 9125; Rheodyne, Cotati, California, USA) connected to a short column (2×30 mM, packed with 5 μM C18 particles), using acetonitrile:H2O (87.5:12.5, vol/vol, containing 0.1% acetic acid) as solvent, at a flow rate of 150 μl/min. Samples were analyzed in the negative ion mode. The ions at m/z 303 and 311 representing the carboxylate anions of AA and D8-AA, respectively, were monitored.
2.6. Measurement of 5-lipoxygenase metabolites by reverse-phase high pressure liquid chromatography
Following stimulation, reactions were stopped and samples were processed either for LC-MS [26] or for reverse phase HPLC analysis, as previously described [27]. RP-HPLC analyses were performed using an on-line extraction procedure with detection limits of 0.2 ng at 280 nm and 1 ng at 229 nm [28].
2.7. Measurement of prostanoids by ELISA
Production of PGE2 was measured using commercial ELISA kits (Cayman Chemical Co., Ann Arbor, MI). Cross-reactivities were <0.04% for 6-keto PGF1α, and <0.01% for LTB4, TXB2, and AA.
2.8. Immunoblots
Cell incubations were stopped in an ice-cold water bath and samples were briefly centrifuged. Cell pellets were resuspended in 100 μl of ice-cold HEPES-buffered HBSS without Ca2+ (containing the following antiprotease cocktail: 0.2 mg/ml diisopropylfluorophosphate (DFP), 10 μg/ml leupeptin, 10 μg/ml aprotinin); then 150 μl of warmed up (65 °C) 2× sample buffer (125 mM Tris–HCl, pH 6.8, 8% SDS, 10% α-mercaptoethanol, 17% glycerol, with antiprotease cocktail) were added and the mixtures were boiled for 7 min. Aliquots (50 μl) were then subjected to 10% SDS-PAGE and transferred to Immobilon membranes (Millipore Corporation, Bedford, Massachusetts, USA). Equal protein loading and transfer efficiency was visualized by Ponceau Red staining. The membranes were soaked for 30 min at RT in Tris-buffered saline (TBS: 25 mM Tris–HCl pH 7.6, 0.2 M NaCl, 0.15% Tween 20) containing 5% (w/v) dried milk, and exposed for 30 min with the first antibody, according to manufacturers’ instructions. Membranes were then washed twice in TBS, and incubated for 30 min with a 1:10000 dilution of a horseradish peroxidase-linked donkey anti-rabbit, or sheep anti-mouse antibody (Santa Cruz, California, USA). Enzyme expression was revealed with ECL-Plus (NEN-Mandel, Mississauga, Ontario, Canada).
2.9. Densitometry
Immunoblot autoradiograms were digitalized using a snapscan 1236 scanner (Agfa, Woburn, MA). Densitometric analyses of autoradiograms were performed with NIH Image (http://rsb.info.nih.gov). For each immuno-blot, the pixel density (integrated optical density) was determined by selecting a rectangle of identical surface for all determinations, designed to entirely cover the band of interest. Background, which was subtracted from all values, was obtained using an average value from a clear area of the autoradiogram. Integrated optical density was translated as arbitrary units using the following scale: 0=background; 4=theorical maximum pixel density (all pixels at a black value of 255).
2.10. Nitrogen cavitation and Percoll fractionation
The procedure was conducted essentially as described [29], with modifications. All steps were performed at 4°C. Briefly, 4×108 neutrophils in 10 ml of ice-cold relaxation buffer (10 mM HEPES pH 7.30, 0.1 M KCl, 3 mM NaCl, 1.25 mM EGTA and containing the anti-protease cocktail) were pressurized (400 psi, 10 min, with constant stirring) in a nitrogen bomb (Parr Instrument Co., Moline, IL, USA). Cavitates were spun at 400×g, 5 min, to pellet unbroken cells and intact nuclei. Supernatants were laid onto 3×4.5 ml of a Percoll step gradient (1.050, 1.090 and 1.120 g/ml), prior to centrifugation (37,000×g, 30 min) in a Beckman L8-70M ultracentrifuge, using a rotor type 42.1. For each cell preparation, 18 fractions were generated (1 ml each), starting from the bottom of the tube. This procedure allows the distinct separation of azurophil granules, specific granules, a plasma membrane-enriched fraction, and cytosol [29]. Each fraction was re-centrifuged (100,000×g, 90 min) in a Beckman TL 100 ultracentrifuge, using a TL 100.2 rotor, in order to pellet Percoll. Fractions (50 μl) were carefully aspirated with a syringe and processed for western immunoblot analysis.
2.11. Real-time PCR
Reverse-transcription and real-time PCR were performed essentially as described in [30]. Briefly, first strand cDNA synthesis was performed using 1 μg of total RNA with Superscript II (Invitrogen Lifetechnology, Carlsbad, CA, USA) in recommended conditions, using 500 ng of random hexamers. Amplification of neutrophil cDNA was carried out in a Rotor-Gene 3000 operated with Rotor Gene software version 6.0.19 (Corbett Research, Mortlake, 2137 NSW, Australia). Each sample consisted of: 50 ng cDNA, 1.3 mM MgCl2, 0.2 mM dNTP, 500 nM of primers, 0.5 unit of Taq polymerase (Amersham Biosciences) and Sybr Green dye (Molecular Probe, Eugene, OR; 1:30 000 dilution) in a reaction volume of 20 μl. Amplification conditions were as follows: 95 °C (20 s), 58 °C (20 s), 72 °C (20 s); 35 repetitions. Specificity of each reaction was ascertained by performing the Melt® procedure (58–99 °C; 1 °C/5 s) after completion of the amplification protocol, according to the manufacturer’s instructions. Primers used in real-time PCR procedures were, GAPDH: 5′-CGA GAT CCC TCC AAA ATC AA-3′ (forward), 5′-TTC ACA CCC ATG ACG AAC AT-3′ (reverse); mPGES-1: 5′-GGA ACG ACA TGG AGA CCA TCT AC-3′ (forward), 5′-TCC AGG CGA CAA AAG GGT TA-3′ (reverse); COX-1 and COX-2: as described in [31].
2.12. Statistical analysis
Where applicable, statistical analysis was performed by student’s non paired t-test (two-tailed), and significance (*) was considered to be attained when p<0.05.
3. Results
3.1. Liberation of arachidonic acid: role of type IV cPLA2
Regarding the identity of the PLA2(s) isoform(s) responsible for the liberation of arachidonic acid, human neutrophils express type IV cPLA2. In addition, mRNA for groups V and X sPLA2 (GV and GX) have been detected, whereas GIB, GIIA, GIID, GIIE, GIIF, GIII, and GXII sPLA2s were not found [19]. Given the demonstrated and prominent role of type IV cPLA2 in the generation of 5-LO-derived leukotrienes [19], we used an inhibitor displaying highest selectivity for this PLA2 subtype, termed pyrrophenone [32,33] in order to address its implication in the generation of COX-2-derived PGE2. To this end, COX-2 was up-regulated in neutrophils by incubation with a mixture of 1.5 nM GM-CSF and 100 nM TNF-α (GM-CSF/TNF) for 120 min [8] then stimulated with the Ca2+-ionophore A23187 for 15 min, in absence or presence of 100 nM pyrrophenone. These experiments were performed in presence of adenosine deaminase (ADA; 0.1 U/ml), a condition which prevents accumulation of endogenous adenosine in cell suspensions thus minimizing the well-documented modulating effect of adenosine on the generation of lipid mediators by neutrophils [13,34].
Pyrrophenone prevented the generation of 5-LO-derived products, 5-HETE and LTB4, while LY 311727 (50 μM), an inhibitor of GIIA, GV and GX sPLA2s had no significant effect (Fig. 1A), confirming the implication of type IV cPLA2 in that process. In a similar fashion, inhibiting type IV cPLA2 with pyrrophenone prevented the generation of PGE2 by approximately 90% (Fig. 1B, left panel) while inhibiting all of the other PLA2s present in neutrophils with LY 311727 had no significant impact. Similar results were obtained with neutrophils in which COX-2 was up-regulated by fMLP instead of GM-CSF/TNF (data not shown). Also, pyrrophenone had no discernible effect on the generation of PGE2 in response to exogenous AA (Fig. 1B, right panel), excluding possible non-specific interferences of the compound with cellular processes. NS 398, a highly specific inhibitor of the COX-2 isoform, efficiently prevented PGE2 production, but did not significantly affect the production of 5-LO-derived metabolites.
Fig. 1.
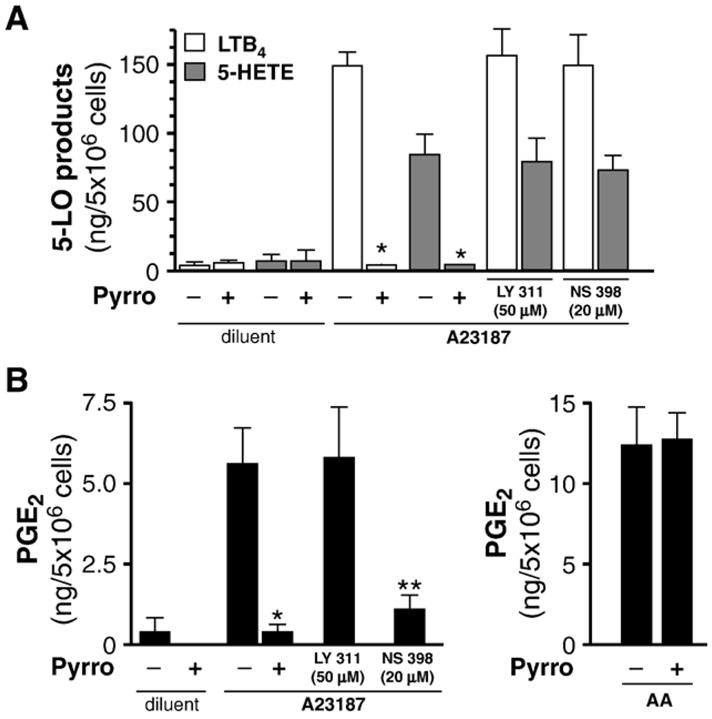
Type IV cPLA2 is chiefly involved in the release of arachidonate transformed in PGE2 through the COX-2 pathway in neutrophils. Cell suspensions, incubated for 120 min with GM-CSF/TNF (1.5 nM/100 nM) in adenosine-free conditions, were stimulated with 100 nM A23187 (or its diluent:DMSO) for 10 min, in absence or presence of the specific cPLA2 inhibitor pyrrophenone (100 nM), an inhibitor of the GIIA, GV and GX sPLA2s, LY 311727 (LY 311; 50 μM), or the specific COX-2 inhibitor, NS 398 (20 μM). (A) Generation of metabolites from the 5-LO (5-HETE and LTB4) pathways were measured by RP-HPLC, as described in the Experimental procedures section. (B) In addition to the conditions described in A), cells were stimulated with exogenous AA (10 μM). PGE2 levels were determined in cell-free supernatants by ELISA, as described in the Experimental procedures section. Results are presented as the mean±SEM, from n=3 separate experiments, each performed with different donors. *Significantly different from cells incubated in absence of pyrrophenone. **Significantly different from cells incubated in absence of NS 398.
Presence of type IV cPLA2 has been observed in distinct subcellular compartments, including the cytosol, plasma and nuclear membranes, as well as the endoplasmic reticulum and Golgi apparatus [35–37]; as such, sub-groups of type IV cPLA2 (s) could be differentially sensitive to inhibition. Along these lines, we conducted experiments designed to compare sensitivity of type IV cPLA2(s) implicated in the generation of LTB4 and of PGE2. GM-CSF/TNF-treated neutrophils were incubated with pyrrophenone at concentrations ranging from 100 pM to 100 nM then stimulated with A23187. Profiles of inhibition were produced for AA, LTB4 and PGE2. All of the profiles were found to be very similar with a common IC50 of approximately 4 nM (Fig. 2), supporting the idea that LTB4 and PGE2 originate from a pharmacologically-indistinguishable type IV cPLA2-dependent AA source.
Fig. 2.
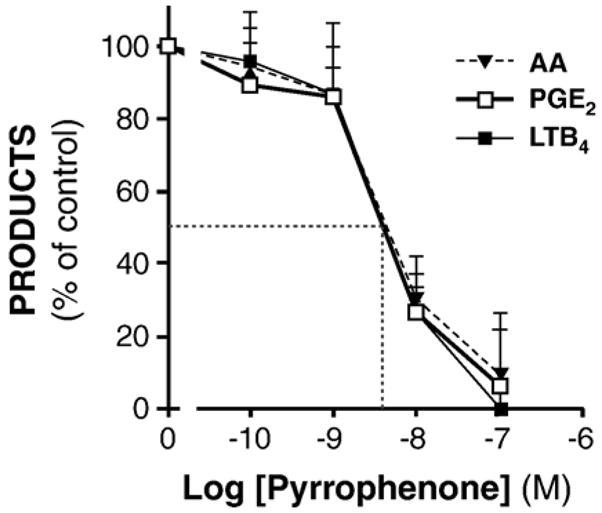
Pyrrophenone-elicited inhibition of AA release, LTB4 and PGE2 formation are pharmacologically indistinguishable. Cells were incubated for 120 min with GM-CSF/TNF in adenosine-free conditions, then stimulated with 100 nM A23187 for 10 min, alone or in presence of indicated concentrations of pyrrophenone. Arachidonate mobilization was determined by LC-MS, as described in the Experimental procedures section, whereas the generation of metabolites from the 5-LO or COX pathways were measured by RP-HPLC or ELISA. For each indicated metabolite, the amounts generated are shown as percentages, relative to samples stimulated in the absence of pyrrophenone (mean±SEM from n=3 experiments, each performed with different donors). Visually-determined IC50 is indicated by a dotted line.
In addition, experiments were performed on cells stimulated with inflammatory agonists acting on cellular receptors. As was observed with the Ca2+-ionophore, presence of pyrrophenone prevented the generation of PGE2 by at least 90% in response to fMLP or opsonized-zymosan particles (Fig. 3). Here also, NS 398 largely prevented PGE2 biosynthesis, confirming paramount implication of COX-2 in this process. Taken together, these results support a central role for type IV cPLA2 in the release of AA destined to the biosynthesis of COX-2-derived PGE2 by neutrophils.
Fig. 3.
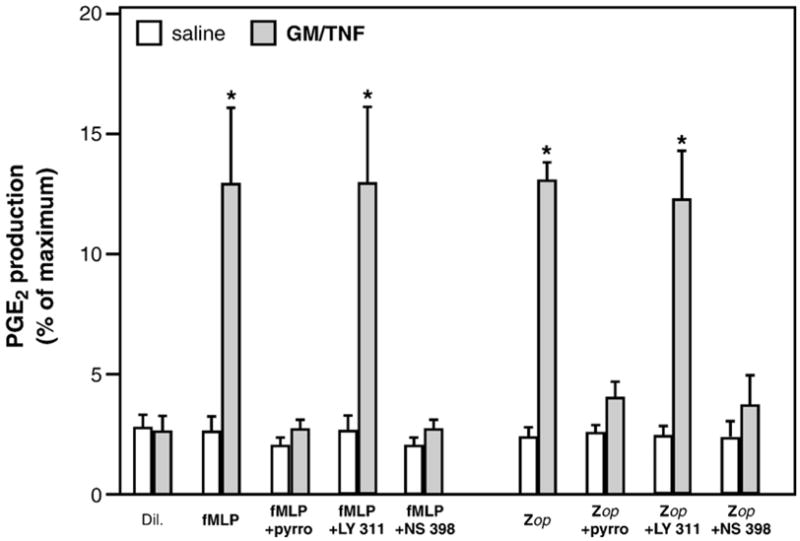
Pyrrophenone inhibits COX-2-derived PGE2 production elicited by inflammatory mediators involving receptor engagement. Cells were treated for 120 min with saline or GM-CSF/TNF in adenosine-free conditions, then stimulated with 100 nM fMLP or opsonized zymosan (Zop), for 15 min. Stimulations were performed in absence or presence of pyrrophenone (100 nM), LY 311727 (50 μM), or NS 398 (20 μM). PGE2 levels were determined in cell-free supernatants by ELISA. Results are expressed as percentages of production, relative to the value obtained in AA-stimulated cells (10 μM; 100%=14.5±3.3 ng/5×106 cells). Results presented are the mean±SEM from at least n=3 experiments, each performed with different donors. *Significantly higher than in saline-treated cells.
3.2. Arachidonic acid utilization
Metabolism of available AA was then assessed through the 5-LO and COX-2 pathways. Accumulation of 5-HPETE is the result of the first enzymatic reaction catalyzed by 5-LO and increased with increasing concentrations of available AA, as measured by the determination of its non-enzymatic conversion to 5-HETE. LTA4 biosynthesis, as assessed by the accumulation of its non-enzymatic conversion to 6-trans-LTB4, followed in contrast a bell-shaped curve peaking at a concentration of 5 μM AA above which it rapidly decreased, remaining minimal up to the highest concentration used (Fig. 4A), as documented earlier [38]. On the other hand, the biosynthesis of PGE2 by neutrophils increased as a simple function of substrate concentration (Fig. 4B). Thus, free AA appears to be differentially metabolized by the two pathways, with higher concentrations eventually favoring the production of PGE2 over that of LTB4.
Fig. 4.
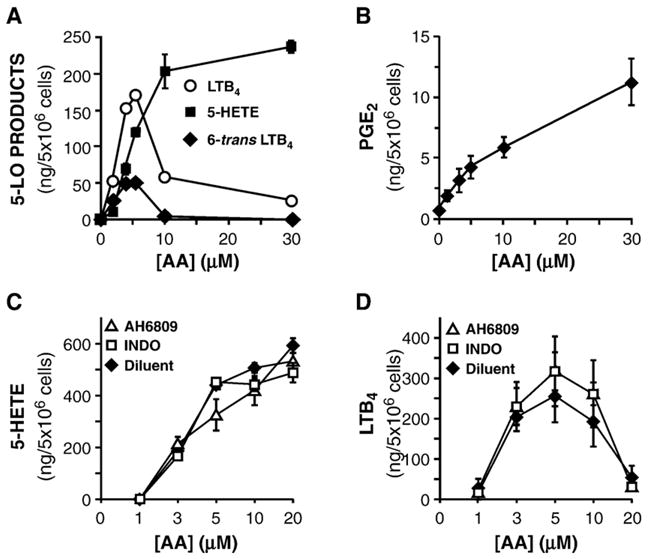
Exogenous arachidonate is utilized differentially by the 5-LO and COX pathways in neutrophils. (A & B) Cells were treated for 60 min with GM-CSF/TNF in adenosine-free conditions, then stimulated with indicated concentrations of arachidonate for 10 min. Generation of 5-HETE, 6-trans-LTB4, and LTB4 (A) was determined by RP-HPLC. PGE2 generation (B) was assayed by ELISA. (C & D) Cells were treated as in (A), alone or in the presence of the COX-1/COX-2 inhibitor, indomethacin (INDO; 10 μM), or the non-specific EP receptor antagonist, AH 6809 (10 μM). Generation of 5-HETE (C) and of LTB4 (D) was determined by RP-HPLC. In each panel, results are presented as the mean±SEM from n=3 experiments, each performed with different donors.
PGE2 is well known for its capacity to prevent LTB4 generation from neutrophils [13,16]; thus, in the present set of experiments where 5-LO- and COX-derived metabolites can be simultaneously produced, extracellular concentrations of PGE2 may potentially reach levels which could explain, at least in part, the blunted biosynthesis of LTB4 obtained when AA concentrations increase. This specific issue was addressed by two pharmacological approaches. Firstly, PGE2 production was blocked with the COX-1/COX-2 inhibitor, indomethacin. Secondly, PGE2 receptor engagement was prevented by AH 6809, a PGE2 receptor antagonist. Blocking PGE2 metabolism by either of these methods did not have any significant impact on the generation of 5-HETE (Fig. 4C) or of LTB4 (Fig. 4D). In the case of the latter, the bell-shaped curve remained largely intact, with the generation of LTB4 still being dramatically blunted at higher AA concentrations. This set of results indicates that, in these experimental conditions, PGE2 is not responsible for the decreased LTB4 generation observed at higher AA concentrations.
3.3. Intracellular calcium and regulation of PGE2 production
Increases in intracellular Ca2+ levels (Ca2+i) play a pivotal role in the generation of eicosanoids, namely in the activation of type IV cPLA2, but also in the activation and translocation of 5-LO to cellular membranes where the enzyme can associate with 5-lipoxygenase activating-protein (FLAP) [39–41]. On the other hand, the implication of Ca2+i in PGE2 biosynthesis is less well understood. In order to clarify this aspect, neutrophils were incubated in normal, or in Ca2+-free conditions, as described earlier [42], treated with GM-CSF/TNF then stimulated, either with the Ca2+-ionophore A23187 (1 μM) or with exogenous AA (10 μM). Ca2+ depletion markedly prevented the production of PGE2 in response to A23187, the inhibition being of approximately 85%. Interestingly, depletion of Ca2+ did not prevent the up-regulation of COX-2 (insert). In response to exogenous AA, in contrast, the generation of PGE2 in normal- and Ca2+-depleted cells was virtually undistinguishable (Fig. 5A, left panel). Similar results were obtained in which COX-2 was up-regulated with fMLP instead of GM-CSF/TNF (data not shown). In an additional set of experiments, cells were treated with GM-CSF/TNF, then stimulated with fMLP: Ca2+-depletion in these conditions also significantly decreased PGE2 production, albeit in a more modest fashion (Fig. 5A, right panel). Together, these results clearly indicate that an elevation of Ca2+i is pivotal for the availability of AA (e.g. activation of type IV cPLA2) but, in contrast to its actions on 5-LO activation, Ca2+ does not impact on COX-2 expression or activity.
Fig. 5.
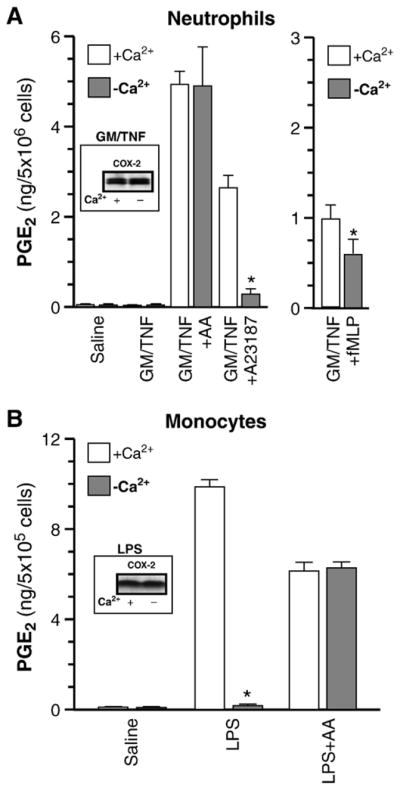
Elevation in intracellular Ca2+ is required for AA availability, but not for COX-2 up-regulation. (A) Cell suspensions were incubated either for 60 min with GM-CSF/TNF in normal, or in Ca2+-free (incubated in HBSS supplemented with 3 mM EGTA) conditions. Cells were stimulated with arachidonic acid (AA) or with A23187 for 10 min (left panel), or with 100 nM fMLP (right panel). PGE2 levels in cell-free supernatants were determined by ELISA. Results presented are the mean±SEM from n=3 experiments, each performed with different donors. *Significantly lower than in normal conditions. Insert: neutrophils were incubated with GM-CSF/TNF in normal, or Ca2+-free conditions then processed for the determination of COX-2 protein levels by western immunoblotting. (B) Monocytes were treated with 2 μg/ml LPS for 16 hr in normal, or Ca2+-free (medium supplemented with 5 mM EGTA) conditions. PGE2 levels in cell-free supernatants were determined by ELISA. AA stimulation: cells were washed twice with PBS, fresh medium (normal or Ca2+-free) was added, then cells were stimulated with 10 μM AA for an additional 60 min. Results presented are the mean ± SEM from n = 3 experiments, each performed with different donors. *Significantly lower than in normal conditions. Insert: monocytes incubated with LPS in normal, or Ca2+-free conditions, then processed for the determination of COX-2 protein levels by western immunoblotting.
Given this differential implication of Ca2+i on 5-LO and COX-2 pathways observed in neutrophils, it was of interest to validate these findings in the other circulating leukocyte type in which COX-2 can be up-regulated, monocytes. To this end, monocytes were treated with LPS, a condition sufficient to elicit COX-2-dependent PGE2 release [25]. In these cells, Ca2+ depletion essentially obliterated the COX-2-derived PGE2 generation, while it did not affect monocyte response to exogenous AA (Fig. 5B). COX-2 up-regulation in monocytes was not altered by the absence of Ca2+ (insert), entirely concurring with results obtained in neutrophils. In both leukocyte types, presence of 20 μM NS-398 consistently prevented the production of PGE2 production by more than 90% (data not shown) once more confirming a predominantly COX-2-mediated event, as reported earlier [8,25].
3.4. Neutrophil-derived eicosanoids vs. COX-2
Next, the main eicosanoids generated by neutrophils, PGE2, TXA2 and LTB4, were assessed for their involvement in the COX-2 pathway. In experiments designed to determine their impact on COX-2 up-regulation, PGE2 or the stable analog of TXA2, carbocyclic TXA2, had no detectable effect on COX-2 expression at any of the monitored time points (Fig. 6A). LTB4, in contrast, was able to up-regulate COX-2 (Fig. 6B); its action appeared to be linked to specific engagement of its receptor (BLTR1) as evidenced by the use of a structural isomer showing 20–50 times less affinity for BLTR1: 6-trans-LTB4 [43] which, in comparison to LTB4, poorly induced COX-2. When used with fMLP, LTB4 had an additive effect (Fig. 6C). Together, these results indicate that LTB4, but neither PGE2 nor TXA2, may positively regulate COX-2 protein levels. In cells treated with LTB4 for 120 min, stimulation with A23187 or with AA led to significantly-creased PGE2 generation over untreated cells (Fig. 6D). PGE2 production was about half of that obtained in cells treated with fMLP instead.
Fig. 6.
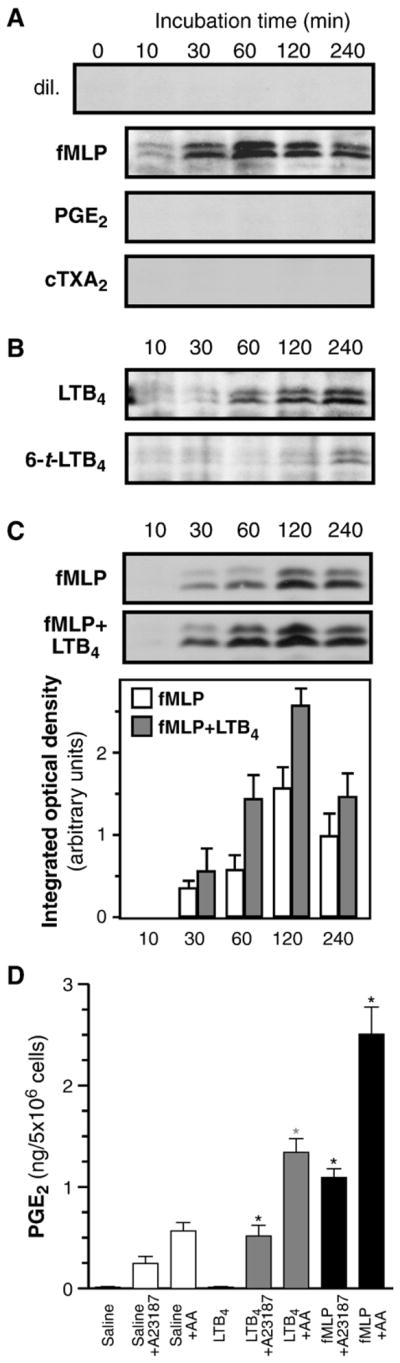
LTB4 up-regulates COX-2 protein expression in neutrophils through engagement of its receptor. (A) Cells were incubated with indicated agonists (each used at a concentration of 100 nM) for the indicated times (B) Cells were incubated with LTB4 or with its less-active analog 6-trans-LTB4, (each used at a concentration of 100 nM) for the indicated times. (C) Cells were incubated with fMLP, alone or in combination with LTB4 for the indicated times. Samples were processed for the determination of COX-2 by western immunoblotting, as described in the experimental procedures section. In each panel, shown are immunoblots obtained in one experiment, typical of three separate experiments, each performed with different donors. In panel C, COX-2 densitometry analyzes were performed as described in Experimental procedures; results represent the average (n=3, ±SEM) from data obtained with different 3 donors. (D) Cells were treated with saline, LTB4 (100 nM) or fMLP (100 nM), then stimulated with AA or A23187. PGE2 levels in cell-free supernatants were determined by ELISA. Results presented are the mean±SEM from n=3 experiments, each performed with different donors. *Significantly higher than in untreated cells.
In a separate set of experiments, GM-CSF/TNF-treated neutrophils were stimulated either with LTB4, or cTXA2 (both at 300 nM), and resulting PGE2 generation was determined. Neither compound was able to elicit detectable PGE2 production (data not shown). Thus, of the main eicosanoids that neutrophils generate, only LTB4 appears to be able to up-regulate COX-2 expression, while none of the assayed compounds stimulate the generation of PGE2.
3.5. Constitutive expression of mPGE synthase-1 in neutrophils
In neutrophils, the final step in prostanoid production is the transformation of PGH2 into PGE2 and TXA2. Production of the former can happen either non-enzymatically or through one of several PGE-synthases (PGES), while the latter is formed by the action of thromboxane synthase. To our knowledge, the PGES isoform(s) contributing to the enzymatic transformation of PGH2 into PGE2 in neutrophils have not been documented. We considered here the three known human PGES isoforms: mPGES-1, mPGES-2 and cPGES and assessed their mRNA expression by real-time PCR [44]. Of these, only mPGES-1 was detected in neutrophils. Its mRNA expression was increased in GM-CSF/TNF-treated cells, approximately 2 fold over basal levels at t=15 min (Fig. 7A). At the protein level, on the other hand, mPGES-1 was readily detected in unstimulated cells and its expression was not significantly increased, either by fMLP, GM-CSF/TNF or LPS treatment, be that at t=2 h (Fig. 7B) or t=4 h (data not shown). Moreover, activation of the A2A adenosine receptor, which potentiates COX-2 expression in these conditions [13,30], also failed to significantly alter the expression of mPGES-1 (Fig. 7C). Subcellular fractionation experiments, using the well-characterized nitrogen-bomb disruption technique coupled to fractionation on Percoll cushion [29], revealed a co-localization for mPGES-1 and COX-2 in fractions which also contain the Golgi apparatus and endoplasmic reticulum (Fig. 7D), consistent with the reported functional coupling of these enzymes [21]. Furthermore, thromboxane synthase, also constitutively-expressed in neutrophils [13], was found in these same fractions. Thus, it appears that mPGES-1 is the only PGES isoform present in neutrophils, that it is readily expressed in freshly-isolated cells, and localized in vicinity of other enzymes of the prostanoid biosynthetic machinery.
Fig. 7.
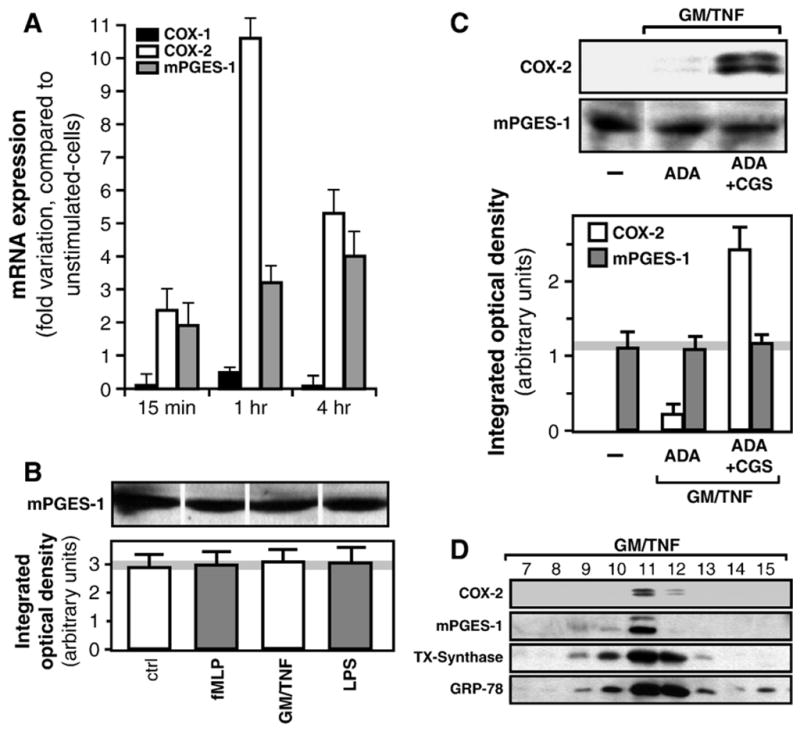
Microsomal PGES-1 is constitutively expressed in neutrophils and co-localizes with cyclooxygenase-2. (A) Cells were incubated with saline, or with GM-CSF/TNF for indicated times, than processed for the determination of COX-1, COX-2 and mPGES-1 mRNA expression by real-time PCR, as described in the Experimental procedures section. Results are expressed in fold-variations of mRNA expression, compared to that observed in saline-treated cells, and values are normalized for GAPDH. Results are presented as the mean±SD from two experiments performed with different donors. (B) Cell suspensions were incubated for 120 min with fMLP, or LPS, or GM-CSF/TNF, and samples were processed for the determination of mPGES-1 by western immunoblotting, as described in the Experimental procedures section. Shown is one immunoblot, typical of three experiments, each performed with different donors, as well as densitometric measurements (mean±SEM) obtained from these 3 donors. (C) Cell suspensions were incubated for 120 min with GM-CSF/TNF, alone or in combination with adenosine deaminase (ADA) and with the specific A2A receptor agonist, CGS 21680 (CGS). Samples were processed for the determination of COX-2 and mPGES-1 by western immunoblotting. Shown is one immunoblot, typical of three experiments, each performed with different donors, as well as densitometric measurements (mean±SEM) obtained from these 3 donors. (D) Cells were incubated for 120 min with GM-CSF/TNF, then subjected to cell cavitation by the nitrogen bomb technique and separated into 18 fractions by centrifugation on Percoll gradient, as described in the Experimental procedures section. Samples processed for the determination of COX-2, mPGES-1 and thromboxane (TX)-Synthase expression by western immunoblotting. GRP-78 was used as a marker of ER and Golgi. Fractions 7 to 15 are shown.
Steps governing the biosynthesis of eicosanoids by neutrophils, including some of the main characterizations made in the present study, are schematized in Fig. 8.
Fig. 8.
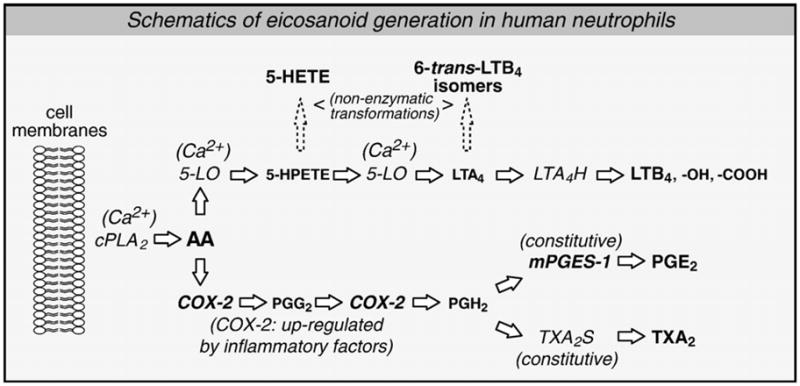
Updates in the characterization of the COX-2 pathways in human neutrophils. In response to a stimulus, membrane-esterified arachidonic acid (AA) is mobilized by type IV cytosolic phospholipase A2 (cPLA2) in a Ca2+-dependent fashion. AA can be utilized either by 5-lipoxygenase (5-LO) or by the inducible cyclooxygenase isoform (COX-2). 5-LO pathway: 5-LO, a Ca2+-dependent enzyme, catalyzes two reactions: transformation of AA into 5-hydroperoxyeicosatetranoic acid (5-HPETE), and dehydration of the latter in leukotriene (LT)A4. LTA4 is metabolized in the active metabolite LTB4 by the LTA4 hydrolase. Neutrophils also express an omega carboxylase which sequentially transforms LTB4 into less active metabolites: 20-OH- and 20-COOH-LTB4. COX-2 pathway: COX-2, which can be up-regulated in neutrophils, also catalyses two reactions by which AA is transformed into prostaglandin (PG)H2. PGH2 can then be metabolized into the active metabolite PGE2 by the microsomal PGE2 synthase (mPGES)-1, or into TXA2 by TXA2 synthase (TXA2S), both of which being constitutively-expressed in neutrophils. Thus, COX-2 expression is held to be the main limiting factor for the biosynthesis of prostanoids in these cells. Available information indicates that LTB4, PGE2 and TXA2 are the main eicosanoids produced by human neutrophils, in vitro.
4. Discussion
In the present study, we characterized important aspects of the COX-2-derived PGE2 biosynthesis. The first aspect was PLA2-mediated liberation of AA. Human neutrophils express group IV, V and X PLA2s [19]; group IV cPLA2 has been identified earlier as the main isoform implicated in leukotriene and platelet-activating factor synthesis [45]. Comprehensive data presented herein extend a central role for this same isoform in the generation of PGE2 as well. Indeed, specific inhibition of type IV cPLA2 prevented PGE2 production by neutrophils, be that in response to pharmacological Ca2+ influx (A23187), or to the engagement of cell surface receptors (fMLP, Zop), while inhibition of other PLA2 isotypes potentially present in neutrophils had no discernible effect. Moreover, inhibition of LTB4 and PGE2 biosynthesis by pyrrophenone were pharmacologically indistinguishable, consistent with a common cPLA2 providing AA to both the 5-LO and COX pathways.
Our present observations indicate that regulatory mechanisms defining the generation of LTB4 and PGE2 in neutrophils may be found downstream of AA release. A first hint of these mechanisms could be obtained by treating neutrophils with exogenous AA. Increasing concentrations resulted in proportional amounts of PGE2 produced. In sharp contrast, generation of LTB4 was markedly reduced at AA concentrations higher than 5 μM. Conversion of AA to 5-HPETE followed a function of AA availability while the transformation of 5-HPETE to LTA4 followed a bell-shaped curve, indicating that the second enzymatic reaction catalyzed by 5-LO, but not the first, is affected at higher AA concentrations. Although the explanation for this inhibition is not clear, high concentrations of AA were previously reported to bind to FLAP [46] and to inhibit LT biosynthesis [38]. Also, in a recent study where 5-lipoxygenase-activating protein (FLAP) was shown to be present in neutrophil membranes as monomers and homodimers, and where the functional importance of the dimer (over the monomer) in LT biosynthesis was highlighted [47], it was observed that high AA concentrations decreased FLAP dimer levels, supporting the possibility that high concentrations of AA could disrupt or destabilize the FLAP dimer, an effect which correlates with the suppression of 5-LO product biosynthesis by the fatty acid. In the present study, we also assessed the possibility that PGE2 formed in these experiments may loop back through an autocrine effect on EP receptors present at the cell surface and inhibit LTB4 generation. However, inhibition of PGE2 production or blockage of PGE2 receptors failed to prevent the inhibition of LTB4 biosynthesis observed at higher AA concentrations, indicating that PGE2 is not involved in the inhibition of LTB4 generation in this system. While the peculiar profile of LTB4 synthesis in response to exogenous AA requires further investigation, results obtained herein clearly indicate that PGE2 formed during these incubations is not responsible for blocking LTB4 formation. The modulation in the profile of lipid mediators observed at different concentrations of available AA, coupled with the distinct (and often opposite) physiological actions of LTB4 and PGE2, may have profound consequences on the evolution of an inflammatory response.
A second hint of the regulatory steps governing PGE2 synthesis in neutrophils was gained by looking into the role of intracellular Ca2+. Apart from exogenous AA, Ca2+-ionophore was the strongest inducer for PGE2 production, further supporting a role for the Ca2+-dependent cPLA2. Depletion of Ca2+ had a lesser impact in the case of fMLP than with A23187, but that impact was still significant. This differential impact may reflect the relative influences of intracellular and extracellular Ca2+ in the process of cPLA2 activation elicited by a Ca2+-ionophore or by a receptor-mediated event. It seemed clear, however, that the impact of Ca2+ on the COX-2 pathway was limited at the step of substrate availability, since the expression and activity of COX-2 were found to be largely Ca2+-independent. Observations made with neutrophils found confirmation in human monocytes thereby generalizing this differential involvement of Ca2+ in the 5-LO and COX pathways in leukocytes.
Cross-talk between the two pathways was documented, namely on the potential of the main eicosanoids produced by neutrophils to impact on either pathway. It is already well characterized that LTB4 production can be inhibited by pre-treating the cells with PGE2 [13,16]. However, when PGE2 and LTB4 were allowed to be produced simultaneously, as was the case in the present study, little – if any – effect on the 5-LO pathway could be observed. Shunting of the PGE2 pathway, either by blocking its production or by blocking its receptors, had no significant effect, positive or negative, on the generation of 5-HETE and LTB4, clearly indicating that EP receptors need to be engaged prior to cell stimulation in order to have an impact of the LT-generating machinery. On the other hand, LTB4, but not PGE2 or TXA2, increased COX-2 protein levels, indicating that one of the early signals leading to COX-2 up-regulation and increased PGE2 biosynthesis is a product of the 5-LO pathway: LTB4.
Once AA is converted to PGH2 by COX, it can be transformed to PGE2 either non-enzymatically, or through the activity of a PGE synthase (PGES). In neutrophils, only one of the three known human PGES isoforms, mPGES-1, was detected at the mRNA level. Moreover, at the protein level, mPGES-1 was readily detected in resting neutrophils; surprisingly, its expression was not significantly altered by any of the treatments. In a number of studies, mPGES-1 has been reported to be up-regulated and preferentially coupled to COX-2 [21]. In spite of this remarkable apparent difference between cell types, the relatively robust level of mPGES-1 protein expression in resting neutrophils, coupled with basal constitutive levels of COX-2 in these cells [23], is in good accordance with the observed capacity of neutrophils to rapidly generate PGE2 from either endogenous or exogenous AA sources [13]. In addition, mPGES-1 was found to co-localize with COX-2, in endoplasmic reticulum/Golgi containing fractions, a result which, at least circumstantially, supports their functional coupling in neutrophils. Presence of thromboxane-synthase in these same fractions further strengthened this concept. While it remains possible that other PGES isoforms, which expression might be modulated by inflammatory factors, be involved, our data nonetheless point for a central implication of the mPGES-1 in the COX-2-derived generation of PGE2 in neutrophils, as reported in other cell types [21].
In summary, we provided comprehensive evidence for a central implication of the cPLA2 type IV isoform for the biosynthesis of PGE2 in human neutrophils. Downstream of cPLA2, studies on AA trafficking indicated little cross-talk between the 5-LO and COX pathways, except for the fact that LTB4 could increase COX-2 protein levels. Increase in Ca2+i is required at several steps in the 5-LO pathway; in contrast, its implication appears limited to cPLA2 activation in the COX-2 pathway in leukocytes. Microsomal PGES-1 isoform was found to be constitutively expressed in neutrophils and co-localized with COX-2, supporting the hypothesis of a functional coupling for the enzymatic conversion of PGH2 into PGE2. Taken together, results presented herein elucidate important aspects of the metabolism of AA through the COX-2 pathway by further defining the generation of PGE2 in neutrophils.
Acknowledgments
This work is supported by grants from the Canadian Institutes of Health Research (CIHR, to MP; grants nos. MOP-64315 & NSM-72200). MP is the recipient of a New Investigator Scholarship (CIHR—The Arthritis Society of Canada). NF is the recipient of a post-doctoral fellowship from the CIHR. MSO is the recipient of a studentship from the Canadian Arthritis Network. Authors wish to thank Dr. K. Seno (Shionogi Research Laboratories) for providing pyrrophenone.
Abbreviations
- 5-LO
5-lipoxygenase
- AA
arachidonic acid
- COX
cyclooxygenase
- fMLP
n-formyl-Methionyl-Leucyl-Phenylalanine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HBSS
Hank’s balanced salt solution
- LC-MS
liquid chromatography-mass spectrometry
- LPS
lipopolysaccharide
- LT
leukotriene
- PG
prostaglandin
- PLA2
phospholipase A2
- RP-HPLC
reverse phase-high performance liquid chromatography
- TNF-α
tumor necrosis factor-α
- TX
thromboxane
References
- 1.McColl SR, Beauseigle D, Gilbert C, Naccache PH. Priming of the human neutrophil respiratory burst by granulocyte-macrophage colony-stimulating factor and tumor necrosis factor-alpha involves regulation at a post-cell surface receptor level. Enhancement of the effect of agents which directly activate G proteins. J Immunol. 1990;145:3047–3053. [PubMed] [Google Scholar]
- 2.Edwards SW. Biochemistry and Physiology of the Neutrophil. Cambridge University Press; 1994. [Google Scholar]
- 3.Cassatella MA. Neutrophil-derived proteins: selling cytokines by the pound. Adv Immunol. 1999;73:369–509. doi: 10.1016/s0065-2776(08)60791-9. [DOI] [PubMed] [Google Scholar]
- 4.Waksman Y, Golde DW, Savion N, Fabian I. Granulocyte-macrophage colony-stimulating factor enhances cationic antimicrobial protein synthesis by human neutrophils. J Immunol. 1990;144:3437–3443. [PubMed] [Google Scholar]
- 5.Fasano MB, Wells JD, Mccall CE. Clin Immunol Immunopathol. 1998;87:304–308. doi: 10.1006/clin.1998.4545. [DOI] [PubMed] [Google Scholar]
- 6.Maloney CG, Kutchera WA, Albertine KH, McIntyre TM, Prescott SM, Zimmerman GA. Inflammatory agonists induce cyclooxygenase type 2 expression by human neutrophils. J Immunol. 1998;160:1402–1410. [PubMed] [Google Scholar]
- 7.Niiro H, Otsuka T, Izuhara K, Yamaoka K, Ohshima K, Tanabe T, Hara S, Nemoto Y, Tanaka Y, Nakashima H, Niho Y. Regulation by interleukin-10 and interleukin-4 of cyclooxygenase-2 expression in human neutrophils. Blood. 1997;89:1621–1628. [PubMed] [Google Scholar]
- 8.Pouliot M, Gilbert C, Borgeat P, Poubelle PE, Bourgoin S, Creminon C, Maclouf J, McColl SR, Naccache PH. Expression and activity of prostaglandin endoperoxide synthase-2 in agonist-activated human neutrophils. FASEB J. 1998;12:1109–1123. doi: 10.1096/fasebj.12.12.1109. [DOI] [PubMed] [Google Scholar]
- 9.Busse WW. Leukotrienes and inflammation. Am J Respir Crit Care Med. 1998;157:S210–S213. (discussion S247–8) [PubMed] [Google Scholar]
- 10.Katori M. Pharmacology of prostaglandins; their profile and characterization in the body. Nippon Yakurigaku Zasshi. 1989;94:159–171. doi: 10.1254/fpj.94.159. [DOI] [PubMed] [Google Scholar]
- 11.Lehmeyer JE, Johnston RB., Jr Effect of anti-inflammatory drugs and agents that elevate intracellular cyclic AMP on the release of toxic oxygen metabolites by phagocytes: studies in a model of tissue-bound IgG. Clin Immunol Immunopathol. 1978;9:482–490. doi: 10.1016/0090-1229(78)90144-7. [DOI] [PubMed] [Google Scholar]
- 12.O’Flaherty JT, Kreutzer DL, Ward PA. Effect of prostaglandins E1, E2 and F2alpha on neutrophil aggregation. Prostaglandins. 1979;17:201–210. doi: 10.1016/0090-6980(79)90039-x. [DOI] [PubMed] [Google Scholar]
- 13.Pouliot M, Fiset ME, Masse M, Naccache PH, Borgeat P. Adenosine up-regulates cyclooxygenase-2 in human granulocytes: impact on the balance of eicosanoid generation. J Immunol. 2002;169:5279–5286. doi: 10.4049/jimmunol.169.9.5279. [DOI] [PubMed] [Google Scholar]
- 14.Rivkin I, Rosenblatt J, Becker EL. The role of cyclic AMP in the chemotactic responsiveness and spontaneous motility of rabbit peritoneal neutrophils. The inhibition of neutrophil movement and the elevation of cyclic AMP levels by catecholamines, prostaglandins, theophylline and cholera toxin. J Immunol. 1975;115:1126–1134. [PubMed] [Google Scholar]
- 15.Zurier RB, Weissmann G, Hoffstein S, Kammerman S, Tai HH. Mechanisms of lysosomal enzyme release from human leukocytes: II. Effects of cAMP and cGMP, autonomic agonists, and agents which affect microtubule function. J Clin Invest. 1974;53:297–309. doi: 10.1172/JCI107550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ham EA, Soderman DD, Zanetti ME, Dougherty HW, McCauley E, Kuehl FA., Jr Inhibition by prostaglandins of leukotriene B4 release from activated neutrophils. Proc Natl Acad Sci U S A. 1983;80:4349–4353. doi: 10.1073/pnas.80.14.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakraborti S. Phospholipase A(2) isoforms: a perspective. Cell Signal. 2003;15:637–665. doi: 10.1016/s0898-6568(02)00144-4. [DOI] [PubMed] [Google Scholar]
- 18.Marshall J, Krump E, Lindsay T, Downey G, Ford DA, Zhu P, Walker P, Rubin B. Involvement of cytosolic phospholipase A2 and secretory phospholipase A2 in arachidonic acid release from human neutrophils. J Immunol. 2000;164:2084–2091. doi: 10.4049/jimmunol.164.4.2084. [DOI] [PubMed] [Google Scholar]
- 19.Degousee N, Ghomashchi F, Stefanski E, Singer A, Smart BP, Borregaard N, Reithmeier R, Lindsay TF, Lichtenberger C, Reinisch W, Lambeau G, Arm J, Tischfield J, Gelb MH, Rubin BB. Groups IV, V, and X phospholipases A2s in human neutrophils: role in eicosanoid production and gram-negative bacterial phospholipid hydrolysis. J Biol Chem. 2002;277:5061–5073. doi: 10.1074/jbc.M109083200. [DOI] [PubMed] [Google Scholar]
- 20.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 21.Murakami M, Kudo I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog Lipid Res. 2004;43:3–35. doi: 10.1016/s0163-7827(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 22.Yu CL, Huang MH, Kung YY, Tsai CY, Tsai YY, Tsai ST, Huang DF, Sun KH, Han SH, Yu HS. Interleukin-13 increases prostaglandin E2 (PGE2) production by normal human polymorpho-nuclear neutrophils by enhancing cyclooxygenase 2 (COX-2) gene expression. Inflamm Res. 1998;47:167–173. doi: 10.1007/s000110050312. [DOI] [PubMed] [Google Scholar]
- 23.Gilbert C, Poubelle PE, Borgeat P, Pouliot M, Naccache PH. Crystal-induced neutrophil activation: VIII. Immediate production of prostaglandin E2 mediated by constitutive cyclooxygenase 2 in human neutrophils stimulated by urate crystals. Arthritis Rheum. 2003;48:1137–1148. doi: 10.1002/art.10851. [DOI] [PubMed] [Google Scholar]
- 24.Böyum A. Isolation of mononuclear cells and granulocytes from human blood: isolation of mononuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest, Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- 25.Pouliot M, Baillargeon J, Lee JC, Clel LG, James MJ. Inhibition of prostaglandin endoperoxide synthase-2 expression in stimulated human monocytes by inhibitors of p38 mitogen-activated protein kinase. J Immunol. 1997;158:4930–4937. [PubMed] [Google Scholar]
- 26.Surette ME, Palmantier R, Gosselin J, Borgeat P. Lipopolysaccharides prime whole human blood and isolated neutrophils for the increased synthesis of 5-lipoxygenase products by enhancing arachidonic acid availability: involvement of the CD14 antigen. J Exp Med. 1993;178:1347–1355. doi: 10.1084/jem.178.4.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonald PP, McColl SR, Naccache PH, Borgeat P. Activation of the human neutrophil 5-lipoxygenase by leukotriene B4. Br J Pharmacol. 1992;107:226–232. doi: 10.1111/j.1476-5381.1992.tb14491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgeat P, Picard S, Vallerand P, Bourgoin S, Odeimat A, Sirois P, Poubelle PE. Automated on-line extraction and profiling of lipoxygenase products of arachidonic acid by high-performance liquid chromatography. Methods Enzymol. 1990;187:98–116. doi: 10.1016/0076-6879(90)87014-t. [DOI] [PubMed] [Google Scholar]
- 29.Borregaard N, Heiple JM, Simons ER, Clark RA. Subcellular localization of the b-cytochrome component of the human neutrophil microbicidal oxidase: translocation during activation. J Cell Biol. 1983;97:52–61. doi: 10.1083/jcb.97.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cadieux JS, Leclerc P, St-Onge M, Dussault AA, Laflamme C, Picard S, Ledent C, Borgeat P, Pouliot M. Potentiation of neutrophil cyclooxygenase-2 by adenosine: an early anti-inflammatory signal. J Cell Sci. 2005;118:1437–1447. doi: 10.1242/jcs.01737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McColl SR, St-Onge M, Dussault AA, Laflamme C, Bouchard L, Boulanger J, Pouliot M. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J. 2006;20:187–189. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T. Pyrrolidine inhibitors of human cytosolic phospholipase A2: Part 2. synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘pyrrophenone’. Bioorg Med Chem Lett. 2001;11:587–590. doi: 10.1016/s0960-894x(01)00003-8. [DOI] [PubMed] [Google Scholar]
- 33.Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, Seno K. Characterization of a novel inhibitor of cytosolic phospholipase A2alpha, pyrrophenone. Biochem J. 2002;363:727–735. doi: 10.1042/0264-6021:3630727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krump E, Picard S, Mancini J, Borgeat P. Suppression of leukotriene B4 biosynthesis by endogenous adenosine in ligand-activated human neutrophils. J Exp Med. 1997;186:1401–1406. doi: 10.1084/jem.186.8.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy R. The role of cytosolic phospholipase A2-alfa in regulation of phagocytic functions. Biochim Biophys Acta. 2006;1761:1323–1334. doi: 10.1016/j.bbalip.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Pouliot M, McDonald PP, Krump E, Mancini JA, McColl SR, Weech PK, Borgeat P. Colocalization of cytosolic phospholipase A2, 5-lipoxygenase, and 5-lipoxygenase-activating protein at the nuclear membrane of A23187-stimulated human neutrophils. Eur J Biochem. 1996;238:250–258. doi: 10.1111/j.1432-1033.1996.0250q.x. [DOI] [PubMed] [Google Scholar]
- 37.Nahas N, Waterman WH, Sha’afi RI. Granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes phosphorylation and an increase in the activity of cytosolic phospholipase A2 in human neutrophils. Biochem J. 1996;313(Pt 2):503–508. doi: 10.1042/bj3130503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surette ME, Krump E, Picard S, Borgeat P. Activation of leukotriene synthesis in human neutrophils by exogenous arachidonic acid: inhibition by adenosine A(2a) receptor agonists and crucial role of autocrine activation by leukotriene B(4) Mol Pharmacol. 1999;56:1055–1062. doi: 10.1124/mol.56.5.1055. [DOI] [PubMed] [Google Scholar]
- 39.Kargman S, Rousseau P, Reid GK, Rouzer CA, Mancini JA, Rands E, Dixon RA, Diehl RE, Leveille C, Nathaniel D, et al. Leukotriene synthesis in U937 cells expressing recombinant 5-lipoxygenase. J Lipid Mediat. 1993;7:31–45. [PubMed] [Google Scholar]
- 40.Kargman S, Rouzer CA. Studies on the regulation, biosynthesis, and activation of 5-lipoxygenase in differentiated HL60 cells. J Biol Chem. 1989;264:13313–13320. [PubMed] [Google Scholar]
- 41.Rouzer CA, Rands E, Kargman S, Jones RE, Register RB, Dixon RA. Characterization of cloned human leukocyte 5-lipoxygenase expressed in mammalian cells. J Biol Chem. 1988;263:10135–10140. [PubMed] [Google Scholar]
- 42.Krump E, Pouliot M, Naccache PH, Borgeat P. Leukotriene synthesis in calcium-depleted human neutrophils: arachidonic acid release correlates with calcium influx. Biochem J. 1995;310(Pt 2):681–688. doi: 10.1042/bj3100681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin V, Ronde P, Unett D, Wong A, Hoffman TL, Edinger AL, Doms RW, Funk CD. Leukotriene binding, signaling, and analysis of HIV coreceptor function in mouse and human leukotriene B4 receptor-transfected cells. J Biol Chem. 1999;274:8597–8603. doi: 10.1074/jbc.274.13.8597. [DOI] [PubMed] [Google Scholar]
- 44.Dussault AA, Pouliot M. Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol Proced Online. 2006;8:1–10. doi: 10.1251/bpo114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mancini JA, Abramovitz M, Cox ME, Wong E, Charleson S, Perrier H, Wang Z, Prasit P, Vickers PJ. 5-lipoxygenase-activating protein is an arachidonate binding protein. FEBS Lett. 1993;318:277–281. doi: 10.1016/0014-5793(93)80528-3. [DOI] [PubMed] [Google Scholar]
- 47.Plante H, Picard S, Mancini J, Borgeat P. 5-Lipoxygenase-activating protein homodimer in human neutrophils: evidence for a role in leukotriene biosynthesis. Biochem J. 2006;393:211–218. doi: 10.1042/BJ20060669. [DOI] [PMC free article] [PubMed] [Google Scholar]